
Abstract
Fruit characteristics of sweet watermelon are largely the result of human selection. Here we report an improved watermelon reference genome and whole-genome resequencing of 414 accessions representing all extant species in the Citrullus genus. Population genomic analyses reveal the evolutionary history of Citrullus, suggesting independent evolutions in Citrullus amarus and the lineage containing Citrullus lanatus and Citrullus mucosospermus. Our findings indicate that different loci affecting watermelon fruit size have been under selection during speciation, domestication and improvement. A non-bitter allele, arising in the progenitor of sweet watermelon, is largely fixed in C. lanatus. Selection for flesh sweetness started in the progenitor of C. lanatus and continues through modern breeding on loci controlling raffinose catabolism and sugar transport. Fruit flesh coloration and sugar accumulation might have co-evolved through shared genetic components including a sugar transporter gene. This study provides valuable genomic resources and sheds light on watermelon speciation and breeding history.
Similar content being viewed by others


Main
Watermelon (Citrullus lanatus, 2n = 2 × = 22) is one of the most popular fruit crops worldwide. It belongs to the Citrullus genus of the Cucurbitaceae family, and originated in Africa1. Watermelon has been domesticated for more than 4,000 years, and has been improved by domestication and modern breeding from wild watermelons with small fruits harboring hard, pale-colored and bitter- or bland-tasting flesh, into modern sweet watermelons carrying large fruits with crisp sweet and red flesh and a thin rind2. A watermelon fruit captured in a seventeenth-century painting by Italian artist Giovanni Stanchi displayed unevenly colored pinkish flesh, a thick rind and many dark-colored seeds, which may represent a sweet watermelon in the midst of its domestication. Modern breeding of sweet watermelon has focused primarily on fruit quality traits, such as sugar content, flesh color and rind pattern, resulting in a narrow genetic base of sweet watermelon3. How natural and human selections leading to marked phenotypic changes have shaped the watermelon genome remains largely unknown.
The genus Citrullus contains seven extant species4. The only diecious and most morphologically unique species, Citrullus naudinianus, is commonly found in sub-Saharan Africa3,5. Citrullus ecirrhosus and Citrullus rehmii are adapted to a desert environment and are endemic to southern Africa. Citrullus colocynthis is grown for its medicinal properties and seed oil, and is widely distributed in northern Africa and southwestern and central Asia2. The wild form of Citrullus amarus can be found in southern Africa, and the cultivated types are grown throughout the Mediterranean region, where they are used for jam and animal fodder and as a source of water5. Citrullus mucosospermus is mainly grown for seed consumption and is now distributed in western Africa5. In addition, C. colocynthis, C. amarus and C. mucosospermus have been used in breeding programs to identify new sources of disease and pest resistance for the improvement of sweet watermelon3.
Weak reproductive barriers have obscured the taxonomy of Citrullus species2,4. Previous studies aiming to elucidate the relationship between the seven Citrullus species are mainly based on genetic diversity present at a limited number of nuclear and plastid loci4,5. A high-resolution genome variation map is essential for a better understanding of the evolution and divergence of Citrullus species. In this study, we first assembled an improved genome sequence of the watermelon cultivar ‘97103’ using PacBio long reads combined with BioNano optical and Hi-C chromatin interaction maps. We then resequenced the genomes of 414 watermelon accessions representing all seven extant Citrullus species and performed population genomic analyses and genome-wide association studies (GWAS) for several important fruit quality traits. Our study identifies a number of candidate loci associated with fruit quality traits and provides insights into the speciation and domestication of the modern sweet watermelon.
Results
An improved watermelon reference genome
The genome of watermelon cultivar ‘97103’ was previously assembled using Illumina short reads6. To improve its quality, we assembled the ‘97103’ genome de novo using PacBio long reads, combined with BioNano optical and Hi-C chromatin interaction maps. In total, 20.3 Gb of PacBio sequences were generated with an N50 length of 10.8 kb, covering approximately 47.2× of the watermelon genome. The PacBio assembly had a total size of 359.8 Mb, containing 367 contigs with an N50 size of 2.3 Mb. In total, 410.7 Gb cleaned BioNano optical map data were generated and assembled de novo into BioNano genome maps, which were used to connect PacBio assembled contigs, resulting in 149 scaffolds with an N50 size of 21.9 Mb and a cumulative length of 365.1 Mb. Furthermore, approximately 135.2 million cleaned Hi-C reads were generated, of which roughly 92.1 million (68.1%) were uniquely mapped to the assembly, resulting in a final set of approximately 69.5 million valid read pairs that were used to generate contact information (Supplementary Table 1). The Hi-C contact information, combined with previously published genetic maps7,8,9, was used to order and orient the scaffolds into chromosome-scale pseudomolecules. Finally, 31 scaffolds with a total size of 362.7 Mb (99.3% of the assembly) were clustered into 11 chromosomes ranging from 27.1 to 37.9 Mb in length (Extended Data Figs. 1–3). Comparison with the previous assembly of ‘97103’ suggested high collinearity between the two assemblies (Extended Data Fig. 4). Comprehensive assessment indicated that the quality of this new assembly was high and substantially improved compared to the previous assembly (Supplementary Note, Supplementary Tables 2,3 and Extended Data Figs. 5–7). Approximately 55.55% of the assembly was annotated as repeat sequences, a substantially higher percentage than in the previous assembly (46.60%) (Supplementary Table 4), and 22,596 high-confidence genes were predicted in the assembly (Supplementary Note). This much improved ‘97103’ genome provides a robust reference for watermelon research and genetic improvement.
Genome variation map and phylogeny of Citrullus species
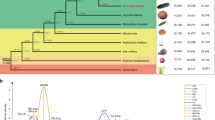
In total, 414 Citrullus accessions collected in various geographic regions (Fig. 1a) were selected for genome resequencing, including 15 C. colocynthis, 31 C. amarus, 19 C. mucosospermus, 345 C. lanatus (258 cultivars and 87 landraces), two C. rehmii, one C. ecirrhosus and one C. naudinianus accessions (Supplementary Table 5). These accessions were sequenced to an average depth of 14.5× and coverage of 92.2% of the ‘97103’ genome. In total, 19,725,853 SNPs were identified, of which 1,100,803 were located in coding regions, causing 502,028 nonsynonymous mutations, 589,735 synonymous mutations, 1,031 start codon changes and 6,808 stop codon changes. Furthermore, 6,675,290 small indels were identified, of which 56,115 were located in coding regions.

a, Geographic distribution of resequenced Citrullus accessions. The diameter of the circle is proportional to the number of accessions, maximized at the size for 100 accessions. The world map was generated using the R package ‘rworldmap’ (v1.3-6, https://cran.r-project.org/web/packages/rworldmap/index.html). b, Neighbor-joining phylogenetic tree of Citrullus accessions and model-based clustering with K from 2 to 4. Colors of branches in the tree indicate different species (matching the colors shown in a). Two C. lanatus accessions from Sudan located in the deepest branch of the C. lanatus clade are indicated by the arrow. c, Principal component analysis of Citrullus accessions excluding C. naudinianus (left), and of C. mucosospermus and C. lanatus accessions (right). PC1, first principal component; PC2, second principal component. d, Schematic representation of Patterson’s D test of gene flow between Citrullus species. Red arrows represent gene flow between lineages. P values for significant deviations of D from zero are shown near the dots representing D values. The bars represent standard errors. CA, C. amarus; CC, C. colocynthis; CE, C. ecirrhosus; CL_CUL, C. lanatus cultivar; CL_LR, C. lanatus landrace; CM, C. mucosospermus; CN, C. naudinianus.
