
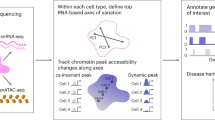
Abstract
A hallmark of the immune system is the interplay among specialized cell types transitioning between resting and stimulated states. The gene regulatory landscape of this dynamic system has not been fully characterized in human cells. Here we collected assay for transposase-accessible chromatin using sequencing (ATAC-seq) and RNA sequencing data under resting and stimulated conditions for up to 32 immune cell populations. Stimulation caused widespread chromatin remodeling, including response elements shared between stimulated B and T cells. Furthermore, several autoimmune traits showed significant heritability in stimulation-responsive elements from distinct cell types, highlighting the importance of these cell states in autoimmunity. Allele-specific read mapping identified variants that alter chromatin accessibility in particular conditions, allowing us to observe evidence of function for a candidate causal variant that is undetected by existing large-scale studies in resting cells. Our results provide a resource of chromatin dynamics and highlight the need to characterize the effects of genetic variation in stimulated cells.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
Our RNA-seq, ATAC-seq and ChIP-seq data have been deposited with the Gene Expression Omnibus (GEO): RNA-seq data (accession no. GSE118165); ATAC-seq data (accession no. GSE118189); ChIP-seq data (accession no. GSE126505). Progenitor data have been deposited with the GEO under accession no. GSE74912. Additional supplementary information can be retrieved from the Pritchard Lab Data website (http://web.stanford.edu/group/pritchardlab/dataArchive.html).
References
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Pickrell, J. K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94, 559–573 (2014).
Hu, X. et al. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am. J. Hum. Genet. 89, 496–506 (2011).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Trynka, G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 45, 124–130 (2013).
Chen, L. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414.e24 (2016).
Chun, S. et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet. 49, 600–605 (2017).
Kim-Hellmuth, S. et al. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 8, 266 (2017).
Alasoo, K. et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 50, 424–431 (2018).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
Ye, C. J. et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014).
Simeonov, D. R. et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017).
Huang, H. et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178 (2017).
Onengut-Gumuscu, S. et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 47, 381–386 (2015).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Moskowitz, D. M. et al. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2, eaag0192 (2017).
van der Veeken, J. et al. Memory of inflammation in regulatory T cells. Cell 166, 977–990 (2016).
He, B. et al. CD8+ T cells utilize highly dynamic enhancer repertoires and regulatory circuitry in response to infections. Immunity 45, 1341–1354 (2016).
Yu, B. et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol. 18, 573–582 (2017).
Leslie, R., O’Donnell, C. J. & Johnson, A. D. GRASP: analysis of genotype–phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 30, i185–i194 (2014).
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Gate, R. E. et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat. Genet. 50, 1140–1150 (2018).
Hess, K. et al. Kinetic assessment of general gene expression changes during human naive CD4+ T cell activation. Int. Immunol. 16, 1711–1721 (2004).
Diehn, M. et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl Acad. Sci. USA 99, 11796–11801 (2002).
Trickett, A. & Kwan, Y. L. T cell stimulation and expansion using anti-CD3/CD28 beads. J. Immunol. Methods 275, 251–255 (2003).
Wortis, H. H., Teutsch, M., Higer, M., Zheng, J. & Parker, D. C. B-cell activation by crosslinking of surface IgM or ligation of CD40 involves alternative signal pathways and results in different B-cell phenotypes. Proc. Natl Acad. Sci. USA 92, 3348–3352 (1995).
Van Belle, K. et al. Comparative in vitro immune stimulation analysis of primary human B cells and B cell lines. J. Immunol. Res. 2016, 5281823 (2016).
Hodgkin, P. D., Go, N. F., Cupp, J. E. & Howard, M. Interleukin-4 enhances anti-IgM stimulation of B cells by improving cell viability and by increasing the sensitivity of B cells to the anti-IgM signal. Cell. Immunol. 134, 14–30 (1991).
Rieckmann, J. C. et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 18, 583–593 (2017).
Degner, J. F. et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–394 (2012).
Kilpinen, H. et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342, 744–747 (2013).
Kasowski, M. et al. Extensive variation in chromatin states across humans. Science 342, 750–752 (2013).
McVicker, G. et al. Identification of genetic variants that affect histone modifications in human cells. Science 342, 747–749 (2013).
Neph, S. et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 (2012).
van de Geijn, B., McVicker, G., Gilad, Y. & Pritchard, J. K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063 (2015).
