
Abstract
Most genetic susceptibility to cutaneous melanoma remains to be discovered. Meta-analysis genome-wide association study (GWAS) of 36,760 cases of melanoma (67% newly genotyped) and 375,188 controls identified 54 significant (P < 5 × 10−8) loci with 68 independent single nucleotide polymorphisms. Analysis of risk estimates across geographical regions and host factors suggests the acral melanoma subtype is uniquely unrelated to pigmentation. Combining this meta-analysis with GWAS of nevus count and hair color, and transcriptome association approaches, uncovered 31 potential secondary loci for a total of 85 cutaneous melanoma susceptibility loci. These findings provide insights into cutaneous melanoma genetic architecture, reinforcing the importance of nevogenesis, pigmentation and telomere maintenance, together with identifying potential new pathways for cutaneous melanoma pathogenesis.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
Genome-wide summary statistics for the confirmed meta-analysis have been made publicly available at dbGaP (phs001868.v1.p1), with the exclusion of self-reported data from 23andMe and UK Biobank. Results for SNPs with a fixed or random P < 5 × 10−7 from the total meta-analysis are reported in Supplementary Table 7. The total meta-analysis includes self-reported cutaneous melanoma GWAS data from the UK Biobank and 23andMe. The raw genetic and phenotypic UK Biobank data used in this study, which were used under license, are available from: http://www.ukbiobank.ac.uk/. The genome-wide summary statistics from 23andMe data were obtained under a data transfer agreement. Further information about obtaining access to the 23andMe summary statistics is available from https://research.23andme.com/collaborate/. Source data for Fig. 2, Extended Data Figs. 4–6 and Supplementary Figs. 2 and 3 are available with the paper.
References
Karimkhani, C. et al. The global burden of melanoma: results from the global burden of disease study 2015. Br. J. Dermatol. 177, 134–140 (2017).
Secretan, B. et al. WHO International agency for research on cancer monograph working group a review of human carcinogens—Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 10, 1033–1034 (2009).
Ford, D. et al. Risk of cutaneous melanoma associated with a family history of the disease. Int. J. Cancer 62, 377–381 (1995).
Olsen, C. M., Carroll, H. J. & Whiteman, D. C. Familial melanoma: a meta-analysis and estimates of attributable fraction. Cancer Epidemiol. Biomark. Prev. 19, 65–73 (2010).
Olsen, C. M., Carroll, H. J. & Whiteman, D. C. Estimating the attributable fraction for melanoma: a meta-analysis of pigmentary characteristics and freckling. Int. J. Cancer 127, 2430–2445 (2010).
Chang, Y. M. et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int. J. Cancer 124, 420–428 (2009).
Olsen, C. M., Carroll, H. J. & Whiteman, D. C. Estimating the attributable fraction for cancer: a meta-analysis of nevi and melanoma. Cancer Prev. Res. 3, 233–245 (2010).
Bataille, V. et al. Nevus size and number are associated with telomere length and represent potential markers of a decreased senescence in vivo. Cancer Epidemiol. Biomark. Prev. 16, 1499–1502 (2007).
Han, J. et al. A prospective study of telomere length and the risk of skin cancer. J. Invest. Dermatol. 129, 415–421 (2009).
Green, A. C. & Olsen, C. M. Increased risk of melanoma in organ transplant recipients: systematic review and meta-analysis of cohort studies. Acta Derm. Venereol. 95, 923–927 (2015).
Kamb, A. et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet. 8, 23–26 (1994).
Berwick, M. et al. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol. Biomark. Prev. 15, 1520–1525 (2006).
Robles-Espinoza, C. D. et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 46, 478–481 (2014).
Shi, J. et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 46, 482–486 (2014).
Palmer, J. S. et al. Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 66, 176–186 (2000).
Landi, M. T. et al. MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J. Natl. Cancer Inst. 98, 144–145 (2005).
Brown, K. M. et al. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat. Genet. 40, 838–840 (2008).
Bishop, D. T. et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 41, 920–925 (2009).
Amos, C. I. et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 20, 5012–5023 (2011).
Barrett, J. H. et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 43, 1108–1113 (2011).
Macgregor, S. et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat. Genet. 43, 1114–1118 (2011).
Iles, M. M. et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet. 45, 428–432 (2013).
Law, M. H. et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 47, 987–995 (2015).