Phylogenetic relationships between the Citrullus accessions were inferred using 89,914 SNPs at fourfold degenerate sites. The placement of the seven species in the phylogenetic tree (Fig. 1b) was largely consistent with the previously reported phylogeny4,5, with the most morphologically distinct Citrullus species, C. naudinianus, sister to the other six species, followed by C. colocynthis and C. rehmii. However, C. ecirrhosus was sister to C. amarus, C. mucosospermus and C. lanatus, instead of being most closely related to C. amarus, as proposed previously4. Two C. lanatus accessions collected in Sudan (PI 481871 and PI 254622) were placed in the deepest branch of the C. lanatus clade (Fig. 1b), supporting the idea that the primitive watermelons from Sudan and neighboring countries of northeastern Africa may be the closest to the progenitor of the sweet watermelon2,5,10. Twelve accessions were clustered into unexpected species groups and were therefore excluded from downstream analyses (Supplementary Table 5).
Phylogenetic and population structure inferences revealed a close relationship and shared ancestry between C. mucosospermus and C. lanatus (Fig. 1b,c and Extended Data Fig. 8), suggesting that they were derived from the same ancestral population and perhaps domesticated for different purposes: one for seed consumption and the other for fruit flesh. Furthermore, the fixation index (FST) between C. mucosospermus and C. lanatus was only 0.299, whereas the pairwise FST between C. amarus and the other species ranged from 0.509 to 0.686 (Extended Data Fig. 9). Gene flow analysis further suggested admixture between C. mucosospermus and C. lanatus (Fig. 1d). Gene flow was also present between C. ecirrhosus and C. mucosospermus, and between C. ecirrhosus and C. amarus, whereas no significant gene flow was detected between C. amarus and C. mucosospermus (Fig. 1d). Combined with previous findings on genome organization differences between C. amarus and C. lanatus shown by ribosomal DNA chromosome landmarks6,11 and indicated by non-Mendelian segregation in genetic populations derived from crosses between C. amarus and C. lanatus12, these results suggest that C. amarus and the lineage including C. mucosospermus and C. lanatus might have been derived from different ancestral populations or evolved independently after divergence.
Nucleotide diversities (π) in four Citrullus species with multiple accessions were estimated. C. colocynthis (π = 6.75 × 10−3) and C. amarus (π = 2.28 × 10−3) had much greater nucleotide diversity than C. mucosospermus (π = 0.792 × 10−3) and C. lanatus landraces (π = 0.56 × 10−3) and cultivars (π = 0.548 × 10−3). This suggests that C. colocynthis and C. amarus, which can be intercrossed with C. lanatus, are valuable resources for expanding the genetic base for watermelon improvement. The linkage disequilibrium (LD) decay rates were highest in C. colocynthis and C. amarus, followed by C. mucosospermus and C. lanatus landraces, and lowest in C. lanatus cultivars (Extended Data Fig. 10), correlating with level of nucleotide diversity. The LD extended further in C. lanatus cultivars than in landraces, suggesting a possible bottleneck during the improvement of sweet watermelon.
GWAS
The variation map at single-base resolution empowered GWAS for seven important fruit quality traits in watermelon (Supplementary Table 6). In total, 43 association signals were identified, of which eight overlapped with previously identified QTLs. A peak strongly associated with flesh sweetness (measured by soluble solid content (SSC)) was identified within the previously reported QTL, QBRX2-1 (ref. 9), which harbors the sugar transporter gene ClTST2 (Cla97C02G036390, Fig. 2a). Two additional regions strongly associated with flesh sweetness were found on chromosome 10 (Fig. 2b), in agreement with previous GWAS and QTL studies13,14. These two regions contained the sucrose synthase gene Cla97C10G194010 and the raffinose synthase gene Cla97C10G196740, which contribute to the synthesis of sucrose and raffinose, respectively, the dominant metabolites transported in watermelon fascicular phloem tissues15. Two signals significantly associated with flesh color were detected on chromosomes 2 and 4 (Fig. 2c), with the one on chromosome 4 overlapping with the flesh color QTL FC4.1 (ref. 16) and harboring a lycopene β-cyclase gene (LCYB, Cla97C04G070940). In total, 14 signals associated with fruit shape were detected, with the strongest signal near the ClFS1 (Cla97C03G066390) gene, which is known to control fruit elongation17, and overlapping with fruit shape QTLs Qfsi3, FSI3.1 and FSI3.2 (refs. 9,16) (Fig. 2d). Three peaks highly associated with rind color and rind stripe were found on chromosomes 4, 6 and 8, corresponding to the rind trait loci, Dgo, S and D, respectively18 (Fig. 2e,f). Candidate genes in these peaks included Cla97C08G161570, which encodes a chloroplastic 2-phytyl-1,4-beta-naphthoquinone methyltransferase that is required for the conversion of 2-phytyl-1,4-beta-naphthoquinol to phylloquinone in photosystem I (ref. 19), and Cla97C04G068530, which encodes a magnesium-chelatase subunit H involved in chlorophyll synthesis20. The strongest signal associated with rind stripe was found in a WD40-repeat gene, Cla97C06G126710. In total, 13 regions were found to be associated with seed coat color (Fig. 2g). The strongest associated SNP on chromosome 3 overlapped with the seed coat color QTL qrc-c8-1 (ref. 21) and was located in Cla97C03G057100, which encodes a polyphenol oxidase that polymerizes o-quinones to produce black, brown or red pigments22.

a–f, Manhattan plots of GWAS of sugar content using the watermelon population grown in Xinxiang (a) and Yanqing (b), and Manhattan plots of GWAS of flesh color (c), fruit shape (d), rind stripe (e), rind color (f) and seed color (g). For sugar content, phenotypic data of the population grown in Xinxiang and the population grown in Yanqing were analyzed separately for GWAS, whereas for other traits, phenotypic data from the two populations were combined for the analyses. Known QTL regions are highlighted in light green. Significant GWAS signals are indicated by vertical black arrows. Candidate genes are indicated by diagonal arrows. Gray horizontal dashed lines indicate the Bonferroni-corrected significance thresholds of GWAS (α = 0.05 and α = 0.01, respectively). CHLH, magnesium-chelatase subunit H; ClFS1, FRUIT SHAPE 1 (encoding an IQ domain protein); ClTST2, tonoplast sugar transporter; LCYB, lycopene β-cyclase; MENG, chloroplastic 2-phytyl-1,4-beta-naphthoquinone methyltransferase; PPO, polyphenol oxidase; RFS, raffinose synthase; SUS, sucrose synthase; WDR, WD40-repeat protein.
We then investigated the expression of these candidate genes in the flesh and rind of ‘97103’ and in the flesh of a wild watermelon, C. amarus ‘PI 296341-FR’, during their fruit development using RNA sequencing (RNA-seq) data that we generated previously23. Except Cla97C03G057100, which was associated with seed color, all other genes were abundantly expressed in the flesh and rind, with those associated with flesh sweetness being expressed at much higher levels in the flesh of ‘97103’ than in that of ‘PI 296341-FR’, further supporting their potential roles in these fruit quality traits (Supplementary Table 7).
Evolution and domestication of fruit quality traits
To investigate how environment and human selection have shaped the genomes of Citrullus species, we searched for signatures of selection in the watermelon genome. C. mucosospermus is the closest relative of C. lanatus. Although C. mucosospermus has been domesticated for seed consumption, its fruit flesh characteristics such as sweetness and coloration have not been subjected to human selection. Therefore, C. mucosospermus was used as a representative of the progenitor of C. lanatus. C. mucosospermus was first compared with C. colocynthis to uncover genomic regions under selection mostly during speciation from C. colocynthis to C. mucosospermus (referred to hereafter as speciation sweeps). In total, 172 sweeps in C. mucosospermus were identified, covering 14.0 Mb and 416 genes (Fig. 3a, Supplementary Fig. 1 and Supplementary Tables 8,9). C. lanatus landraces was then compared with C. mucosospermus to identify selective sweeps associated with domestication-related traits (domestication sweeps). In total, 151 domestication sweeps with a cumulative size of 24.8 Mb (containing 771 genes) were detected (Fig. 3b, Supplementary Fig. 2 and Supplementary Tables 10,11). Comparison between modern C. lanatus cultivars and landraces identified 125 selective sweeps (17.2 Mb and 667 genes) related to sweet watermelon improvement (improvement sweeps) (Fig. 3c, Supplementary Fig. 3 and Supplementary Tables 12,13). In total, 620 genes in selective sweeps were unique to the improvement process of sweet watermelon (Supplementary Fig. 4). The enriched biological processes for these improvement-specific genes included glucose import and cell wall modification (Supplementary Table 14).