Stephens, M. False discovery rates: a new deal. Biostatistics 18, 275–294 (2017).
Banovich, N. E. et al. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res. 28, 122–131 (2018).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Walsh, A. M. et al. Integrative genomic deconvolution of rheumatoid arthritis GWAS loci into gene and cell type associations. Genome Biol. 17, 79 (2016).
Ardlie, K. G. et al. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Krikos, A., Laherty, C. D. & Dixit, V. M. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, A20, is mediated by κB elements. J. Biol. Chem. 267, 17971–17976 (1992).
Housley, W. J. et al. Genetic variants associated with autoimmunity drive NFκB signaling and responses to inflammatory stimuli. Sci. Transl. Med. 7, 291ra93 (2015).
Calderon, D. et al. Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet. 101, 686–699 (2017).
Bank, S. et al. Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenomics J. 14, 526–534 (2014).
Thomson, W. et al. Rheumatoid arthritis association at 6q23. Nat. Genet. 39, 1431–1433 (2007).
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R. & Pfister, H. UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 20, 1983–1992 (2014).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Schep, A. N. et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 25, 1757–1770 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 (2003).
Acknowledgements
We thank D. Yao for helping process the samples, C. J. Ye and members of the Greenleaf, Marson and Pritchard laboratories for helpful conversations and manuscript feedback. We relied on the Flow Cytometry Core at UCSF, which was supported by Diabetes Research Center grant nos. NIH P30 DK063720 and 1S10OD021822-01, and sequencing data generated by the Stanford Functional Genomics Facility on an Illumina HiSeq 4000 that was purchased with funds from the National Institutes of Health (NIH) under award no. S10OD018220. Sequencing that was generated on an Illumina NovaSeq was supported by the Chan Zuckerberg Biohub. Some of the computing for this project was performed on the Sherlock cluster. Sequencing of the ChIP-seq libraries was supported by the UCSF Center for Advanced Technology. We thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. Support for D.C. was provided by National Library of Medicine training grant no. T15LM007033. A. Mezger is supported by the Swedish Research Council (grant no. 2015-06403). F.B. was supported by the Care-for-Rare Foundation and NIH/National Institute of General Medical Sciences funding for the HIV Accessory & Regulatory Complexes Center (no. P50 GM082250; A.Marson). This work was supported by NIH grants (no. 1R01HG008140—J.P. serves as principal investigator with subcontract to A. Marson and L.A.C. at UCSF; no. P30AR070155 to L.A.C.; no. DP3DK111914-01 to A. Marson; no. P50HG007735 to W.J.G.; no. UM1HG009442 to W.J.G.; no. U19AI057266 to W.J.G.), the Howard Hughes Medical Institute (J.K.P.), the Rheumatology Research Foundation (L.A.C.), the UCSF-Stanford Arthritis Center of Excellence (L.A.C.) (supported in part by the Arthritis Foundation), the Rita Allen Foundation (W.J.G.), the Human Frontiers Science Program grant no. RGY006S (W.J.G.), the Burroughs Wellcome Fund (A. Marson) and the National Multiple Sclerosis Society (A. Marson; no. CA 1074-A-21). Both W.J.G. and A. Marson are supported by the Chan Zuckerberg Biohub. Additionally, A. Marson holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, has received funding from the Innovative Genomics Institute and is supported by the Parker Institute for Cancer Immunotherapy.
Author information
Authors and Affiliations
Contributions
D.C., M.L.T.N., A. Mezger, L.A.C., W.J.G., A. Marson and J.K.P. conceptualized the study. M.L.T.N., A. Mezger, A.K., V.N., N.L., B.W., J.T., F.B. and A.V.P. carried out the investigation. D.C., F.M., D.A.K., Z.G. and J.V.R. carried out the formal analysis. A.K. and F.M. contributed equally to merit second authorship. M.S.A., T.D.B, W.J.G., A. Marson and J.K.P. were responsible for obtaining the resources. L.A.C., W.J.G., A. Marson and J.K.P. acquired the funding. D.C., M.L.T.N. and A. Mezger wrote the original draft. D.C., M.L.T.N., A. Mezger, L.A.C., W.J.G., A. Marson and J.K.P. reviewed and edited the draft. L.A.C., W.J.G., A. Marson and J.K.P. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
Stanford University has filed a provisional patent application on the methods described and W.J.G. is named as an inventor. W.J.G. is a cofounder of Epinomics and consultant for 10x Genomics and Guardant Health. A. Marson is a cofounder of Arsenal Biosciences and Spotlight Therapeutics. A.Marson serves as on the scientific advisory board of PACT Pharma, is an advisor to Trizell and was a former advisor to Juno Therapeutics. The Marson Laboratory has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and a gift from Gilead.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Fig. 1 Gating strategies and surface markers for cell sorting.
