
Abstract
A rare loss-of-function allele p.Arg138* in SLC30A8 encoding the zinc transporter 8 (ZnT8), which is enriched in Western Finland, protects against type 2 diabetes (T2D). We recruited relatives of the identified carriers and showed that protection was associated with better insulin secretion due to enhanced glucose responsiveness and proinsulin conversion, particularly when compared with individuals matched for the genotype of a common T2D-risk allele in SLC30A8, p.Arg325. In genome-edited human induced pluripotent stem cell (iPSC)-derived β-like cells, we establish that the p.Arg138* allele results in reduced SLC30A8 expression due to haploinsufficiency. In human β cells, loss of SLC30A8 leads to increased glucose responsiveness and reduced KATP channel function similar to isolated islets from carriers of the T2D-protective allele p.Trp325. These data position ZnT8 as an appealing target for treatment aimed at maintaining insulin secretion capacity in T2D.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
Individual level data for the human study can only be obtained via the Biobank of The Institute of Health and Welfare in Finland (https://thl.fi/en/web/thl-biobank). Next-generation sequencing data have been deposited in the SRA database (PRJNA563975) and the processed counts data can be found in the Supplementary Dataset 1. The individual processed data from cell lines (Figs. 4 and 5), mice studies (Fig. 6) and human islet work (Fig. 7) are available in the Source Data files. Additional data supporting the findings of this study are available on request from the corresponding author. Source data for Figs. 2 and 4–7 and Extended Data Figs. 2, 3, 6 and 8 are available online.
References
Chabosseau, P. & Rutter, G. A. Zinc and diabetes. Arch. Biochem. Biophys. 611, 79–85 (2016).
Chimienti, F., Devergnas, S., Favier, A. & Seve, M. Identification and cloning of a β-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes 53, 2330–2337 (2004).
Flannick, J. et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 46, 357–363 (2014).
Parsons, D. S., Hogstrand, C. & Maret, W. The C-terminal cytosolic domain of the human zinc transporter ZnT8 and its diabetes risk variant. FEBS J. 285, 1237–1250 (2018).
Sladek, R. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885 (2007).
Lemaire, K. et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl Acad. Sci. USA 106, 14872–14877 (2009).
Pound, L. D. et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem. J. 421, 371–376 (2009).
Wijesekara, N. et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 53, 1656–1668 (2010).
Mitchell, R. K. et al. Molecular genetic regulation of Slc30a8/ZnT8 reveals a positive association with glucose tolerance. Mol. Endocrinol. 30, 77–91 (2016).
Kleiner, S. et al. Mice harboring the human SLC30A8 R138X loss-of-function mutation have increased insulin secretory capacity. Proc. Natl Acad. Sci. USA 115, E7642–E7649 (2018).
Groop, L. et al. Metabolic consequences of a family history of NIDDM (the Botnia study): evidence for sex-specific parental effects. Diabetes 45, 1585–1593 (1996).
Rezania, A. et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133 (2014).
Miyaoka, Y., Chan, A. H. & Conklin, B. R. Using digital polymerase chain reaction to detect single-nucleotide substitutions induced by genome editing. Cold Spring Harb. Protoc. https://doi.org/10.1101/pdb.prot086801 (2016).
Scharfmann, R. et al. Development of a conditionally immortalized human pancreatic β cell line. J. Clin. Invest. 124, 2087–2098 (2014).
Li, D. et al. Imaging dynamic insulin release using a fluorescent zinc indicator for monitoring induced exocytotic release (ZIMIR). Proc. Natl Acad. Sci. USA 108, 21063–21068 (2011).
Hastoy, B. et al. Electrophysiological properties of human beta-cell lines EndoC-βH1 and -βH2 conform with human beta-cells. Sci Rep 8, 16994 (2018).
Nicolson, T. J. et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58, 2070–2083 (2009).
Vinkenborg, J. L. et al. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 6, 737–740 (2009).
Flannick, J. et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019).
Tamaki, M. et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Invest. 123, 4513–4524 (2013).
Hardy, A. B. et al. Effects of high-fat diet feeding on Znt8-null mice: differences between β-cell and global knockout of Znt8. Am. J. Physiol. Endocrinol. Metab. 302, E1084–E1096 (2012).