a–d, Selective sweeps in C. mucosospermus compared with C. colocynthis (speciation sweeps) (a), in C. lanatus landrace compared with C. mucosospermus (domestication sweeps) (b), in C. lanatus cultivar compared with C. lanatus landrace (improvement sweeps) (c) and in C. amarus compared with C. colocynthis (speciation sweeps) (d). XP-CLR scores are plotted across the 11 watermelon chromosomes. Known QTL regions are shown in green text. Candidate genes in the selective regions are indicated by black arrows. Red bars at the bottom of each plot indicate candidate selective sweeps in the watermelon genome. BEBT, benzyl alcohol O-benzyltransferase; BGAL, β-galactosidase; Chr, chromosome; ClTST2, tonoplast sugar transporter; CRTISO, carotenoid isomerase; HSF, heat stress transcription factor; LAX, auxin transporter; PE, pectinesterase; PG, polygalacturonase; PMT, polyol/monosaccharide transporter; RLK, receptor-like protein kinase; SLO1, SLOW GROWTH 1 (encoding an E motif-containing pentatricopeptide repeat protein); STP, sugar transporter; XTH, xyloglucan endotransglucosylase/hydrolase. Fruit weight QTLs: Qfwt2-1, Qfwt3 and Qfwt5-1. Flesh sweetness QTLs: FCE10.1, QBrix6, QBRX2-1 and Qfru2-3. Disease resistance QTLs: Fo-1.3 and Qfon2-2.
Fruit size
The large size of harvestable plant organs is one of the most important characteristics that farmers choose when keeping and propagating seeds. The average fruit weight of C. colocynthis accessions in this study was approximately 0.21 kg. By contrast, C. amarus, C. mucosospermus and C. lanatus landraces and cultivars produced larger fruits with average weights of approximately 3.3, 1.7, 3.7 and 3.4 kg, respectively (Supplementary Fig. 5). Five watermelon fruit weight QTLs, Qfwt2-1, Qfwt2-2, Qfwt3, Qfwt5-1 and Qfwt5-2, have been identified using segregating populations9,24. However, whether these loci have contributed to watermelon fruit size enlargement during speciation, domestication or improvement remains unknown.
Qfwt2-1 and Qfwt3 overlapped with both speciation and domestication sweeps (Fig. 3a,b). Another fruit weight QTL, Qfwt5-1, was found to be under selection only during watermelon improvement (Fig. 3c). Qfwt2-2 and Qfwt5-2 were not found in domestication or improvement sweeps, indicating their potential in the future improvement of sweet watermelon fruit size.
The fruits of C. amarus are larger than those of C. colocynthis, suggesting that fruit enlargement has also been selected for in C. amarus after its split from the ancestor of C. mucosospermus and C. lanatus. Among the selective sweeps detected between C. colocynthis and C. amarus, three regions overlapped with Qfwt2-1 and Qfwt3 (Fig. 3d), suggesting their importance in controlling fruit size and parallel evolution of these loci in different Citrullus species.
Fruit taste and flavor
Selection for non-bitter fruits probably occurred during the initial domestication of sweet watermelon. Among the 374 Citrullus accessions evaluated for flesh bitterness, all nine C. colocynthis and 25 C. amarus accessions produced bitter fruits, whereas 12 of 16 C. mucosospermus accessions and all 324 C. lanatus landraces and cultivars accessions had non-bitter fruits (Supplementary Fig. 6). The previously identified bitterness QTL, qbt-c1-1 (ref. 21), contains the ClBt gene (Cla97C01G003400), which encodes a basic helix-loop-helix transcription factor and is homologous to the cucumber bitterness regulatory genes CsBt and CsBl (refs. 25,26). The genomic region near ClBt was highly differentiated between C. lanatus landraces and C. mucosospermus (Supplementary Fig. 7), and genetic diversity was substantially higher in C. mucosospermus than in C. lanatus landraces (Supplementary Fig. 8). At the SNP site leading to a premature stop codon of ClBt associated with non-bitterness25 (Chr01:3,216,322C to T), all C. colocynthis, C. amarus and C. mucosospermus accessions carrying bitter fruits had the homozygous bitter allele (C), whereas the homozygous non-bitterness allele (T) was found in all 12 non-bitter C. mucosospermus and all C. lanatus accessions, suggesting that this non-bitterness allele arose in the progenitor of C. lanatus and is fixed in sweet watermelons. Interestingly, the expression of ClBt was not detectable in the fruit flesh and rind (Supplementary Table 15). Exploring the public RNA-seq datasets in the Cucurbit Genomics Database27 revealed that the expression of ClBt was detected in the leaf but not in the flower, fruit, seed or root tissues, suggesting that the mechanisms by which ClBt regulates watermelon fruit bitterness may be complicated.
Improvement of fruit flesh sweetness is an important focus in modern watermelon breeding. The average SSC of C. colocynthis, C. mucosospermus and C. lanatus landraces and cultivars was about 1.6, 3.4, 8.3 and 10.1 °Brix, respectively (Supplementary Fig. 9). Stachyose and raffinose, in addition to sucrose, are the major sugars translocated in cucurbits28. Stachyose and raffinose, arriving at the fruit sink, are rapidly metabolized into disaccharides and monosaccharides by α-galactosidases29. The fruit of C. colocynthis had low levels of disaccharides and monosaccharides, but abundant raffinose and stachyose, whereas the fruit of C. lanatus showed an opposite pattern (Supplementary Fig. 10). An alkaline α-galactosidase gene, Cla97C04G070460 (ClAGA2), which is orthologous to melon CmAGA2 (ref. 30), was found in a genomic region with greatly reduced nucleotide diversity in C. mucosospermus and C. lanatus compared with C. colocynthis (Supplementary Fig. 11). The expression of ClAGA2 in the flesh of ‘97103’ was substantially increased during fruit development, and was much higher than that in the rind of ‘97103’ and the flesh of ‘PI 296341-FR’ (Supplementary Fig. 12a). To functionally characterize the role of ClAGA2 in fruit flesh sugar accumulation, we generated ClAGA2-mutated C. lanatus lines using CRISPR–Cas9 (Supplementary Fig. 12b,c). The mutants showed substantially reduced soluble sugars, glucose, fructose and sucrose contents in fruit flesh, but had increased raffinose content (Supplementary Fig. 12d). Together, these results indicate that ClAGA2 contributes to sugar accumulation in watermelon fruit flesh through facilitating the metabolism of raffinose into glucose, fructose and sucrose, and could have already been selected for early on in the progenitor of sweet watermelon, as indicated by the already reduced genetic diversity in C. mucosospermus.
QTLs Qfru2-3, QBRX2-1 and QBrix6, which are known to control fruit flesh sugar content9,24, overlapped with domestication sweeps (Fig. 3b). Based on the identified improvement sweeps, QBRX2-1 was also under selection during the breeding of modern cultivars, whereas QBrix6 was probably only selected for during domestication (Fig. 3c and Supplementary Table 15). In addition, a sweetness locus, FCE10.1 (ref. 14), was found in the improvement sweeps (Fig. 3c). Several fruit quality-related genes were found in these and other sweeps, indicating their potential contribution to the aromatic flavor, texture and nutritional profiles of cultivated watermelon fruit (Supplementary Note).
