
Abstract
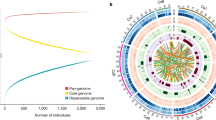
We report a map of 4.97 million single-nucleotide polymorphisms of the chickpea from whole-genome resequencing of 429 lines sampled from 45 countries. We identified 122 candidate regions with 204 genes under selection during chickpea breeding. Our data suggest the Eastern Mediterranean as the primary center of origin and migration route of chickpea from the Mediterranean/Fertile Crescent to Central Asia, and probably in parallel from Central Asia to East Africa (Ethiopia) and South Asia (India). Genome-wide association studies identified 262 markers and several candidate genes for 13 traits. Our study establishes a foundation for large-scale characterization of germplasm and population genomics, and a resource for trait dissection, accelerating genetic gains in future chickpea breeding.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The data that support the findings of this study have been deposited in the NCBI under accession code SRA: SRP096939; BioProject: PRJNA362278, and these data are also available in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP0000370.
References
Mba, C., Guimaraes, E. P. & Ghosh, K. Re-orienting crop improvement for the changing climatic conditions of the 21st century. Agri. & Food Sec. 1, 7 (2012).
Ritchie, H. & Roser, M. Micronutrient deficiency. Our World in Data https://ourworldindata.org/micronutrient-deficiency (2017).
Atlin, G. N., Cairns, J. E. & Das, B. Rapid breeding and varietal replacement are critical to adaptation of cropping systems in the developing world to climate change. Glob. Food Sec. 12, 31–37 (2017).
Abbo, S., Berger, J. & Turner, N. C. Viewpoint: evolution of cultivated chickpea: four bottlenecks limit diversity and constrain adaptation. Funct. Plant Biol. 30, 1081–1087 (2003).
Varshney, R. K. et al. Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor. Appl. Genet. 127, 445–462 (2014).
Thudi, M. et al. Understanding the genetic architecture of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping. PLoS ONE 9, e96758 (2014).
Upadhyaya, H. D. et al. Genomic tools and germplasm diversity for chickpea improvement. Plant Genet. Resour. 9, 45–58 (2011).
Zhou, Z. et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414 (2015).
3K RGP. The 3,000 rice genomes project. GigaScience 3, 7 (2014).
Varshney, R. K. et al. Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 49, 1082–1088 (2017).
Varshney, R. K. et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 35, 969–976 (2017).
Fang, L. et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49, 1089–1098 (2017).
Romero Navarro, J. A. et al. A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nat. Genet. 49, 476–480 (2017).
Xu, X. et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 30, 105–111 (2012).
Lam, H. M. et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059 (2010).
Varshney, R. K., Nayak, S. N., May, G. D. & Jackson, S. A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530 (2009).
Upadhyaya, H. D. et al. Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol. 8, 106 (2008).
Thudi, M. et al. Recent breeding programs enhanced genetic diversity in both desi and kabuli varieties of chickpea (Cicer arietinum L.). Sci. Rep. 6, 38636 (2016).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Mace, E. S. et al. Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum. Nat. Commun. 4, 2320 (2013).
Pritchard, J. K., Stephens, M. & Donnelly, P. J. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Penmetsa, R. V. et al. Multiple post-domestication origins of kabuli chickpea through allelic variation in a diversification-associated transcription factor. New Phytol. 211, 1440–1451 (2016).
Roorkiwal, M. et al. Exploring germplasm diversity to understand the domestication process in Cicer spp. using SNP and DArT markers. PLoS ONE 9, e102016 (2014).
Barrett, J. C. et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Gore, M. A. et al. A first-generation haplotype map of maize. Science 326, 1115–1117 (2009).
Branca, A. et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc. Natl Acad. Sci. USA 108, E864–E870 (2011).
von Wettberg, E. J. et al. Ecology and community genomics of an important crop wild relative as a prelude to agricultural innovation. Nat. Commun. 9, 649 (2018).
Eldon, B., Birkner, M., Blath, J. & Freund, F. Can the site-frequency spectrum distinguish exponential population growth from multiple-merger coalescents? Genetics 199, 841–856 (2015).
Ferretti, L., Ledda, A., Wiehe, T., Achaz, G. & Ramos-Onsins, S. E. Decomposing the site frequency spectrum: the impact of tree topology on neutrality tests. Genetics 207, 229–240 (2017).
Hudson, R. R. et al. Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589 (1992).
Vavilov, N. I. Centres of origin of cultivated plants. Bull. Appl. Bot. Genet. Plant Breed. 16, 1–248 (1926).
Whitlock, M. C. & McCauley, D. E. Indirect measures of gene flow and migration: FST does not=1/(4Nm + 1). Heredity 82, 117–125 (1999).