Ransohoff, K. J. et al. Two-stage genome-wide association study identifies a novel susceptibility locus associated with melanoma. Oncotarget 8, 17586–17592 (2017).
Yokoyama, S. et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480, 99–103 (2011).
Bertolotto, C. et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480, 94–98 (2011).
Duffy, D. L. et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat. Commun. 9, 4774 (2018).
Zhang, T. et al. Cell-type-specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res. 28, 1621–1635 (2018).
Elder, D. E., Massi, D., Willemze, R. & Scolyer, R. WHO Classification of Skin Tumours (International Agency for Research on Cancer, 2018).
Higgins, J. P. T. & Thompson, S. G. Quantifying heterogeneity in a meta-analysis. Stat. Med. 21, 1539–1558 (2002).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Zhang, Y., Qi, G., Park, J.-H. & Chatterjee, N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326 (2018).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Duffy, D. L. et al. Multiple pigmentation gene polymorphisms account for a substantial proportion of risk of cutaneous malignant melanoma. J. Invest. Dermatol. 130, 520–528 (2010).
Duffy, D. L. et al. IRF4 variants have age-specific effects on nevus count and predispose to melanoma. Am. J. Hum. Genet. 87, 6–16 (2010).
Delgado, D. A. et al. Genome-wide association study of telomere length among South Asians identifies a second RTEL1 association signal. J. Med. Genet. 55, 64–71 (2018).
Iles, M. M. et al. The effect on melanoma risk of genes previously associated with telomere length. J. Natl. Cancer Inst. 106, dju267 (2014).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
GTEx Consortium Human genomics: the genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Visconti, A. et al. Genome-wide association study in 176,678 Europeans reveals genetic loci for tanning response to sun exposure. Nat. Commun. 9, 1684 (2018).
Choi, J. et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nat. Genet. 49, 1326–1335 (2017).
Li, F. P. & Fraumeni, J. F. Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 71, 747–752 (1969).
Curiel-Lewandrowski, C., Speetzen, L. S., Cranmer, L., Warneke, J. A. & Loescher, L. J. Multiple primary cutaneous melanomas in Li–Fraumeni syndrome. Arch. Dermatol. 147, 248–250 (2011).
Beausejour, C. M. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 22, 4212–4222 (2003).
Kuilman, T., Michaloglou, C., Mooi, W. J. & Peeper, D. S. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Aoude, L. G. et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J. Natl. Cancer Inst. 107, dju408 (2015).
Rafnar, T. et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet. 41, 221–227 (2009).
Rachakonda, S. et al. Telomere length, telomerase reverse transcriptase promoter mutations, and melanoma risk. Genes Chromosomes Cancer 57, 564–572 (2018).
Derheimer, F. A. & Kastan, M. B. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 584, 3675–3681 (2010).
Demenais, F. et al. A linkage study between HLA and cutaneous malignant melanoma or precursor lesions or both. J. Med. Genet. 21, 429–435 (1984).
Bale, S. J. et al. Hereditary malignant melanoma is not linked to the HLA complex on chromosome 6. Int. J. Cancer 36, 439–443 (1985).
Holland, E. A., Beaton, S. C., Kefford, R. F. & Mann, G. J. Linkage analysis of familial melanoma and chromosome 6 in 14 Australian kindreds. Genes Chromosomes Cancer 19, 241–249 (1997).
Barger, B. O., Acton, R. T., Soong, S. J., Roseman, J. & Balch, C. Increase of HLA-DR4 in melanoma patients from Alabama. Cancer Res. 42, 4276–4279 (1982).
Rovini, D., Sacchini, V., Codazzi, V., Vaglini, M. & Illeni, M. T. HLA antigen frequencies in malignant melanoma patients. A second study. Tumori 70, 29–33 (1984).
Hors, J. et al. in Histocompatibility Testing 1984 (ed. Albert, E. D.) 407–410 (Springer, 1984).
Lee, J. E., Reveille, J. D., Ross, M. I. & Platsoucas, C. D. HLA-DQB1* 0301 association with increased cutaneous melanoma risk. Int. J. Cancer 59, 510–513 (1994).
Muto, M. et al. HLA class I polymorphism and the susceptibility to malignant melanoma. Tissue Antigens 47, 447–449 (1996).
Kageshita, T. et al. Molecular genetic analysis of HLA class II alleles in Japanese patients with melanoma. Tissue Antigens 49, 466–470 (1997).