Li, L., Bai, S. & Sheline, C. T. hZnT8 (Slc30a8) transgenic mice that overexpress the R325W polymorph have reduced islet Zn2+ and proinsulin levels, increased glucose tolerance after a high-fat diet, and altered levels of pancreatic zinc binding proteins. Diabetes 66, 551–559 (2017).
Merriman, C., Huang, Q., Rutter, G. A. & Fu, D. Lipid-tuned zinc transport activity of human ZnT8 protein correlates with risk for type-2 diabetes. J. Biol. Chem. 291, 26950–26957 (2016).
Gerber, P. A. et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic beta cells. Diabetologia 57, 1635–1644 (2014).
Wong, W. P. et al. Exploring the association between demographics, SLC30A8 genotype, and human islet content of zinc, cadmium, copper, iron, manganese and nickel. Sci. Rep. 7, 473 (2017).
Vergnano, A. M. et al. Zinc dynamics and action at excitatory synapses. Neuron 82, 1101–1114 (2014).
Ferrer, R., Soria, B., Dawson, C. M., Atwater, I. & Rojas, E. Effects of Zn2+ on glucose-induced electrical activity and insulin release from mouse pancreatic islets. Am. J. Physiol. 246, C520–C527 (1984).
Bloc, A., Cens, T., Cruz, H. & Dunant, Y. Zinc-induced changes in ionic currents of clonal rat pancreatic β-cells: activation of ATP-sensitive K+ channels. J. Physiol 529, 723–734 (2000).
Zhang, Q. et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 18, 871–882 (2013).
Michael, D. J. et al. Pancreatic beta-cells secrete insulin in fast- and slow-release forms. Diabetes 55, 600–607 (2006).
Kirchhoff, K. et al. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia 51, 597–601 (2008).
Majithia, A. R. et al. Association of the SLC30A8 missense polymorphism R325W with proinsulin levels at baseline and after lifestyle, metformin or troglitazone intervention in the diabetes prevention program. Diabetologia 54, 2570–2574 (2011).
Jainandunsing, S. et al. A stable isotope method for in vivo assessment of human insulin synthesis and secretion. Acta. Diabetol. 53, 935–944 (2016).
Ivanova, A. et al. Age-dependent labeling and imaging of insulin secretory granules. Diabetes 62, 3687–3696 (2013).
Abecasis, G. R., Cardon, L. R. & Cookson, W. O. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 66, 279–292 (2000).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Isomaa, B. et al. A family history of diabetes is associated with reduced physical fitness in the prevalence, prediction and prevention of diabetes (PPP)–Botnia study. Diabetologia 53, 1709–1713 (2010).
Ahlqvist, E. et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 6, 361–369 (2018).
Sluiter, W. J., Erkelens, D. W., Reitsma, W. D. & Doorenbos, H. Glucose tolerance and insulin release, a mathematical approach I. Assay of the beta-cell response after oral glucose loading. Diabetes 25, 241–244 (1976).
Mohandas, C. et al. Ethnic differences in insulin secretory function between black African and white European men with early type 2 diabetes. Diabetes Obes. Metab. 20, 1678–1687 (2018).
Navalesi, R., Pilo, A. & Ferrannini, E. Kinetic analysis of plasma insulin disappearance in nonketotic diabetic patients and in normal subjects. A tracer study with 125I-insulin. J. Clin. Invest. 61, 197–208 (1978).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
van de Bunt, M. et al. Insights into islet development and biology through characterization of a human iPSC-derived endocrine pancreas model. Islets 8, 83–95 (2016).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Krentz, N. A. J. et al. Phosphorylation of NEUROG3 links endocrine differentiation to the cell cycle in pancreatic progenitors. Dev. Cell 41, 129–142.e6 (2017).
Perez-Alcantara, M. et al. Patterns of differential gene expression in a cellular model of human islet development, and relationship to type 2 diabetes predisposition. Diabetologia 61, 1614–1622 (2018).
Harries, L. W., Hattersley, A. T. & Ellard, S. Messenger RNA transcripts of the hepatocyte nuclear factor-1α gene containing premature termination codons are subject to nonsense-mediated decay. Diabetes 53, 500–504 (2004).
Merriman, C., Li, H., Li, H. & Fu, D. Highly specific monoclonal antibodies for allosteric inhibition and immunodetection of the human pancreatic zinc transporter ZnT8. J. Biol. Chem. 293, 16206–16216 (2018).