Flesh color
Different from wild and primitive watermelons that produce pale-colored mature fruit, sweet watermelons produce abundant carotenoids in fruit flesh and accumulate them in chromoplasts during ripening, leading to a spectrum of flesh colors such as red, orange and yellow31. Phytoene synthase (PSY) is the first rate-limiting enzyme in the carotenogenesis pathway and defines the size of the carotenoid pool32. A PSY1 gene, Cla97C01G008760, within the flesh color QTL qFC.1 (ref. 33) was highly expressed in fruit flesh and its expression levels positively correlated with increased lycopene accumulation during fruit ripening23 (Supplementary Fig. 13a). Cla97C01G008760 was located in a genomic region that was highly differentiated between C. lanatus landraces and C. mucosospermus (Supplementary Figs. 7,13b). These results suggest that the regulation of PSY1 expression might contribute to the transition from pale-colored to red, orange or yellow flesh by increasing total carotenoid production in the ripening fruit of sweet watermelon.
Our GWAS analysis identified a strong signal on chromosome 4 associated with flesh color, which contained the LCYB gene Cla97C04G070940 (Fig. 2c). The most highly associated SNP (Chr04:15442987) was located in LCYB, and leads to an amino acid change from a conserved phenylalanine to valine (F226V)34. All 209 red- or pink-fleshed C. lanatus accessions carried the F226V substitution in LCYB, whereas the 14 orange- and 20 yellow-fleshed C. lanatus accessions and all C. colocynthis, C. amarus and C. mucosospermus accessions had the wild-type allele. These results suggested that this mutation in LCYB, which possibly leads to increased lycopene accumulation, was selected for and largely fixed in sweet watermelon, resulting in the red flesh color in most modern cultivars.
A second GWAS signal associated with flesh color was found on chromosome 2 within the sweetness QTL QBRX2-1 (Fig. 2c); this QTL harbors the candidate gene ClTST2, which, when overexpressed, causes elevated sugar levels in fruit and also leads to flesh color development35. A previous study showed that an elevated sugar level activates the watermelon fruit chromoplast-localized phosphate transporter ClPHT4;2 (Cla97C10G205070), the function of which is necessary for carotenoid accumulation in fruit flesh36. It is possible that the sugar transporter gene ClTST2 in the sweetness QTL QBRX2-1, which was selected for during both domestication and improvement (Fig. 3b,c), could promote carotenoid accumulation through ClPHT4;2 by increasing sugar content in fruit flesh. Taken together, these results suggest that pathways controlling vibrant fruit flesh color and increased sweetness, two highly correlated traits (Supplementary Fig. 14), might be interconnected through shared genetic components.
Resistance evolution in C. amarus and introgression into C. lanatus
Modern sweet watermelons are susceptible to many diseases and pests. C. amarus has been used in watermelon breeding as a source of resistance to Fusarium wilt, powdery mildew and nematodes, and tolerance to drought3. To investigate genome changes during evolution that may underlie the disease resistance present in C. amarus, we searched for signatures of selection in C. amarus from C. colocynthis and identified 151 selective sweeps, covering 10.9 Mb and 364 genes (Fig. 3d, Supplementary Fig. 15 and Supplementary Tables 16,17). Among these selective sweeps, 71 selective sweeps containing 146 genes were unique to C. amarus and not under selection during the speciation of C. mucosospermus from C. colocynthis, including four encoding receptor-like kinases (Supplementary Table 18). It is worth noting that the Fusarium oxysporum race 1 resistance locus Fo-1.3 (ref. 8) and the Fusarium oxysporum race 2 resistance locus qFon2-2 (ref. 7) overlapped with these unique sweeps.
Introgression of genomic regions containing beneficial alleles from wild relatives has contributed to the improvement of cultivated crops37,38. We identified an introgression from C. amarus to C. lanatus on chromosome 6 at around 28.3–29.3 Mb (Supplementary Fig. 16a), containing 136 genes, including genes encoding acetolactate synthase, purple acid phosphatase, calcium-binding protein, glyoxal oxidase and receptor-like protein kinases that may function in powdery mildew resistance or tolerance to hypoxia or low phosphate conditions (Supplementary Table 19). Moreover, a genome introgression from C. colocynthis to C. lanatus was identified at 9.66–10.32 Mb of chromosome 4 (Supplementary Fig. 16b), overlapping with the Fusarium oxysporum race 1 resistance QTL Fo-1.4 (ref. 8). These results imply that genomic regions of C. colocynthis and C. amarus related to disease resistance might have been selected for and used in the improvement of elite watermelon cultivars.
Discussion
The improved watermelon ‘97103’ reference genome and resequencing of 414 accessions enabled the capture of genetic variations among the seven extant species of Citrullus and the reconstruction of their divergence history. Using GWAS, genomic regions associated with key fruit quality traits were identified, providing useful information for watermelon breeding and further identification of causal variations. Signatures of selection in the watermelon genome were revealed for speciation, domestication and improvement of the watermelon, the fruit characteristics of which changed during speciation and more significantly after domestication (Fig. 4). The ancestor of sweet watermelon probably produces small and bitter fruits, which can be seen in the extant wild watermelons such as C. colocynthis. As shown in Fig. 4, fruit size enlargement occurred in C. lanatus and C. mucosospermus and in C. amarus lineages, involving different but overlapping sets of QTLs. Flesh bitterness is a protective trait for wild watermelon but undesirable for humans and was selected against during the domestication process. Accompanying the disappearance of flesh bitterness, fruits of watermelon landraces became sweet. The alkaline α-galactosidase ClAGA2, which functions in phloem unloading of stachyose and raffinose in the watermelon fruit sink and rapid metabolism of these oligosaccharides, was under selection in the progenitor of sweet watermelon, providing an important prerequisite for increased soluble sugar levels in fruit flesh. The sugar transporter ClTST2 has been selected for during both domestication and improvement, facilitating sugar accumulation in the vacuoles of fruit flesh cells. Fruit flesh coloration in sweet watermelon has been realized during domestication by first expanding the total carotenoid pool via the key biosynthetic enzyme PSY, and then by increasing carotenoid accumulation indirectly through ClTST2. Red fruit flesh color was later selected for in sweet watermelons by maintaining an amino acid substitution in the lycopene metabolism enzyme LCYB. The potential dual function of ClTST2 in sugar accumulation and flesh coloration may have provided an easily accessible target for human selection, especially after the mutation in LCYB arose and lycopene became the dominant carotenoid, because the more intense red color can serve as a straightforward indicator of sweeter taste. Collectively, our findings shed important light on the evolution and domestication history of watermelons and reveal genome bases underlying the formation of fruit quality traits in sweet watermelon. The resources generated in this study provide a genomic framework for future germplasm use and watermelon improvement.

The featured fruit characteristics are shown below the fruit images. The direction of the speciation, domestication, improvement and introgression processes are indicated by the arrows. The selected loci and candidate genes involved in each process are shown above and below the arrows, respectively. Scale bar, 5 cm. ALS, acetolactate synthase; BEBT, benzyl alcohol O-benzoyltransferase; BGAL, β-galactosidase; ClAGA2, alkaline α-galactosidase; ClBt, bitter fruit bHLH transcription factor; ClTST2, tonoplast sugar transporter; CML, calmodulin; CRTISO, carotenoid isomerase; GLOX, glyoxal oxidase; HSF, heat stress transcription factor; LAX, auxin transporter; LCYB, lycopene β-cyclase; PAP, purple acid phosphatase; PE, pectinesterase; PG, polygalacturonase; PMT, polyol/monosaccharide transporter; PSY1, phytoene synthase 1; RLK, receptor-like protein kinase; SLO1, SLOW GROWTH 1 (encoding an E motif-containing pentatricopeptide repeat protein); STP, sugar transporter; XTH, xyloglucan endotransglucosylase/hydrolase.