Bateman, A. C., Turner, S. J., Theaker, J. M. & Howell, W. M. HLA-DQB1*0303 and *0301 alleles influence susceptibility to and prognosis in cutaneous malignant melanoma in the British Caucasian population. Tissue Antigens 52, 67–73 (1998).
Lombardi, M. L. et al. Molecular analysis of HLA DRB1 and DQB1 polymorphism in Italian melanoma patients. J. Immunother. 21, 435–439 (1998).
Luongo, V. et al. HLA allele frequency and clinical outcome in Italian patients with cutaneous melanoma. Tissue Antigens 64, 84–87 (2004).
Campillo, J. A. et al. HLA class I and class II frequencies in patients with cutaneous malignant melanoma from southeastern Spain: the role of HLA-C in disease prognosis. Immunogenetics 57, 926–933 (2006).
Planelles, D. et al. HLA class II polymorphisms in Spanish melanoma patients: homozygosity for HLA-DQA1 locus can be a potential melanoma risk factor. Br. J. Dermatol. 154, 261–266 (2006).
Jin, Y. et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 48, 1418–1424 (2016).
Jin, Y. et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 44, 676–680 (2012).
Curran, K. et al. Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev. Biol. 344, 107–118 (2010).
Thomas, A. J. & Erickson, C. A. FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development 136, 1849–1858 (2009).
Kumano, K. et al. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 21, 70–78 (2008).
Schouwey, K., Larue, L., Radtke, F., Delmas, V. & Beermann, F. Transgenic expression of notch in melanocytes demonstrates RBP-Jkappa-dependent signaling. Pigment Cell Melanoma Res. 23, 134–136 (2010).
Zabierowski, S. E. et al. Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells 29, 1752–1762 (2011).
Falchi, M. et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat. Genet. 41, 915–919 (2009).
Garraway, L. A. et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122 (2005).
Abel, E. V. & Aplin, A. E. FOXD3 is a mutant B-RAF-regulated inhibitor of G(1)-S progression in melanoma cells. Cancer Res. 70, 2891–2900 (2010).
Weiss, M. B., Abel, E. V., Dadpey, N. & Aplin, A. E. FOXD3 modulates migration through direct transcriptional repression of TWIST1 in melanoma. Mol. Cancer Res. 12, 1314–1323 (2014).
Golan, T. et al. Interactions of melanoma cells with distal keratinocytes trigger metastasis via notch signaling inhibition of MITF. Mol. Cell 59, 664–676 (2015).
Cronin, J. C. et al. SOX10 ablation arrests cell cycle, induces senescence, and suppresses melanomagenesis. Cancer Res. 73, 5709–5718 (2013).
Guilford, P. et al. E-cadherin germline mutations in familial gastric cancer. Nature 392, 402–405 (1998).
Hansford, S. et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 1, 23–32 (2015).
Kim, H. C. et al. The E-cadherin gene (CDH1) variants T340A and L599V in gastric and colorectal cancer patients in Korea. Gut 47, 262–267 (2000).
Tang, A. et al. E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J. Cell Sci. 107(Pt 4), 983–992 (1994).
Hsu, M. Y., Wheelock, M. J., Johnson, K. R. & Herlyn, M. Shifts in cadherin profiles between human normal melanocytes and melanomas. J. Investig. Dermatol. Symp. Proc. 1, 188–194 (1996).
Study, C. et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 40, 1426–1435 (2008).
Wagner, R. Y. et al. Altered E-cadherin levels and distribution in melanocytes precede clinical manifestations of vitiligo. J. Invest. Dermatol. 135, 1810–1819 (2015).
Padmanaban, V. et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444 (2019).
Peña-Chilet, M. et al. Genetic variants in PARP1 (rs3219090) and IRF4 (rs12203592) genes associated with melanoma susceptibility in a Spanish population. BMC Cancer 13, 160 (2013).
Law, M. H. et al. Meta-analysis combining new and existing data sets confirms that the TERT-CLPTM1L locus influences melanoma risk. J. Invest. Dermatol. 132, 485–487 (2012).
Antonopoulou, K. et al. Updated field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma: the MelGene database. J. Invest. Dermatol. 135, 1074–1079 (2015).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
DerSimonian, R. & Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 7, 177–188 (1986).
Machiela, M. J. & Chanock, S. J. LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 34, 887–889 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
DeLuca, D. S. et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 99, 1245–1260 (2016).