Ravassard, P. et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest. 121, 3589–3597 (2011).
Thomsen, S. K. et al. Systematic functional characterization of candidate causal genes for type 2 diabetes risk variants. Diabetes 65, 3805–3811 (2016).
Chandra, V. et al. RFX6 regulates insulin secretion by modulating Ca2+ homeostasis in human β cells. Cell Rep. 9, 2206–2218 (2014).
Ramracheya, R. et al. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes 59, 2198–2208 (2010).
Ottosson-Laakso, E. et al. Glucose-induced changes in gene expression in human pancreatic islets: causes or consequences of chronic hyperglycemia. Diabetes 66, 3013–3028 (2017).
Fadista, J. et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl Acad. Sci. USA 111, 13924–13929 (2014).
Rosengren, A. H. et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 327, 217–220 (2010).
Taneera, J. et al. Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Hum. Mol. Genet. 24, 1945–1955 (2015).
Bonetti, S. et al. Variants of GCKR affect both β-cell and kidney function in patients with newly diagnosed type 2 diabetes: the Verona newly diagnosed type 2 diabetes study 2. Diabetes Care 34, 1205–1210 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Acknowledgements
We thank the Botnia Study Group for recruiting and studying the participants, J. J. Holst for measuring GLP-1 concentrations and L. Boselli for carrying out mathematical modeling of the OGTT studies. We thank D. Fu (Department of Physiology, The Johns Hopkins School of Medicine) for providing monoclonal anti-ZnT8 antibody. We thank W.-h. Li (Departments of Cell Biology and of Biochemistry, University of Texas Southwestern Medical Center) for providing zinc probe ZIMIR. We thank E. Na for her help with the mouse immunohistochemistry and histology, and C. Green and the Chromosome Dynamics & Genome Engineering Cores at the Wellcome Centre for Human Genetics for support with karotyping and genome editing (funded by the Welcome Trust grant no. 203141). We thank the Sequencing Unit core facility at FIMM Technology Centre supported by University of Helsinki and Biocenter Finland. The Botnia and the PPP-Botnia studies (L.G., T.T.) have been financially supported by grants from Folkhälsan Research Foundation, the Sigrid Juselius Foundation, the Academy of Finland (grant nos 263401, 267882, 312063, to L.G., 312072, to T.T. and 317599, to O.P.D.), Nordic Center of Excellence in Disease Genetics, EU (no. EXGENESIS, EUFP7-MOSAIC FP7-600914), Ollqvist Foundation, Swedish Cultural Foundation in Finland, Finnish Diabetes Research Foundation, Foundation for Life and Health in Finland, Signe and Ane Gyllenberg Foundation, Finnish Medical Society, Paavo Nurmi Foundation, Helsinki University Central Hospital Research Foundation, Perklén Foundation, Närpes Health Care Foundation and Ahokas Foundation, as well as by the Ministry of Education in Finland, Municipal Heath Care Center and Hospital in Jakobstad and Health Care Centers in Vasa, Närpes and Korsholm. The work described in this paper has been supported with funding from collaborative agreements with Pfizer Inc., as well as with Regeneron Genetics Center LLC. The work was also supported by Hjelt Foundation (L.G. and C.B.W.) and Rhapsody (L.G.). J.O.L. was supported by Vinnova (Sweden’s Innovation Agency) (grant no. 2015-01549), Swedish Diabetes Foundation, Albert Påhlsson Foundation, Hjelt Foundations, Crafoord Foundation, Royal Physiographic Society in Lund, Swedish Foundation for Strategic Research (grant no. IRC15-0067), Swedish Research Council (grant no. 2009-1039, Strategic research area Exodiab). E.A. was supported by Crafoord Foundation, Påhlsson Foundation, Swedish Research Council (grant no. Dnr: 2017-02688). O.H. was supported by Diabetes Research Foundation. R.C.B. was