Representative flow cytometry plots of the sorting panels for each population of immune cells. Monocytes, bulk CD4+ T and B cells were positive enriched with magnetic beads prior to sorting. Pan T cells, DCs and NK cells were negatively enriched.
Supplementary Fig. 2 Overview of ATAC-seq and RNA-seq data.
a, Enrichment of ATAC-seq reads relative to RefSeq TSSs for B cells (top) and CD4+ T cells (bottom). b, Examples of gene expression across lineages, y-axis represents log2 (count per million (CPM)). c, Correlogram of Pearson’s R2 for ATAC (left) and RNA (right) for all overlapping samples. d, RNA-seq tSNE of batch corrected read count matrix at protein coding genes. Samples sizes can be found in Supplementary Table 1.
Supplementary Fig. 3 Functional enrichment of differentially accessible regions.
Enrichment analysis of genome-wide overlap between regions within corresponding sets (above) and previously published functional data using the LOLA tool (Sheffield, N.C. et al., Bioinformatics. 32, 587-9, 2016). The heatmap shows the combined rank (lower values correspond to higher enrichments) for the most significantly enriched datasets (see Methods). Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 4 IL23R vignette and gene expression of specific TFs.
a, Vignette highlighting accessibility surrounding IL23R. We have highlighted significant GWAS variants from several autoimmune disorders. b, Gene expression of various important TFs. Sample sizes can be found in Supplementary table 1.
Supplementary Fig. 5 Characterization of ATAC peaks and gene expression.
a, Volcano plot of differentially accessible chromatin regions (see Methods) for mature NK (left) and monocytes (right). b, PCA of resting (blue) and stimulated (red) ATAC-seq samples including the innate cells (triangles). c, MACS2 peak count across different cell types stratified by condition. MACS2 peak counts were normalized for read count and sample quality effects. The dotted lines indicate the median count of accessible regions in stimulated (red) and resting (blue) samples. d, Heatmap of chromVar TF binding deviation scores (see Methods) for several PWMs (y-axis) and samples (x-axis). e, Gene expression of SPI1 separated by lineage. f, Expression of BATF grouped by lineage and condition. Resting samples indicated in blue and stimulated samples in red. g, Expression of BATF target genes in B cells (left) and Th17 precursors (right). Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 6 Examining monocyte stimulation.
a, ATAC-seq profile surrounding the CD14 gene. There are accessibility peaks specific to dendritic cells and monocytes, which express CD14. b, Comparison of stimulation gene expression effects estimated from monocytes from this study and Alasoo, K. et al, Nature Genetics. 50, 424-431, 2018. We list the Pearson’s correlation between the effects estimated from each data source. c, Monocyte-specific PCA plot based on RNA-seq (top) and ATAC-seq (bottom) data. The donor of the specific sample is labeled. Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 7 Surface expression of CD69 of activated samples.
Representative histogram plots showing CD69 expression by flow cytometry. Line indicates gating of CD69 positive cells in reference to negative control. These experiments were not repeated. a, Surface expression of CD69 at 6hrs post stimulation. b, Surface expression of CD69 at 24hrs post stimulation.
Supplementary Fig. 8 Pathway analysis of stimulation-specific genes and correlation between promoter accessibility and gene expression.
a, Heatmap of pathway enrichment analysis results from all significant differentially expressed genes from each cell subset (see Methods). We have included the top 3 pathways from several pathway databases (keg=KEG pathways, rea=REACTOME, BP=GO: biological process, CC=GO:cellular component, MF=GO:molecular function). b, Pearson’s correlation between promoter accessibility and gene expression in resting state samples (top) and stimulated samples (bottom). c, Promoter accessibility boxplot of stimulation-associated genes in samples in a resting state and a stimulated state. Sample sizes can be found in Supplementary Table 1. A two-sided p-value test was performed.