Stacey, S. N. et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 43, 1098–1103 (2011).
Chahal, H. S. et al. Genome-wide association study identifies 14 novel risk alleles associated with basal cell carcinoma. Nat. Commun. 7, 12510 (2016).
Ostrom, Q. T. et al. Sex-specific glioma genome-wide association study identifies new risk locus at 3p21.31 in females, and finds sex-differences in risk at 8q24.21. Sci. Rep. 8, 7352 (2018).
Melin, B. S. et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 49, 789–794 (2017).
Zhou, Y., Wu, H., Zhao, M., Chang, C. & Lu, Q. The Bach family of transcription factors: a comprehensive review. Clin. Rev. Allergy Immunol. 50, 345–356 (2016).
Milovic-Holm, K., Krieghoff, E., Jensen, K., Will, H. & Hofmann, T. G. FLASH links the CD95 signaling pathway to the cell nucleus and nuclear bodies. EMBO J. 26, 391–401 (2007).
Acknowledgements
NCI: This study was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute (NCI), National Institutes of Health (NIH) and Department of Health and Human Services (DHHS). AOCS/OCAC/SEARCH: AOCS/OCAC/SEARCH is accessible via European Genome–Phenome Archive. We acknowledge their support and data, and the contribution of the study nurses, research assistants and all clinical and scientific collaborators in generation of these data. We also acknowledge their funding sources: OCAC (NIH grant no. U19CA148112), SEARCH team (Cancer Research UK grant no.C490/A16561), AOCS (US Army Medical Research and Material Command under grant no. DAMD17‐01‐1‐0729, The Cancer Council Victoria, Queensland Cancer Fund, The Cancer Council New South Wales, The Cancer Council South Australia, The Cancer Foundation of Western Australia, The Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC) (grant nos. ID400413 and ID400281, as well as support from S. Boldeman, the Agar family, Ovarian Cancer Action (UK), Ovarian Cancer Australia and the Peter MacCallum Foundation). MelaNostrum Consortium: We thank the participants of the MelaNostrum Consortium from Italy (Genoa, L’Aquila, Rome, Padua, Milan, Florence and Bergamo), Spain (Valencia and Barcelona), Greece (Athens) and Cyprus (Nicosia) who provided data and biospecimens for this study. The Consortium is partially supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, NCI, NIH, DHHS. Funding for the University of Genoa and Genetics of Rare Cancers, Ospedale Policlinico San Martino came from Italian Ministry of Health 5 × 1000 per la Ricerca Corrente to Ospedale Policlinico San Martino and AIRC IG 15460. The research at the Melanoma Unit in Barcelona was supported by the Spanish Fondo de Investigaciones Sanitarias grant nos. PI15/00716 and PI15/00956 cofinanced by FEDER ‘Una manera de hacer Europa’; CIBER de Enfermedades Raras of the Instituto de Salud Carlos III, Spain, cofinanced by European Development Regional Fund ‘A way to achieve Europe’ ERDF; AGAUR 2014_SGR_603 of the Catalan Government, Spain; European Commission, contract no. LSHC-CT-2006-018702 (GenoMEL) and by the European Commission under the 7th Framework Programme, Diagnostics; ‘Fundació La Marató de TV3’ grant no. 201331-30, Catalonia, Spain; ‘Fundación Científica de la Asociación Española Contra el Cáncer’ grant no. GCB15152978SOEN, Spain, and CERCA Programme/Generalitat de Catalunya. Melanoma research at the Department of Dermatology, University of L’Aquila, Italy was supported by the Italian Ministry of the University and Scientific Research (PRIN-2012 grant no. 2012JJX494). Q-MEGA/QTWIN: The Q-MEGA/QTWIN study was supported by the Melanoma Research Alliance, the NIH NCI (grant nos. CA88363, CA83115, CA122838, CA87969, CA055075, CA100264, CA133996 and CA49449), the NHMRC (grant nos. 200071, 241944, 339462, 380385, 389927, 389875, 389891, 389892,389938, 443036, 442915, 442981, 496610, 496675, 496739, 552485, 552498 and APP1049894), the Cancer Councils New South Wales, Victoria and Queensland, the Cancer Institute New South Wales, the Cooperative Research Centre for Discovery of Genes for Common Human Diseases, Cerylid Biosciences (Melbourne), the Australian Cancer Research Foundation, The Wellcome Trust (grant no. WT084766/Z/08/Z) and donations from N. and S. Hawkins. S. MacGregor acknowledges fellowship support from the Australian National Health and Medical Research Council and from the Australian Research Council.