Redden, R. J. & Berger, J. D. History and origin of chickpea. in Chickpea Breeding and Management (eds Yadav, S. S. et al.) 1–13 (C.A.B. International, 2007).
McCouch, S. et al. Agriculture: feeding the future. Nature 499, 23–24 (2014).
Kamran, A. et al. The effect of VRN1 genes on important agronomic traits in high-yielding Canadian soft white spring wheat. Plant Breed. 133, 321–326 (2014).
Samineni, S. et al. Vernalization response in chickpea is controlled by a major QTL. Euphytica 207, 453–461 (2016).
van der Maesen, L. J. G. in The Chickpea (eds Saxena, M. C. & Singh, R. B.) 11–34 (C.A.B. International, 1987).
Hoffmann, A. A. & Sgrò, C. M. Climate change and evolutionary adaptation. Nature 470, 479–485 (2011).
Rafalski, J. A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 13, 174–180 (2010).
Kashiwagi, J. et al. Traits of relevance to improve yield under terminal drought stress in chickpea (C. arietinum L.). Field Crops Res. 145, 88–95 (2013).
Lynch, J. P. Steep, cheap and deep: an ideotype to optimize water and N acquisition by maize root systems. Ann. Bot. 112, 347–357 (2013).
Li, R. et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Li, S. et al. SOAPindel: Efficient identification of Indels from short paired reads. Genome Res. 23, 195–200 (2013).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigen analysis. PLoS Genet. 2, 2074–2093 (2006).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Felsenstein, J. Phylip: phylogeny inference package (version 3.2). Cladistics 5, 164–166 (1989).
Tamura, K. et al. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 (2007).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014).
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360 (2010).
Lipka, A. E. et al. GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399 (2012).
Tang, Y. et al. GAPIT Version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9, 2 (2016).
Liu, X., Huang, M., Fan, B., Buckler, E. S. & Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12, e1005767 (2016).
Acknowledgements
R.K.V. acknowledges the funding support from CGIAR Generation Challenge Programme, Department of Science and Technology Government of India under the Australia-India Strategic Research Fund, Ministry of Agriculture and Farmers Welfare, Government of India and Bill & Melinda Gates Foundation, USA. Shenzhen Municipal Government of China (grant no. JCYJ20150831201643396 and no. JCYJ20170817145512476 under the Basic Research Program) and the Guangdong Provincial Key Laboratory of Genome Read and Write (grant no. 2017B030301011) are acknowledged to provide support to X.X. and X.L. This work has been undertaken as part of the CGIAR Research Program on Grain Legumes and Dryland Cereals. ICRISAT is a member of the CGIAR Consortium.
Author information
Authors and Affiliations
Contributions
R.K.V. conceived and designed the experiments. R.K.V., X.X. and X.L. coordinated sequencing and genome analysis. W.H., W.Y., J.J., H.D.U., N.P.S., S.K.C., G.V.P.R.N., L.K., A.F., K.K.B.P., P.M.G. and S.M.S. performed the experiments. W.H., P.B., A.R., D.D., V.G., A.W.K., H.D.U., J.C. and Y.V. performed statistical analysis. R.K.V., M.T., M.R., A.C., P.C., C.B., S.T., J.W., S.-H.L., D.E., K.K.B.P., R.V.P., J.C., H.T.N., K.H.M.S., T.D.C., T.S., E.v.W., Y.V., X.X. and X.L. analyzed the data. R.K.V., H.D.U., A.C., P.M.G., N.P.S., S.K.C., G.P.D., D.X., J.W., X.X. and X.L. contributed to the reagents, materials and analysis tools. R.K.V., M.T., M.R., W.H. and X.L. wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15 and Supplementary Note
Supplementary Tables
Supplementary Tables 1–29
Rights and permissions
About this article

Cite this article
Varshney, R.K., Thudi, M., Roorkiwal, M. et al. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet 51, 857–864 (2019). https://doi.org/10.1038/s41588-019-0401-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0401-3
This article is cited by
-
Genome-wide association study, population structure, and genetic diversity of the tea plant in Guizhou Plateau
BMC Plant Biology (2024)
-
Greater ecophysiological stress tolerance in the core environment than in extreme environments of wild chickpea (Cicer reticulatum)
Scientific Reports (2024)
-
A genomic variation map provides insights into peanut diversity in China and associations with 28 agronomic traits
Nature Genetics (2024)
-
Meta-QTL analysis enabled identification of candidate genes and haplotypes for enhancing biotic stress resistance in chickpea
Journal of Plant Biochemistry and Biotechnology (2024)
-
Dissecting chickpea genomic loci associated with the root penetration responsive traits in compacted soil
Planta (2024)