Supplementary Fig. 9 Stimulation-specific chromatin imbalance in Th1 precursor cells.
a, Changes in the proportion of reads mapping to the reference allele at heterozygous sites as a function of stimulation condition and PWM-predicted JUNB, BACH1 and FOSL1 binding affinity. The PWM associated with each TF is visualized below. We display the p-value computed from a Kruskal-Wallis test. b, Similar to Fig. 4d but displaying sharing of allele-specific chromatin effects for each donor. c, Similar to (b) but visualized as a boxplot. Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 10 GWAS enrichment at chromatin accessible sites.
a, Significance of heritability enrichment between samples and complex traits (see Methods). b, Trait heritability enrichment of samples from various lineage and condition classes. Resting samples indicated in blue and stimulated samples in red. Innate cells were excluded. c, Heatmap showing top 100 accessible peaks (y-axis) for lineage clusters (labeled) that are unique for different cell lineages conditions across samples (x-axis). Increasingly accessible regions are colored red corresponding to a larger scaled intensity value (see Methods). d, Definition of peak profiles. Peak profiles are labeled on the y-axis, which are defined by accessibility in various lineage-condition groups on the x-axis. Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 11 Additional GWAS and eQTL enrichment in sites of allele-specific chromatin.
a, Similar to Fig. 6a, however we included an additional null set of SNPs (green). The new null set represent the entire set of heterozygous variants where we computed a p-value. b, Similar to Fig. 6b, however we used eQTLs from naïve T cells that were previously published (Chen, L. et al., Cell. 167, 1398-1414 e24, 2016). Sample sizes can be found in Supplementary Table 1. We computed p-values with a two-sided Mann–Whitney U test.
Supplementary Fig. 12 ATAC-seq profile of TNFAIP3 locus.
Visualization of chromatin accessibility surrounding the TNFAIP3 gene, across all samples. Darker colored regions indicate increased accessibility of resting (blue) and stimulated (red) samples. A box highlights the candidate GWAS variant.
Supplementary Fig. 13 Putative regulation of TFNAIP3.
a, UC-associated variants that overlap accessible regions at the TNFAIP3 locus and the distribution of stimulation-effects across all cell types. The variants rs9321624, rs9321625, and rs9402908 overlap the same accessible region. b, The distribution of PWM-predicted NFκB1 binding affinity across all heterozygous sites from four donors. The reference allele at chr6:138002175 is represented by the solid vertical line, and the alternative is represented by the dotted vertical line. c, Gene expression measured for NFκB1 grouped by lineage and condition. Sample sizes can be found in Supplementary Table 1.
Supplementary Fig. 14 Proposed regulatory model.
a, CD4+ T cells at rest, TNFAIP3 is constitutively expressed and candidate region is inaccessible. TNFAIP3 inactivates NFκB1. b, Upon initial stimulation, expression of TNFAIP3 is decreased (resulting in activation of NFκB1) and the candidate region becomes accessible. c, We propose that NFκB1 binds to the candidate variant to enhance the expression of TNFAIP3. d, Illustration of the overall regulatory network, by which rs6927172 has an effect on autoimmune disease risk.
Supplementary information
Supplementary Text and Figures
Supplementary Note and Supplementary Figs. 1–14.
Supplementary Table 1
Additional supplementary tables. Miscellaneous collection of tables with additional study information.
Dataset 1
Differentiation ATAC-seq peaks. Significant differentiation-associated ATAC-seq peaks.
Dataset 2
Differentiation genes. Significant differentiation-associated RNA-seq genes.
Dataset 3
Stimulation ATAC-seq peaks. Significant stimulation-associated ATAC-seq peaks.
Dataset 4
Stimulation genes. Significant stimulation-associated RNA-seq genes.
Dataset 5
Aggregated GWAS and functional information. Table with significant GWAS, variants with allele-specific chromatin accessibility that disrupt PWM-predicted transcription factor binding, regions of differential accessibility and nearby differentially expressed genes.
Rights and permissions
About this article

Cite this article
Calderon, D., Nguyen, M.L.T., Mezger, A. et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat Genet 51, 1494–1505 (2019). https://doi.org/10.1038/s41588-019-0505-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0505-9
This article is cited by
-
Mapping the functional impact of non-coding regulatory elements in primary T cells through single-cell CRISPR screens
Genome Biology (2024)
-
InPACT: a computational method for accurate characterization of intronic polyadenylation from RNA sequencing data
Nature Communications (2024)
-
Mapping the epigenomic landscape of human monocytes following innate immune activation reveals context-specific mechanisms driving endotoxin tolerance
BMC Genomics (2023)
-
High-capacity sample multiplexing for single cell chromatin accessibility profiling
BMC Genomics (2023)
-
Characterizing cis-regulatory elements using single-cell epigenomics
Nature Reviews Genetics (2023)
