
Abstract
After a decade of genome-wide association studies (GWASs), fundamental questions in human genetics, such as the extent of pleiotropy across the genome and variation in genetic architecture across traits, are still unanswered. The current availability of hundreds of GWASs provides a unique opportunity to address these questions. We systematically analyzed 4,155 publicly available GWASs. For a subset of well-powered GWASs on 558 traits, we provide an extensive overview of pleiotropy and genetic architecture. We show that trait-associated loci cover more than half of the genome, and 90% of these overlap with loci from multiple traits. We find that potential causal variants are enriched in coding and flanking regions, as well as in regulatory elements, and show variation in polygenicity and discoverability of traits. Our results provide insights into how genetic variation contributes to trait variation. All GWAS results can be queried and visualized at the GWAS ATLAS resource (https://atlas.ctglab.nl).
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

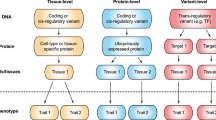
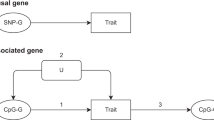
Data availability
All publicly available GWAS summary statistics (original) files curated in this study are accessible from the original links provided at https://atlas.ctglab.nl. GWAS summary statistics for 600 traits from UK Biobank performed in this study are also provided at https://atlas.ctglab.nl and an archived file will be made available upon publication from https://ctg.cncr.nl/software/summary_statistics.
Change history
06 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Edwards, A. O. et al. Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–425 (2005).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Lander, E. S. Initial impact of the sequencing of the human genome. Nature 470, 187–197 (2011).
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Henderson, P. & Stevens, C. The role of autophagy in Crohn’s disease. Cells 1, 492–519 (2012).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Gaulton, K. J. et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet. 47, 1415–1425 (2015).
Canela-Xandri, O., Rawlik, K. & Tenesa, A. An atlas of genetic associations in UK Biobank. Nat. Genet. 50, 1593–1599 (2018).
Timpson, N. J., Greenwood, C. M. T., Soranzo, N., Lawson, D. J. & Richards, J. B. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 19, 110–124 (2018).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Wray, N. R., Wijmenga, C., Sullivan, P. F., Yang, J. & Visscher, P. M. Common disease is more complex than implied by the core gene omnigenic model. Cell 173, 1573–1580 (2018).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Goh, K. et al. The human disease network. Proc. Natl Acad. Sci. USA 104, 8685–8690 (2007).
Polderman, T. J. C. et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Bulik-sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Solovieff, N., Cotsapas, C., Lee, P. H., Purcell, S. M. & Smoller, J. W. Pleiotropy in complex traits: challenges and strategies. Nat. Rev. Genet. 14, 483–495 (2013).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
The GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Lee, S., Abecasis, G. R., Boehnke, M. & Lin, X. Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23 (2014).
van de Bunt, M., Cortes, A., Brown, M. A., Morris, A. P. & McCarthy, M. I. Evaluating the performance of fine-mapping strategies at common variant GWAS loci. PLoS Genet. 11, e1005535 (2015).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Speed, D. et al. Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992 (2017).
Speed, D. & Balding, D. J. SumHer better estimates the SNP heritability of complex traits from summary statistics. Nat. Genet. 51, 277–284 (2019).
Holland, D. et al. Beyond SNP heritability: polygenicity and discoverability estimated for multiple phenotypes with a univariate gaussian mixture model. Preprint at https://doi.org/10.1101/133132 (2018).
Frei, O. et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun. 10, 2417 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700,000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Visscher, P. M. et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 10, e1004269 (2014).
Benner, C. et al. Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet. 101, 539–551 (2017).
Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Tak, Y. G. & Farnham, P. J. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin 8, 57 (2015).
Acknowledgements
We thank all consortiums and all other individual laboratories for making GWAS summary statistics publicly available. We also thank P. Visscher and N. Wray for their thoughtful suggestions and discussions. We additionally thank A. Dale for his suggestions. This work was funded by the Netherlands Organization for Scientific Research (grant nos. NWO VICI 453-14-005 and NWO VIDI 452-12-014).
Author information
Authors and Affiliations
Contributions
D.P. designed the study. K.W. curated the database and performed analyses. T.J.C.P. assisted with harmonization of phenotype labels of the database. S.S. performed quality control on the UK Biobank data and wrote the analysis pipeline for UKB analyses. M.U.M. assisted with the fine-mapping analyses. C.d.L. assisted with the discussion of SNP heritability estimates with different models. O.F. and O.A.A. developed software the MiXeR and assisted with the analyses. S.v.d.S. and B.M.N. discussed and provided valuable suggestions for analyses. K.W. and D.P. wrote the paper. All authors critically reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note and Supplementary Figs. 1–12
Supplementary Tables 1–26
Supplementary Tables 1–26
Rights and permissions
About this article

Cite this article
Watanabe, K., Stringer, S., Frei, O. et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet 51, 1339–1348 (2019). https://doi.org/10.1038/s41588-019-0481-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0481-0
This article is cited by
-
Human genetic associations of the airway microbiome in chronic obstructive pulmonary disease
Respiratory Research (2024)
-
Transcriptome-wide association studies associated with Crohn’s disease: challenges and perspectives
Cell & Bioscience (2024)
-
Prioritizing susceptibility genes for the prognosis of male-pattern baldness with transcriptome-wide association study
Human Genomics (2024)
-
Defining type 2 diabetes polygenic risk scores through colocalization and network-based clustering of metabolic trait genetic associations
Genome Medicine (2024)
-
Genetic overlap between Alzheimer’s disease and immune-mediated diseases: an atlas of shared genetic determinants and biological convergence
Molecular Psychiatry (2024)