Methods
Plant materials and phenotyping
Watermelon accessions were obtained from the Germplasm Bank of National Engineering Research Center for Vegetables of China, the National Mid-term Genebank for Watermelon and Melon of China and The U.S. National Plant Germplasm System. For de novo assembly, seedlings of watermelon cultivar ‘97103’ were grown in a greenhouse and transferred to a dark room for 24 h before sample collection. For genome resequencing and phenotyping, 117 watermelon accessions were planted, in triplicate, in a randomized block design, in Yanqing Experiment Station (40° 46′ N, 115° 90′ E) of the National Engineering Research Center for Vegetables, and another 298 watermelon accessions were planted in Xinxiang Experiment Station (35° 18′N, 113° 55′E) of the Zhengzhou Fruit Research Institute of Chinese Academy of Agricultural Sciences in 2015. All accessions were sown on 1 May and fruits were harvested at the end of August. One fruit per plant was harvested 30 d after self-pollination. Each fruit was cut lengthwise, photographed and sampled to evaluate flesh sweetness, bitterness, fruit shape, rind color, rind stripe, seed color and flesh color. Plants from some accessions showed abnormal fruit growth and were therefore excluded from the phenotypic analysis. Flesh SSC was measured using a hand-held digital PAL-1 refractometer (Atago). Fruit bitterness was determined by tasting as described by Zhang et al.39. Briefly, a small piece of flesh from the mature fruit was tasted by three people trained to detect bitterness, and the fruit samples were categorized into bitter and non-bitter groups. Fruit weight, length and width were recorded, and fruit shapes were categorized into spheroidal and elongated groups on the basis of the ratio of fruit length to width. Rind color, rind stripe, seed color and flesh color were determined by visual observation. Rind colors were categorized into yellow, white, light green, middle green, green and dark green. Rind stripes were categorized into no-stripe, netted, narrow, middle and wide stripe. Seed coat colors were divided into red, rufous and non-red. Flesh colors were divided into white, pale yellow, yellow, orange, pink and red.
Genome library construction and sequencing
For PacBio sequencing, high molecular weight (HMW) DNA was extracted from young fresh leaves of ‘97103’ with the cetyltrimethylammonium bromide method40. The DNA was randomly sheared to fragments with an average size of 20 kb using g-TUBE (Covaris) and the sheared DNA was used to construct the PacBio SMRT libraries following the standard SMRT bell construction protocol (PacBio). The libraries were sequenced using the P6-C4 chemistry on a PacBio Sequel sequencing platform (PacBio) at Nextomics Biosciences (Wuhan, China).
Hi-C libraries were prepared following the proximo Hi-C plant protocol (Phase Genomics) and sequenced on an Illumina HiSeq X platform (Illumina) at Nextomics Biosciences.
For genome resequencing, DNA was extracted using the Plant DNA Mini Kit (Aidlab Biotechnologies). Illumina DNA libraries with an insert size of approximately 400 bp were constructed using the TruSeq Nano DNA High Throughput Library Prep Kit following the manufacturer’s instructions (Illumina), and sequenced on an Illumina HiSeq X or HiSeq 2000 platform at Berry Genomics (Beijing, China).
BioNano data generation
HMW DNA was extracted from young fresh leaves of ‘97103’ using a BioNano Prep Plant DNA isolation kit (BioNano Genomics). The DNA was purified on Percoll (Sigma) cushions, washed by HB+ buffer and embedded in an agarose layer, digested with Nb.BssSI and labeled with IrysPrep labeling mix and Taq polymerase according to standard BioNano protocols (BioNano Genomics). The molecules were counterstained using the protocol provided with the IrysPrep reagent kit. Samples were then loaded into IrysChips (BioNano Genomics) and imaged on an Irys imaging instrument (BioNano Genomics) at Nextomics Biosciences.
Transcriptome sequencing
Watermelon ‘97103’ plants were grown in a greenhouse. Six different tissues (apical point, carpopodium, fruit flesh, stem, leaf and root) were sampled at four time points: 10, 18, 26 and 34 d after pollination. Each sample had three biological replicates. Total RNA was extracted from these samples using the QIAGEN RNeasy Plant Mini Kit (QIAGEN). Strand-specific RNA-seq libraries were prepared as described by Zhong et al.41 and sequenced on an Illumina HiSeq 2000 platform (Illumina). Trimmomatic42 (v0.36) was used to trim low-quality and adapter sequences. Reads aligned to sequences in the SILVA rRNA database43 were removed.
For PacBio Iso-Seq, full-length complementary DNA was synthesized from a mixture of total RNA from the six tissues using the SMARTer PCR cDNA Synthesis Kit (Takara Bio). The cDNA product was filtered using the BluePippin DNA Size Selection System (Sage Science). The Iso-Seq libraries were constructed following the standard SMRT bell construction protocol (PacBio) and sequenced on the PacBio RS II platform (PacBio) at Nextomics Biosciences.
De novo assembly of the ‘97103’ genome
PacBio SMRT reads were de novo assembled into contigs using Falcon44 (v1.8.7) with the following parameters: ‘length_cutoff = 4000 length_cutoff_pr = 4000 pa_HPCdaligner_option=‘-v -w8 -M24 -e.75 -k18 -h280 -l2800’ ovlp_HPCdaligner_option=‘-v -k18 -h180 -e.96 -l2800’ falcon_sense_option = ‘-output_multi -min_idt 0.75 -min_cov 2 -max_n_read 300’ overlap_filtering_setting = ‘-max_diff 50 -max_cov 70 -min_cov 1 -bestn 10’’. PacBio reads were aligned to the raw assembly using BLASR (v5.1)45 with parameters ‘-bam -bestn 5 -minMatch 18 -nproc 6 -minSubreadLength 1000 -minAlnLength 500 -minPctSimilarity 70 -minPctAccuracy 70 -hitPolicy randombest -randomSeed 1’, followed by correction of errors in the assembled contigs with Arrow (v2.2.2; PacBio). Illumina reads were then aligned to the assembled contigs using BWA-MEM (v0.7.12)46 with default parameters, and the resulting alignments were used to further polish the assembled contigs using Pilon47 (v1.22) with parameters ‘-diploid -fix bases -mindepth 10’. The assembled contigs were then compared against the NCBI non‐redundant nucleotide database, and those with more than 95% of their length similar to sequences of microorganisms, mitochondria or chloroplasts were considered contaminants and removed.
Raw BioNano data were cleaned by removing molecules matching any of the following rules: length less than 150 kb, molecule signal-to-noise ratio less than 2.75 and label signal-to-noise ratio less than 2.75, or label intensity greater 0.8. De novo assembly of BioNano molecules into genome maps was performed using the script pipelineCL.py in the BioNano Solve package v3.0 (BioNano Genomics) with parameters ‘-d -U -N 6 -y -i 3 -F 1 -a optArgs.xml’. Hybrid scaffolds were assembled from PacBio assembly and BioNano genome maps using the script hybridScaffold.pl in the Solve package with parameters ‘-c hybridScaffold_config_aggressive.xml -u CACGAG -B 2 -N 2 -f’.
Hi-C read pairs were filtered using programs ‘filter_data_parallel’ with parameters ‘-y -B 50 -a 3 -b 2 -c 3 -d 2 -l 400 -q 33’ and ‘duplication150’ with default parameters, in the SOAPdenovo2 package (r240)48. The cleaned Hi-C read pairs were aligned to the hybrid scaffolds using Bowtie2 (v2.3.2)(ref. 49) with parameters ‘-very-sensitive -L 30 -score-min L,-0.6,-0.2 -end-to-end -reorder -phred33-quals’. On the basis of the alignments, the hybrid scaffolds were further assembled into super-scaffolds using SALSA (v2.2)50 with parameters ‘-e GATC -i 2’. Finally, genetic maps7,8,9 were used to generate pseudochromosomes.