supported by Italian Ministry of University and Research (grant no. PRIN 2015373Z39_004) and University of Parma Research Funds. G.R. was supported by a Wellcome Trust Senior Investigator Award (no. WT098424AIA), MRC Programme grants (nos. MR/R022259/1, MR/J0003042/1, MR/L020149/1) and Experimental Challenge Grant (no. DIVA, MR/L02036X/1), MRC (grant no. MR/N00275X/1), Diabetes UK (grant nos. BDA/11/0004210, BDA/15/0005275, BDA 16/0005485) and Imperial Confidence in Concept grants and a Royal Society Wolfson Research Merit Award. A.L.G. is a Wellcome Trust Senior Fellow in Basic Biomedical Science. M.I.M. and P.R. are Wellcome Senior Investigators. This work was funded in Oxford by the Wellcome Trust (grant nos. 095101 and 200837 to A.L.G., 098381 to M.I.M., 106130 to A.L.G. and M.I.M., 203141 to A.L.G., B.D. and M.I.M., 203141 to. M.I.M. and 090531 to P.R.), Medical Research Council (grant no. MR/L020149/1 to M.I.M., A.L.G. and P.R.), European Union Horizon 2020 Programme (T2D Systems) (A.L.G.) and NIH (grant nos. U01-DK105535 and U01-DK085545 to M.I.M. and A.L.G.). The research was funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (A.L.G., M.I.M., P.R.). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
M.L., L.S., T.T. and L.G. conducted the human study. E.A., O.H., A.B., O.P.D. and J.F. analyzed the genotype data. M.L., O.P.D., M.T., E.B., R.C.B., T.T. and L.G. analyzed the human data. B.H., A.G., N.L.B., S.K.T., M.v.d.B., V.C., O.P.D., T.O. and A.L.G. characterized the human β cell model. N.A.J.K., F.A., N.L.B., B.C., D.M., P.K., B.D., O.P.D., A.S., M.I.M. and A.L.G. characterized the human iPSC-derived model. U.K., R.B.P., O.P.D., B.H., A.J.P., I.S., R.R., I.A., P.R., M.I.M. and A.L.G. characterized the human islets. S.K., D.G. and J.G. characterized the Slc30a8 p.Arg138* mice. D.J., J.O.L., P.C., A.T., R.C., A.-M.R., J.B. and G.A.R. characterized the rat insulinoma cell line. M.I.M., A.L.G., T.T. and L.G. supervised the project. O.P.D., M.L., B.H., N.A.J.K., S.K., P.R., C.B.W., A.L.G., T.T. and L.G. wrote the manuscript. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
L.G. has received research funding from Pfizer Inc., Regeneron Pharmaceuticals, Eli Lilly and Astra Zeneca. N.L.B. and M.v.d.B are now employees of Novo Nordisk, although all experimental work was carried out under employment at the University of Oxford. A.L.G. has received honoraria from Novo Nordisk and Merck. M.I.M. serves on advisory panels for Pfizer, Novo Nordisk, Zoe Global; has received honoraria from Pfizer, Novo Nordisk and Eli Lilly; has stock options in Zoe Global; has received research funding from Abbvie, Astra Zeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, Novo Nordisk, Pfizer, Roche, Sanofi Aventis, Servier, Takeda. G.A.R. is a consultant for Sun Pharma and has received grant funding from Servier. J.O.L. has received research funding from Pfizer Inc. and Novo Nordisk A/S.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Families of SLC30A8-p.Arg138* carriers involved in genotype-based recall for human in vivo study.
Families (≥ 2 members per family) of SLC30A8-p.Arg138* carriers participated in genotype-based recall (test meal) study. To protect anonymity of the carriers, the gender of the offspring is not revealed and some pedigrees have been split to smaller nuclear families. The carrier status of p.Arg138* is shown by yellow (p.Arg138Arg) or magenta (p.Arg138*), that of p.Trp325Arg by vertical (p.Arg325Arg), horizontal (p.Trp325Trp) or diagonal (p.Trp325Arg) lines. The white color (with no lines) represents individuals with unknown status for genotype and phenotype.
Extended Data Fig. 2 Association of SLC30A8 p.Arg138* and p.Trp325Arg with free fatty acids, hormones, and insulin clearance during test meal.