Please see the Supplementary Note for additional acknowledgments.
Author information
Authors and Affiliations
Consortia
Contributions
M.T.L., M.M.I. and M.H.L. conceptualized and designed the project. D.T.B., S. MacGregor, M.T.L. and S.J.C. provided funding support. M.T.L., D.T.B., S. MacGregor, M.J.M., J.S., M.M.I. and M.H.L. interpreted the results and supervized the study. M.J.M., M.T.L., M.M.I., K.B., J.C. and M.H.L. wrote the manuscript. J.S., M.M.I., K.B., T.Z., J.C., D.L.D. and M.H.L. analyzed the data. A.J. Stratigos, P. Ghiorzo, S.P. and E.N. coordinated the study and collected the data. M.T.L., D.T.B., S. MacGregor, M.J.M., A.J. Stratigos, P. Ghiorzo, M.B., D.C., J.C., M.C.F., T.Z., M.R., A.J.T., C.M., J. Martinez, A. Hadjisavvas, L.S., I.S., R.S., X.R.Y., A.M.G., M.P., K.P.K., L.P., P.Q., C.P., L.C., M.Z., P. Gimenez-Xavier, A.R., L.E., S. Manoukian, L.R., B.H.S., M.A.L., L.D.R., D.M., M. Mandala, K.K., L.A.A., C.I.A., P.A.A., M.A., E.A., H.P.S., V.B., B.D., L.M.B., K.P.B., W.V.C., V.C., J.E.C., T.D., M.F., S.F., E.F., S.S., P. Galan, Z.G., E.M.G., S.G., A.G., N.A.G., J. Hansson, M. Harland, J. Harris, P.H., A. Henders, M. Hočevar, V.H., D.H., C.I., R.K., J. Lang, G.M.L., J.E.L., X.L., J. Lubiński, R.M.M., M. Malt, J. Malvehy, K.M., H.M., A.M., E.K.M., R.E.N., S.N., D.R.N., H.O., N.O., L.G.F., J.A.P., A.A.Q., G.L.R., J.R., C. Requena, C. Rowe, N.J.S., M. Sanna, D.S., H.S., L.A.S., M. Smithers, F.S., A.J. Swerdlow, N.V.D.S., N.A.K., A. Visconti, L. Wallace, S.V.W., L. Wheeler, R.A.S., A. Hutchinson, K.J., M. Malasky, A. Vogt, W.Z., K.A.P., D.E.E., J. Han, B.H., N.K.H., P.A.K., C.B., G.W.M., C.M.O., C.H., A.M.D., N.G.M., E.E., G.J.M., G.L., P.D.P.P., D.F.E., J.H.B., A.E.C., G.A., D.L.D., D.C.W., H.G., A.D.N., M.A.T., J.A.N.B., K.P., S.J.C., K.M.B., F.D., S.P., E.N., J.S., M.M.I. and M.H.L. participated in data collection, results interpretation and manuscript review.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quantile-Quantile plot of total CM meta-analysis.
Quantile-quantile plots of negative log10 two-sided P value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS listed in Supplementary Table 1. All confirmed and self-report cases are included, with a total sample size of 36,760 melanoma cases and 375,188 controls.
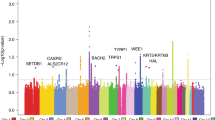
Extended Data Fig. 2 Manhattan plots of melanoma risk loci from total and confirmed-only GWAS meta-analyses.
Negative log10 two-sided P value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS (y-axis) are plotted by their chromosome position. The confirmed-only analysis included 30,134 cases with histopathologically confirmed CM, and 81,415 controls. The total CM meta-analysis includes all confirmed and self-report cases, with a total sample size of 36,760 CM cases and 375,188 controls. Multiple-testing corrected genome-wide significance threshold was P<5×10−8. We display in order the total CM meta-analysis without limiting the y-axis; the pathologically confirmed CM cases only meta-analysis with the y-axis limited to 1×10−25 and without a limit to more clearly display loci other than MC1R.
Extended Data Fig. 3 Quantile-Quantile plot of confirmed-only CM meta-analysis.
Quantile-quantile plots of negative log10 two-sided P value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS listed in Supplementary Table 1. Only cases with histopathologically confirmed CM are included, with a total sample size of 30,134 melanoma cases and 81,415 controls.
