
Abstract
The oocyte epigenome plays critical roles in mammalian gametogenesis and embryogenesis. Yet, how it is established remains elusive. Here, we report that histone-lysine N-methyltransferase SETD2, an H3K36me3 methyltransferase, is a crucial regulator of the mouse oocyte epigenome. Deficiency in Setd2 leads to extensive alterations of the oocyte epigenome, including the loss of H3K36me3, failure in establishing the correct DNA methylome, invasion of H3K4me3 and H3K27me3 into former H3K36me3 territories and aberrant acquisition of H3K4me3 at imprinting control regions instead of DNA methylation. Importantly, maternal depletion of SETD2 results in oocyte maturation defects and subsequent one-cell arrest after fertilization. The preimplantation arrest is mainly due to a maternal cytosolic defect, since it can be largely rescued by normal oocyte cytosol. However, chromatin defects, including aberrant imprinting, persist in these embryos, leading to embryonic lethality after implantation. Thus, these data identify SETD2 as a crucial player in establishing the maternal epigenome that in turn controls embryonic development.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
The generated and analyzed datasets in the current study are available in the Gene Expression Omnibus with accession number GSE112835.
References
Bernstein, B. E., Meissner, A. & Lander, E. S. The mammalian epigenome. Cell 128, 669–681 (2007).
Margueron, R. & Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 11, 285–296 (2010).
Brookes, E. & Shi, Y. Diverse epigenetic mechanisms of human disease. Annu. Rev. Genet. 48, 237–268 (2014).
Xu, Q. & Xie, W. Epigenome in early mammalian development: inheritance, reprogramming and establishment. Trends Cell Biol. 28, 237–253 (2018).
Tadros, W. & Lipshitz, H. D. The maternal-to-zygotic transition: a play in two acts. Development 136, 3033–3042 (2009).
Bartolomei, M. S. & Ferguson-Smith, A. C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 3, a002592 (2011).
Arima, T. et al. Loss of the maternal imprint in Dnmt3L mat-/- mice leads to a differentiation defect in the extraembryonic tissue. Dev. Biol. 297, 361–373 (2006).
Branco, M. R. et al. Maternal DNA methylation regulates early trophoblast development. Dev. Cell 36, 152–163 (2016).
Inoue, A., Jiang, L., Lu, F. & Zhang, Y. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev. 31, 1927–1932 (2017).
Inoue, A., Jiang, L., Lu, F., Suzuki, T. & Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424 (2017).
Kelsey, G. & Feil, R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. Lond. B 368, 20110336 (2013).
Brind’Amour, J. et al. LTR retrotransposons transcribed in oocytes drive species-specific and heritable changes in DNA methylation. Nat. Commun. 9, 3331 (2018).
Kobayashi, H. et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8, e1002440 (2012).
Shirane, K. et al. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 9, e1003439 (2013).
Chotalia, M. et al. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 23, 105–117 (2009).
Zhang, B. et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557 (2016).
Dahl, J. A. et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552 (2016).
Hanna, C. W. et al. MLL2 conveys transcription-independent H3K4 trimethylation in oocytes. Nat. Struct. Mol. Biol. 25, 73–82 (2018).
Zheng, H. et al. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol. Cell 63, 1066–1079 (2016).
Ooi, S. K. et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717 (2007).
Zhang, Y. et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253 (2010).
Bartke, T. et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 (2010).
Brinkman, A. B. et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 22, 1128–1138 (2012).
Reddington, J. P. et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 14, R25 (2013).
Wachter, E. et al. Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3, e03397 (2014).
Wozniak, G. G. & Strahl, B. D. Hitting the ‘mark’: interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta 1839, 1353–1361 (2014).
Hu, M. et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc. Natl Acad. Sci. USA 107, 2956–2961 (2010).
Mukai, M. et al. H3K36 trimethylation-mediated epigenetic regulation is activated by Bam and promotes germ cell differentiation during early oogenesis in Drosophila. Biol. Open 4, 119–124 (2015).
McKay, D. J. et al. Interrogating the function of metazoan histones using engineered gene clusters. Dev. Cell 32, 373–386 (2015).
Aoshima, K., Inoue, E., Sawa, H. & Okada, Y. Paternal H3K4 methylation is required for minor zygotic gene activation and early mouse embryonic development. EMBO Rep. 16, 803–812 (2015).
Xie, L. et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30, 1473–1484 (2011).
Yuan, W. et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 (2011).
Schmitges, F. W. et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 (2011).
Dhayalan, A. et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285, 26114–26120 (2010).
Rondelet, G., Dal Maso, T., Willems, L. & Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 194, 357–367 (2016).
Yue, F. et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014).
Bourc’his, D., Xu, G. L., Lin, C. S., Bollman, B. & Bestor, T. H. Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539 (2001).
Hata, K., Okano, M., Lei, H. & Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 (2002).
Zhang, Y. et al. Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat. Genet. 50, 96–105 (2018).
Yuan, H. et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Invest. 127, 3375–3391 (2017).