Repeat annotation and gene prediction in the ‘97103’ genome
Miniature inverted-repeat transposable element (MITE) and long terminal repeat (LTR) libraries were constructed by scanning the ‘97103’ genome using MITE-Hunter (v11-2011)51 and LTRharvest (v1.5.9)52, respectively. The identified MITE and LTR libraries were used to mask the ‘97103’ genome with RepeatMasker (v4.0.7; http://www.repeatmasker.org/). The unmasked genome sequences were then fed to RepeatModeler (v1.0.11; http://www.repeatmasker.org/RepeatModeler/) to build a de novo repeat library. Repeat sequences in the MITE, LTR and de novo repeat libraries were combined and compared with the UniProt database53 to remove those homologous to non-TE proteins. The final repeats were classified using the RepeatClassifier program of RepeatModeler and then used to identify repeat sequences in the ‘97103’ genome using RepeatMasker.
Prediction of protein-coding genes was performed on the repeat-masked ‘97103’ genome using Maker (v3.01.02)54. Illumina RNA-seq reads were assembled using Trinity (v2.5.1)55 with the de novo mode and the genome-guided mode, respectively. The resulting two transcriptome assemblies and the PacBio Iso-Seq full-length cDNA sequences were used as transcript evidence. Ab initio gene predictions were performed using Augustus (v3.2.3)56, GeneMark-ET (v4.33)57 and SNAP (v2006-07-28)58. Proteins from SwissProt and from Arabidopsis, watermelon, cucumber and melon were aligned to the ‘97103’ genome using Spaln (v2.1.4)59, and the resulting alignments were used as protein homology evidence. Maker was then run to generate high-confidence gene models by integrating ab initio predictions, transcript mapping evidence and protein homology evidence.
Genome read mapping and variant calling
Raw Illumina reads were processed to remove adapter sequences using Picard v2.0.1 (https://broadinstitute.github.io/picard/). The cleaned reads were aligned to the ‘97103’ genome using BWA-MEM46 (v0.7.12) with default parameters. Duplicated read pairs were marked with Picard (v2.0.1) with the parameter ‘OPTICAL_DUPLICATE_PIXEL_DISTANCE=250’. The HaplotypeCaller function of GATK60 (version 20171018) was then used to generate GVCF files for each accession with parameters ‘-genotyping_mode DISCOVERY -max_alternate_alleles 3 -read_filter OverclippedRead’, followed by population variant calling using the function GenotypeGVCFs of GATK with default parameters. Hard filtering was applied to the raw variant set using GATK, with parameters ‘QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum <−12.5 || ReadPosRankSum <−8.0’ applied to SNPs, and ‘QD < 2.0 || FS > 200.0 || ReadPosRankSum <−20.0’ applied to small indels.
Phylogenetic and population analyses
Bi-allelic SNPs with a missing data rate less than 15% and a minor allele count greater than three were kept for population genomic analyses. Additionally, only SNPs at fourfold degenerated sites (89,914 SNPs) were used to construct a neighbor-joining phylogenetic tree using MEGA7 with 500 bootstraps61. Principal component analysis was performed using Plink62 (v1.9) with the entire set of SNPs (19,725,853 SNPs). Population structure analysis was performed using STRUCTURE63 (v2.3.4). The optimal K, which indicates the most likely number of clusters in the population, was calculated. STRUCTURE analyses were run 20 times for each K value ranging from 2 to 20, using 8,000 randomly selected SNPs at fourfold degenerated sites. After the best K was analyzed, the population structure of the watermelon accessions was inferred using fastStructure64 (v1.0) with all SNPs at fourfold degenerated sites for each K (K = 2–4).
The π and FST were calculated using VCFtools65 (v0.1.15) based on the high-confidence filtered SNPs (19,725,853 SNPs). The π value for each SNP was calculated and the nucleotide diversity level was measured using a 100-kb window with a step size of 10 kb for each sub-population. LD decay was calculated for all pairs of SNPs within 500 kb using PopLDdecay66 (v3.27) with parameters ‘-MaxDist 500 -Het 0.1 -Miss 0.1’.
Putative introgressions between two groups were identified using a likelihood ratio test approach67 with all SNPs. The average ratio of shared variation sites of the two groups was calculated in each of the 200-kb windows with a step size of 20 kb. Regions with ratios of 0.7 or less and five or more SNPs were defined as introgressions.
Identification of selective sweeps
A cross-population composite likelihood ratio test (XP-CLR; v1.0)68 was used to scan the ‘97103’ genome for selective sweeps. Briefly, selective sweeps were identified in the following four comparisons representing different watermelon speciation and breeding processes: C. colocynthis versus C. amarus, C. colocynthis versus C. mucosospermus, C. mucosospermus versus C. lanatus landrace and C. lanatus landraces versus C. lanatus cultivars. Genetic distances between adjacent SNPs were calculated on the basis of proportionally increased physical distance of adjacent surrounding markers in an integrated genetic map9. For each chromosome, the XP-CLR score was calculated with parameter ‘-w1 0.0005 100 100 1 -p0 0.7’. The XP-CLR scores per 100 bp were averaged in each non-overlapping 10-kb window across each chromosome. Adjacent 10-kb windows with an average XP-CLR score no less than 80% of the genome-wide average were joined, and were further merged if two regions were separated by only one low-score 10-kb window. The maximum window-wise average XP-CLR score in a merged region was assigned as the region-wise XP-CLR score. Merged regions with region-wise XP-CLR scores in the top 10% were considered candidate selective sweeps. To improve the accuracy, only candidate selective sweeps with the top 50% of π ratios between the two compared populations were kept.
Gene flow analysis
Potential gene flow between different groups was identified using the ABBA-BABA test69 (also called the D test) with all SNPs. For each group, the D value (sum(ABBA)-sum(BABA))/(sum(ABBA) + sum(BABA)) was weighted using the genotype frequency of the outgroup. Standard errors and significance of the weighted D values were calculated based on Z scores obtained using the jackknife method70.
GWAS
In total, 10,195,082 SNPs with a minor allele frequency of 0.01 or greater and a missing data rate of 50% or less in the entire population were used for GWAS. A kinship (K) matrix generated with the FaST-LMM program (v2.07)71 was used to correct the population structure. GWAS analyses were performed using the linear mixed model algorithm implemented in the FaST-LMM program. The modified Bonferroni correction was used to determine the genome-wide significance thresholds of the GWAS, based on a nominal level of α = 0.05 and α = 0.01 (ref. 72), corresponding raw P values of 4.90 × 10−9 and 9.81 × 10−10, or −log10(P) values of 8.31 and 9.00, respectively.
Metabolite measurement
Fruit sugar content was determined using the ultra-HPLC (UHPLC)-Quadrupole-Orbitrap MS/MS-based method as described by Wang et al.73. Briefly, frozen ground powdered samples (200 mg) were extracted with 1 ml of methanol containing 0.1% formic acid (v/v). The extraction mixture was vortex-mixed and sonicated for 30 min at room temperature (26 ºC) and then centrifuged at 5,000 r.p.m. for 15 min. The supernatant was filtered through a 0.22 μm PVDF syringe filter (Waters) and used for UHPLC–MS/MS analysis of fructose, sucrose, raffinose and stachyose on a DIONEX Ultimate 3000 UHPLC system and Q Exactive Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific).