Extended Data Fig. 4 Distribution of pigmentation polygenic risk scores across melanoma histological subtypes.
The figure shows whether PRS defined based on SNPs associated with hair color differ across CM histological types (Methods; SSM: superficial spreading melanoma; NM: nodular melanoma; LM: lentigo melanoma; Acral: acral lentiginous melanoma). The higher the PRS the lighter the hair color. When comparing subtype 1 vs. subtype 2, we report the effect size for the linear regression of PRS on subtype 1, including study and principal components as covariates to control for population stratification. The regression coefficient, 95% confidence interval, and statistical significance are shown. The positive beta indicates the PRS is higher in subtype 2 (for example, nonacral melanomas). This analysis included 9828 SSM, 2137 NM, 900 LM, 353 acral melanoma cases and 44676 controls. Two-sided t-statistic was used for testing significance. P values reported were not adjusted for multiple comparison.
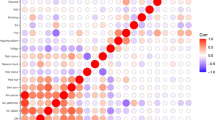
Extended Data Fig. 5 LD score regression plots.
LD score regression was performed for the top 4000 (A) 2000 (B) and 1000 (C) tissue-specific genes from melanocyte and GTEx tissue types (v7 datasets), to assess the enrichment of melanoma heritability in these genomic regions using summary statistics from Total CM GWAS meta-analysis. The level of enrichment and P values are shown, with an FDR = 0.05 cutoff marked as a dashed horizontal line (See Methods for statistical test). Tissue categories are color-coded, and a subset of top individual tissue types are shown on the plot. Tissue types from “Skin” category including melanocytes are highlighted in magenta.
Extended Data Fig. 6 Effect sizes for confirmed-only meta-analysis versus UKBB self-report set.
For each independent genome-wide significant (P<5×10−8) lead SNP from the confirmed-only meta-analysis (30,134 melanoma cases and 81,415 controls), we plot on the Y-axis UK Biobank self-report GWAS (UKBB SR) log(OR) and standard error from a logistic regression GWAS (1,802 self-report CM cases and 7,208 controls) and on the X-axis we plot the log(OR) and standard error from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS for confirmed melanoma cases listed in Supplementary Table 1. We also report the r2 correlation from the linear regression of UKBB SR log(OR) on the confirmed met-analysis estimates, weighted by their standard error.
Supplementary information
Supplementary Information
Supplementary Note and Figs. 1–9
Supplementary Data 1
P value and regional LD for each lead SNP from Supplementary Table 3. Regional association P values and LD patterns for identified CM susceptibility regions. Top panel plots –log10 association P values from the total CM fixed-effects meta-analysis. Blue dot is the most significant regional variant and points above blue line indicate association P < 5× 10−8. Darker shades of red indicate higher LD with most significant regional variant. Bottom panel displays nearby genes. Legend applies to all subsequent plots.
Source data
Source Data Fig. 2
Statistical Source Data for Figure 2: Overlap of loci identified by primary and secondary analyses. Positions for each locus and 0,1 value for detected in the listed analyses as per Fig. 2.
Source Data Extended Data Fig. 4
Statistical source data
Source Data Extended Data Fig. 5
Statistical source data
Source Data Extended Data Fig. 6
Statistical source data
Source Data Supplementary Fig. 2
Statistical source data. A1 is the effect allele for the listed BETA etc.
Source Data Supplementary Fig. 3
Statistical source data
Rights and permissions
About this article

Cite this article
Landi, M.T., Bishop, D.T., MacGregor, S. et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet 52, 494–504 (2020). https://doi.org/10.1038/s41588-020-0611-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-020-0611-8
This article is cited by
-
FORGEdb: a tool for identifying candidate functional variants and uncovering target genes and mechanisms for complex diseases
Genome Biology (2024)
-
Genetic predisposition to childhood obesity does not influence the risk of developing skin cancer in adulthood
Scientific Reports (2024)
-
Polygenic risk scores, radiation treatment exposures and subsequent cancer risk in childhood cancer survivors
Nature Medicine (2024)
-
Colitis-associated carcinogenesis: crosstalk between tumors, immune cells and gut microbiota
Cell & Bioscience (2023)
-
Increase in power by obtaining 10 or more controls per case when type-1 error is small in large-scale association studies
BMC Medical Research Methodology (2023)