Association of SLC30A8 p.Arg138* and p.Trp35Arg variant with a, serum (S)-C-peptide b, S-proinsulin c, plasma (P)-Glucagon d, Total S-GLP-1 e, S-free fatty acid (FFA) concentrations f, Insulin-C-peptide ratio and g, model-based insulin clearance index during test meal. Left panel: Carriers (red, n=50-54) vs. non-carriers (black, n=37-47) of p.Arg138*. Middle panel: Carriers of p.Arg138* (red, n=50-54) vs. p.Arg138Arg having the common risk variant p.Arg325 (blue, n=25-31). Right panel: Carriers of p.Trp325Trp (grey, n=12-16) vs. p.Arg325 (blue, n=25-31). Exact numbers used for genetic association analysis are available in Source Data files. Data are Mean ±SEM; A star (*p < 0.05, ** p < 0.01) indicates significance in family based association (using QTDT35) after 100,000 permutations, adjusted for age, sex and BMI for left panel and age, sex, BMI and genotype of p.Trp325Arg for middle panel. A hash sign (# p < 0.05) indicates significance in QFAM (as implemented in PLINK36) test using 100,000 permutations (see Methods).
Extended Data Fig. 3 Effect of p.Trp325Arg genotype on insulin secretion during intravenous glucose tolerance test (IVGTT) and β-cell sensitivity to glucose during OGTT.
a-b, (a) Serum (S-) insulin concentrations, p.Trp325Trp (grey, n=116) and p.Arg325 (blue, n=733), and (b) S-insulin -plasma glucose ratio, p.Trp325Trp (grey, n=86) and p.Arg325 (blue, n=458) during IVGTT. Data are Mean ± SEM. Analysis was performed using mixed model adjusting for age, sex, BMI and genetic relatedness. * p < 0.05. c, β-cell sensitivity to glucose is presented as insulin secretion rate in response to plasma glucose during oral glucose tolerance test (OGTT) in people with newly diagnosed type 2 diabetes58. Data are Mean ± SEM. Analysis was performed using a generalized linear model (log-transformed data) for repeated measures, adjusting for age, sex and BMI.
Extended Data Fig. 4 Generation of SLC30A8-p.Lys34Serfs50* and SLC30A8-p.Arg138* hiPSC lines.
a, CRISPR-Cas9 strategy to generate SLC30A8-p.Lys34Serfs50* (Exon 2) and SLC30A8-p.Arg138* (Exon 3) hiPSC lines. Orange font highlights the nucleotide changes: c.101-107del; p.Lys34Serfs50* and c.412C>T; p.Arg138*. The gRNA (blue font) and PAM sequences (red font) are indicated on the partial genomic sequence of SLC30A8. b-c, FACS data from undifferentiated b, SLC30A8-p.Arg138* and c, SLC30A8-p.Lys34Serfs50* iPSCs and relevant isotype controls using antibodies against: OCT3/4, SSEA, SOX2, and NANOG. d, Expression of INSULIN in hiPSC-derived beta-like cells. Black bars represent p.Arg138Arg control cells, red bars represent p.Arg138*, and yellow bars represent p.Lys34Serfs50*. (n=6-8 wells from three differentiations) e-g, RNAscope analysis of the number of e, INSULIN- and f, SLC30A8- transcript positive cells in hiPSC-derived beta-like cells. 7-21 image fields were quantified and presented as % of DAPI+ cells. Representative images used for quantification shown in g (scale bar = 50 µm). Data are presented as Mean±SEM. Statistical analysis was performed using the one-way ANOVA and Tukey’s multiple comparison test (n = 5-9 wells from three differentiations, ****p<0.0001).
Extended Data Fig. 5 Confirmation of the ddPCR probe specificity and target SLC30A8 mRNA sequencing.
a, R138 (pGEM_CT) and X138 (pGEM_TT) sequences were inserted in pGEM vector and used as template for digital droplet PCR. Original probe configuration confirmed specificity as R138 droplets were only detected by FAM (CT-FAM; channel 1) and X138 droplets were only detected by VIC (TT-VIC; channel 2). In the swapped probe configuration, FAM and VIC probes were swapped and ddPCR was performed using pGEM_CT or pGEM_TT as template. b, Detection of R138 allele (Channel 1) and X138 allele (Channel 2) using cDNA from the heterozygous hiPSC-derived beta-like cells (B1 clone) as a template. In the swapped probe configuration, FAM and VIC probes were swapped and ddPCR was performed using cDNA from hiPSC-derived beta-like cells as template. c, Depicting the unique sequencing reads coverage at p.Arg138* and p.Ala139Ala obtained by SLC30A8 target mRNAs sequencing in edited clones (B1 and A3) and unedited cells (wildtype).
Extended Data Fig. 6 Silencing of ZnT8 tends to lower granule Zn2+ content.