Functional characterization of ClAGA2
Watermelon plants carrying mutations in the ClAGA2 gene were generated using CRISPR–Cas9 gene editing technology following our previously established protocol74. A DNA construct was designed to target the coding sequence of ClAGA2 using a guide RNA (gRNA), 5′-CTGACCCCAGGATCAGGCCT-3′. The gRNA was cloned into pBSE401 (ref. 75) to be expressed under the Arabidopsis U6 promoter, alongside the Zea mays codon-optimized Cas9 driven by the double CaMV 35S promoter. The binary vector was then transformed into an explant of watermelon cultivar ‘ZZJM’ mediated by the Agrobacterium strain EHA105. Plant regeneration and greenhouse care were performed as described by Tian et al.74. CRISPR–Cas9-positive lines selected on the basis of Basta herbicide resistance were further genotyped using primers flanking the gRNA targeting sequence (Supplementary Table 20). PCR amplicons were used to genotype the ClAGA2 mutant plants by Sanger sequencing. The Cas9-free homozygous Claga2 mutant lines were obtained by selecting against the transgene in the segregating T2 generation using primers binding to the 35S and Cas9 sequences (Supplementary Table 20). Plants with wild-type ClAGA2 in the same segregation population were used as negative controls. Fruit flesh SSC was measured using a sample of juice collected from the center of each watermelon with a hand-held digital PAL-1 refractometer (Atago).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The genome sequence of ‘97013’ has been deposited at DDBJ/ENA/GenBank under the accession AGCB00000000. The version described in this paper is version AGCB02000000. Raw genome and transcriptome sequencing reads have been deposited into the NCBI sequence read archive (SRA) under accessions SRP188834 and SRP192188. The genome sequence of ‘97103’ (version 2) is also available at the Cucurbit Genomics Database (http://cucurbitgenomics.org/organism/21). SNPs and small indels are available at ftp://cucurbitgenomics.org/pub/cucurbit/reseq/watermelon/v2/.
References
Robinson, R. W. & Decker-Walters, D. (eds) S. curcubits 65–97 (CAB International, 1997).
Paris, H. S. Origin and emergence of the sweet dessert watermelon, Citrullus lanatus. An. Bot. 116, 133–148 (2015).
Levi, A. et al. in Genetics and Genomics of Cucurbitacae (eds Grumet, R., Katzir, N. & Garcia-Mas, J.) Vol. 20, 87–110 (Springer, 2017).
Chomicki, G. & Renner, S. S. Watermelon origin solved with molecular phylogenetics including Linnaean material: another example of museomics. New Phytol. 205, 526–532 (2015).
Renner, S. S., Sousa, A. & Chomicki, G. Chromosome numbers, Sudanese wild forms, and classification of the watermelon genus Citrullus, with 50 names allocated to seven biological species. Taxon 66, 1393–1405 (2017).
Guo, S. G. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58 (2013).
Branham, S. E., Levi, A., Farnham, M. W. & Wechter, W. P. A GBS-SNP-based linkage map and quantitative trait loci (QTL) associated with resistance to Fusarium oxysporum f. sp. niveum race 2 identified in Citrullus lanatus var. citroides. Theor. Appl. Genet. 130, 319–330 (2017).
Lambel, S. et al. A major QTL associated with Fusarium oxysporum race 1 resistance identified in genetic populations derived from closely related watermelon lines using selective genotyping and genotyping-by-sequencing for SNP discovery. Theor. Appl. Genet. 127, 2105–2115 (2014).
Ren, Y. et al. An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol. 14, 33 (2014).
Wu S. et al. Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol J. 2019, 1–13 (2019).
Reddy, U. K. et al. Cytomolecular characterization of rDNA distribution in various Citrullus species using fluorescent in situ hybridization. Genet. Resour. Crop Evol. 60, 2091–2100 (2013).
Levi, A., Thomas, C. E., Joobeur, T., Zhang, X. & Davis, A. A genetic linkage map for watermelon derived from a testcross population: (Citrullus lanatus var. citroides × C. lanatus var. lanatus) × Citrullus colocynthis. Theor. Appl. Genet. 105, 555–563 (2002).
Nimmakayala, P. et al. Single nucleotide polymorphisms generated by genotyping by sequencing to characterize genome-wide diversity, linkage disequilibrium, and selective sweeps in cultivated watermelon. BMC Genomics 15, 767 (2014).
Cheng, Y. et al. Construction of a genetic linkage map of watermelon (Citrullus lanatus) using CAPS and SSR markers and QTL analysis for fruit quality traits. Sci. Hortic. 202, 25–31 (2016).
Zhang, B. C., Tolstikov, V., Turnbull, C., Hicks, L. M. & Fiehn, O. Divergent metabolome and proteome suggest functional independence of dual phloem transport systems in cucurbits. Proc. Natl Acad. Sci. USA 107, 13532–13537 (2010).
Liu, S. et al. Development of cleaved amplified polymorphic sequence markers and a CAPS-based genetic linkage map in watermelon (Citrullus lanatus [Thunb.] Matsum. and Nakai) constructed using whole-genome re-sequencing data. Breeding Sci. 66, 244–259 (2016).
Dou, J. L. et al. Genetic mapping reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L.). Theor. Appl. Genet. 131, 947–958 (2018).
Park, S. W., Kim, K. T., Kang, S. C. & Yang, H. B. Rapid and practical molecular marker development for rind traits in watermelon. Hortic. Environ. Biotechnol. 57, 385–391 (2016).
Lohmann, A. et al. Deficiency in phylloquinone (vitamin K1) methylation affects prenyl quinone distribution, photosystem I abundance, and anthocyanin accumulation in the Arabidopsis AtmenG mutant. J. Biol. Chem. 281, 40461–40472 (2006).
Braumann, I., Stein, N. & Hansson, M. Reduced chlorophyll biosynthesis in heterozygous barley magnesium chelatase mutants. Plant Physiol. Bioch. 78, 10–14 (2014).
Li, B. B. et al. Construction of a high-density genetic map and mapping of fruit traits in watermelon (Citrullus lanatus L.) based on whole-genome resequencing. Int. J. Mol. Sci. 19, 3268 (2018).
Mayer, A. M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry 67, 2318–2331 (2006).
Guo, S. G. et al. Comparative transcriptome analysis of cultivated and wild watermelon during fruit development. PLoS One 10, e0130267 (2015).
Sandlin, K. et al. Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. et Nakai]. Theor. Appl. Genet. 125, 1603–1618 (2012).
Zhou, Y. et al. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae. Nat. Plants 2, 16183 (2016).
Shang, Y. et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346, 1084–1088 (2014).
Zheng, Y. et al. Cucurbit genomics database (CuGenDB): a central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 47, D1128–D1136 (2018).
Schaffer, A. A, Pharr, D. M. & Madore, M. A. in Photoassimilate Distribution in Plants and Crops 1st edn (eds Zamski, E. & Schaffer, A. A.) 729–757 (Marcel Dekker, 1996).
Gao, Z. & Schaffer, A. A. A novel alkaline alpha-galactosidase from melon fruit with a substrate preference for raffinose. Plant Physiol. 119, 979–988 (1999).
Carmi, N. et al. Cloning and functional expression of alkaline alpha-galactosidase from melon fruit: similarity to plant SIP proteins uncovers a novel family of plant glycosyl hydrolases. Plant J. 33, 97–106 (2003).
Gusmini, G. & Wehner, T. C. Qualitative inheritance of rind pattern and flesh color in watermelon. J. Hered. 97, 177–185 (2006).
Sun, T. H. et al. Carotenoid metabolism in plants: the role of plastids. Mol. Plant. 11, 58–74 (2018).
Branham, S. E. et al. Genetic mapping of a major codominant QTL associated with beta-carotene accumulation in watermelon. Mol. Breeding 37, 146 (2017).
Bang, H., Kim, S., Leskovar, D. & King, S. Development of a codominant CAPS marker for allelic selection between canary yellow and red watermelon based on SNP in lycopene β-cyclase (LCYB) gene. Mol. Breeding 20, 63–72 (2007).
Ren, Y. et al. A tonoplast sugar transporter underlies a sugar accumulation QTL in watermelon. Plant Physiol. 176, 836–850 (2018).
Zhang, J. et al. High-level expression of a novel chromoplast phosphate transporter ClPHT4;2 is required for flesh color development in watermelon. New Phytol. 213, 1208–1221 (2017).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226 (2014).
Sun, J. et al. Introgression and selection shaping the genome and adaptive loci of weedy rice in northern China. New Phytol. 197, 290–299 (2013).
Zhang, S. P. et al. Localization of a new gene for bitterness in cucumber. J. Hered. 104, 134–139 (2013).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325 (1980).
Zhong, S. et al. High-throughput Illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Luo, R. B. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ghurye, J., Pop, M., Koren, S., Bickhart, D. & Chin, C. S. Scaffolding of long read assemblies using long range contact information. BMC Genomics 18, 527 (2017).
Han, Y. J. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Stanke, M., Tzvetkova, A. & Morgenstern, B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7 (Suppl. 1), S11 (2006).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119 (2014).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Gotoh, O. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Res. 36, 2630–2638 (2008).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M. & Yang, T. L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
McNally, K. L. et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl Acad. Sci. USA 106, 12273–12278 (2009).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Martin, S. H., Davey, J. W. & Jiggins, C. D. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evo. 32, 244–257 (2015).
Efron, B. & Stein, C. The jackknife estimate of variance. Ann. Stat. 9, 586–596 (1981).
Lippert, C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011).
Li, M. X., Yeung, J. M. Y., Cherny, S. S. & Sham, P. C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756 (2012).
Wang, Y. Q., Hu, L. P., Liu, G. M., Zhang, D. S. & He, H. J. Evaluation of the nutritional quality of Chinese kale (Brassica alboglabra Bailey) using UHPLC-Quadrupole-Orbitrap MS/MS-based metabolomics. Molecules 22, 1262 (2017).
Tian, S. W. et al. Efficient CRISPR/Cas9-based gene knockout in watermelon. Plant Cell Rep. 36, 399–406 (2017).
Xing, H. L. et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327 (2014).
Acknowledgements
We thank S. Renner (Ludwig Maximilian University of Munich) for helpful discussion on watermelon taxonomy. This research was supported by grants from the National Natural Science Foundation of China (31930096 to Y.X.), Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2016-ZFRI to W.L.), National Key R&D Program of China (2018YFD0100703 to Y.X. and 2018YFD0100704 to W.L.), Beijing Scholar Program (BSP026 to Y.X.), China Agriculture Research System (CARS-25 to Y.X. and W.L.), Innovation Program of Beijing Academy of Agricultural and Forestry Sciences (KJCX20170102 to Y.X.), Ministry of Science and Technology of China (2016YFD0101007 and 2015BAD02B02 to S.G.), National Natural Science Foundation of China (31801886 to H.S. and 31471893 and 31672178 to W.L.), Agriculture Innovation Consortium (BAIC10-2019 to G.G.), Technology Program of Guangdong (2018B020202007 to S.H.), Leading Talents of Guangdong Province Program (00201515 to S.H.), USDA National Institute of Food and Agriculture Specialty Crop Research Initiative (2015-51181-24285 to Z.F.) and the US National Science Foundation (IOS-1339287 and IOS-1539831 to Z.F.).
Author information
Authors and Affiliations
Contributions
Y.X., W.L., Z.F. and S.H. designed and managed the project. S.G., S.Z., Y.R., J.Z., X.L., A.L. and R.J. collected the samples and extracted DNA and RNA. Y.X., W.L., Z.F., S.H., S.G., S.Z., H.S., C.W. and R.J. coordinated the genome sequencing and resequencing. S.G., H.S., X.W., S.W., S.Z., T.L., Y.R., L.G., J.L., J.Z. and X.L. performed data analyses. H.Z., J.S., G.G., N.H., M.L., Y.D., Y.W., S.T and Y.Z. performed fruit quality trait measurement and analyses. X.Z. designed the project and collected the samples. S.W., S.G., H.S. and X.W. wrote the manuscript. Z.F. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Hi-C interaction heatmap of the ‘97103’ genome (v2).
Color bar at the bottom represents the density of Hi-C interactions, which are indicated number of links (N links) at the 100-kb resolution.
Extended Data Fig. 2 Hi-C interaction heatmap for each of the 11 chromosomes of the ‘97103’ genome (v2).
White crosses in heatmaps are gaps in the ‘97103’ genome assembly introduced during the scaffolding step by BioNano genome maps.
Extended Data Fig. 4 Synteny between the improved (v2) and previous (v1) ‘97103’ genomes.
Each dot represents a homologous region between the two genomes. Purple dots represent forward alignments, while blue dots represent reverse alignments.
Extended Data Fig. 5 Scatterplot of GC content on read depth.
GC content of non-overlapping 500-bp sliding windows in the ‘97130’ v2 assembly and the average per-base sequencing coverage by ‘97103’ Illumina reads are plotted.
Extended Data Fig. 6 Genome coverage evaluated by fully sequenced BACs.
Alignments of fully sequenced BACs, JN402338 (a), JN402339 (b), JX027061 (c) and JX027062 (d), to the ‘97103’ v1 (top) and v2 (bottom) genome assemblies.
Extended Data Fig. 7 Mapping of BioNano maps and mate-pair reads to the ‘97103’ assembly.
a, Alignments of BioNano maps to chromosome 11 of the ‘97103’ assembly. b, c, Alignments of 20-kb insert size mate-pair reads at the potential inversion breakpoint regions. Uniquely aligned read pairs are indicated by orange color. Green lines represent reads aligned to multiple locations.
Extended Data Fig. 8 Principal component analysis (PCA) of Citrullus accessions.
PCA plots of the first three components of Citrullus accessions excluding C. naudinianusand (PC1 × PC3, PC2 × PC3) (a) and C. mucosospermus and C. lanatus accessions (PC1 × PC3, PC2 × PC3) (b), and all accessions in the seven Citrullus species (PC1 × PC2, PC1 × PC3, PC2 × PC3) (c). CA, C. amarus; CC, C. colocynthis; CE, C. ecirrhosus; CL_CUL, C. lanatus cultivar; CL_LR, C. lanatus landrace; CM, C. mucosospermus; CN, C. naudinianus; CR, C. rehmii.
Extended Data Fig. 9 Multidimensional scaling of pairwise FST values between different Citrullus species.
Value near each dot indicates the nucleotide diversity (π) of the corresponding group. CC, C. colocynthis; CA, C. amarus; CM, C. mucosospermus; CL_LR, C. lanatus landrace; CL_CUL, C. lanatus cultivar.
Extended Data Fig. 10 Linkage disequilibrium (LD) decay patterns of different Citrullus species.
The decays of LD (r2) with physical distance (kilobases) for SNPs in five different Citrullus groups are shown. CC, C. colocynthis; CA, C. amarus; CM, C. mucosospermus; CL_LR, C. lanatus landrace; CL_CUL, C. lanatus cultivar.
Supplementary information
Supplementary Information
Supplementary Note and Figs. 1–16
Supplementary Table
Supplementary Tables 1–20
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, S., Zhao, S., Sun, H. et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat Genet 51, 1616–1623 (2019). https://doi.org/10.1038/s41588-019-0518-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0518-4
This article is cited by
-
Promoter variations of ClERF1 gene determines flesh firmness in watermelon
BMC Plant Biology (2024)
-
Fine mapping of ClLOX, a QTL for powdery mildew resistance in watermelon (Citrullus lanatus L.)
Theoretical and Applied Genetics (2024)
-
Callose deposition regulates differences in cotton aphid resistance among six watermelon varieties
Journal of Pest Science (2024)
-
A large presence/absence variation in the promotor of the ClLOG gene determines trichome elongation in watermelon
Theoretical and Applied Genetics (2024)
-
Comparative network analysis reveals the dynamics of organic acid diversity during fruit ripening in peach (Prunus persica L. Batsch)
BMC Plant Biology (2023)