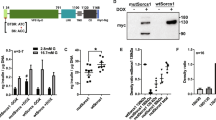
EndoC-βH1 cells were transfected with siRNA control (siCtrl) or targeted against SLC30A8 (siZnT8) for 72 hours prior to imaging. a, ZnT8 knock-down was confirmed at the protein level by Western-blot. b, Control cells were incubated for 20 min. with the membrane-targeted zinc probe ZIMIR15 to monitor zinc secretion after cell stimulation with 20 mM KCl using total internal reflection of fluorescence (TIRF) microscopy (see accompanying movies, Supplementary video 1–4). c, Fluorescence intensity at the membrane was monitored upon time and traces obtained were averaged for cells transfected with siCtrl (14 cells) or with siZnT8 (14 cells). d, Fluorescence intensity increase due to zinc secretion after stimulation with KCl was determined for each cell. An outlier data point in the siZnT8 condition, likely to reflect release from a non-silenced cell, was excluded by Grubb’s test and statistical significance determined by Student’s t-test (Graph Pad Prism 7.0). Scale bar in b, 5 µm. Blots have been cropped and corresponding full blots are available in Source Data files.
Extended Data Fig. 7 RNA (mRNAs) sequencing of SLC30A8 knock down and control EndoC-βH1 cells.
Effect of SLC30A8 knock down (KD) on expression of genes involved in a, proinsulin processing, b, insulin production and β-cell development, c, β-cell excitability and insulin exocytosis (Supplementary Dataset 1). d, Over-representation analysis of differentially expressed genes (red dots) and depiction of most enriched WNT pathway genes (Supplementary Dataset 1) along with a gene set enrichment analysis (GSEA) considering all expressed genes. *p<0.05, ***p<0.001; Differential expression analysis was done using sequencing reads count based data using method similar to Fisher’s Exact test as implemented in edgR software package59 and further corrected for multiple testing using Bonferroni correction (see Supplementary Note).
Extended Data Fig. 8 Expression and localization of p.Arg138* and impact on cytosolic free Zn2+ concentrations in cultured INS1 β-cells.
a-d, Rat INS1e cells were transiently transfected with p.Arg138*-mCherry fusion construct followed by fluorescence microscopy imaging and immunodetection. a, Fusion protein localized to distinct subcellular compartments in INS1e cells at 48 h and 96 h after transfection. b, Expression of mCherry in control INS1e cells indicated cytoplasmic localization. c, Control experiments with immunostaining of p.Arg138* with HA or Myc-His (both are significantly smaller additions than mCherry) confirmed localization of fusion proteins to distinct subcellular compartments in the INS1e cells. d, Immunological detection (anti-mCherry) of the fusion protein at indicated time points after transfection confirms protein expression and indicate protein stability. Tubulin is used as control. e-f, INS1(832/13) cells were transfected constructs expressing p.Arg138*-Myc-His or eCALWY-4, or co-transfected with both, followed by (e) immunostaining or (f) immunofluorescence imaging at 24 h post-transfection using anti-c-Myc antibody. g, Cytosolic free Zn2+ concentrations in INS-1 (832/13) cells. Data are combined from three fully independent experiments. Scale bars are 50 μm (a, b), 10 μm (c) and 25 μm (f). Blots have been cropped and corresponding full blots are available in Source Data files.
Supplementary information
Supplementary Information
Supplementary Tables 1–8 and Note
Supplementary Video 1
Supplementary Video 1
Supplementary Video 2
Supplementary Video 2
Supplementary Video 3
Supplementary Video 3
Supplementary Video 4
Supplementary Video 4
Rights and permissions
About this article

Cite this article
Dwivedi, O.P., Lehtovirta, M., Hastoy, B. et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet 51, 1596–1606 (2019). https://doi.org/10.1038/s41588-019-0513-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0513-9
This article is cited by
-
Defining type 2 diabetes polygenic risk scores through colocalization and network-based clustering of metabolic trait genetic associations
Genome Medicine (2024)
-
Cellular zinc metabolism and zinc signaling: from biological functions to diseases and therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Zinc and iron dynamics in human islet amyloid polypeptide-induced diabetes mouse model
Scientific Reports (2023)
-
Emerging therapeutic options in the management of diabetes: recent trends, challenges and future directions
International Journal of Obesity (2023)
-
Stomach-derived human insulin-secreting organoids restore glucose homeostasis
Nature Cell Biology (2023)