Zuccotti, M., Piccinelli, A., Giorgi Rossi, P., Garagna, S. & Redi, C. A. Chromatin organization during mouse oocyte growth. Mol. Reprod. Dev. 41, 479–485 (1995).
Park, I. Y. et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962 (2016).
Du, Z. et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235 (2017).
Stewart, K. R., Veselovska, L. & Kelsey, G. Establishment and functions of DNA methylation in the germline. Epigenomics 8, 1399–1413 (2016).
Stewart, K. R. et al. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 29, 2449–2462 (2015).
Bošković, A. et al. Analysis of active chromatin modifications in early mammalian embryos reveals uncoupling of H2A.Z acetylation and H3K36 trimethylation from embryonic genome activation. Epigenetics 7, 747–757 (2012).
Ferreira, J. & Carmo-Fonseca, M. Genome replication in early mouse embryos follows a defined temporal and spatial order. J. Cell Sci. 110, 889–897 (1997).
Bouniol, C., Nguyen, E. & Debey, P. Endogenous transcription occurs at the 1-cell stage in the mouse embryo. Exp. Cell Res. 218, 57–62 (1995).
Wu, J. et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 534, 652–657 (2016).
Paynton, B. V. & Bachvarova, R. Polyadenylation and deadenylation of maternal mRNAs during oocyte growth and maturation in the mouse. Mol. Reprod. Dev. 37, 172–180 (1994).
Eckersley-Maslin, M. A., Alda-Catalinas, C. & Reik, W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 19, 436–450 (2018).
Andreu-Vieyra, C. V. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 8, e1000453 (2010).
Kishigami, S. et al. Significant improvement of mouse cloning technique by treatment with trichostatin A after somatic nuclear transfer. Biochem. Biophys. Res. Commun. 340, 183–189 (2006).
Matoba, S. et al. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 159, 884–895 (2014).
Xie, W. et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831 (2012).
Lan, Z. J., Xu, X. & Cooney, A. J. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol. Reprod. 71, 1469–1474 (2004).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Peng, X. et al. TELP, a sensitive and versatile library construction method for next-generation sequencing. Nucleic Acids Res. 43, e35 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Guo, W. et al. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 14, 774 (2013).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003).
Acknowledgements
We thank members of the Xie laboratory for their comments during the preparation of the manuscript. We thank F. Gao for providing the Gdf9-Cre mice. We are thankful for the support provided by the animal core, sequencing core and biocomputing facilities at Tsinghua University. This work was funded by the National Natural Science Foundation of China (grant nos. 31422031 and 31725018 to W.Xie; grant no. 81772938 to L.Li), National Key R&D Program of China (grant no. 2016YFC0900300 to W.Xie), the National Basic Research Program of China (grant no. 2015CB856201 to W.Xie), Beijing Municipal Science & Technology Commission (grant no. Z181100001318006 to W.Xie), Science and Technology Commission of Shanghai Municipality (grant nos. 16140902100 and 18140902700 to L.Li), the THU-PKU Center for Life Sciences (W.Xie), by the Canadian Institutes of Health Research (grant no. MOP-133417 to M.C.L. and grant no. MOP-119357 to L.Lefebvre) and by National Cancer Institute, National Institutes of Health (grant no. CA231993 and CA203012 to C.L.W.). Q.X. is supported by postdoctoral fellowships from the Tsinghua-Peking Joint Center for Life Sciences. L.Li is supported by an Innovation Research Plan from the Shanghai Municipal Education Commission (grant no. ZXGF082101) and funds from State Key Laboratory of Oncogenes and Related Genes (grant no. KF01801). W.Xie is a recipient of an HHMI International Research Scholar award.
Author information
Authors and Affiliations
Contributions
Q.X., Y.X. and W.Xie conceived and designed the project. Q.X. and Y.X. performed the STAR ChIP-seq library construction. Y.X. and Q.W. performed the RNA-seq library construction. Y.Z. performed the STEM-seq experiment. Y.X. analyzed the STAR ChIP-seq and RNA-seq data. Y.X. and Q.W. analyzed the methylome data. Q.X. and Y.X. collected the oocytes and embryos. L.Li and M.W. generated and provided the Setd2loxP/loxP mice. Q.X., Y.X. and Q.W. maintained and genotyped the mice. Z.D. performed mature sperm sorting and B.Z. performed the sperm ChIP-seq. C.H. and J.M. maintained and prepared the PWK/PhJ mice for ICSI. B.Z. and H.Z. provided technical support for the STAR ChIP-seq and data analysis. L.W. and C.L. performed the spindle transfer, ICSI and embryonic culture under the supervision of W.L. G.Y. and W.Xia assisted with chromatin analysis of reconstructed embryos. Q.X. performed the mouse embryo experiments and immunostaining. Q.W. and Y.L. performed the NGS sequencing. C.L.W. and E.J. provided Setd2loxPf/loxP mice, and A.B.B., J.B.A. and L.Lefebvre performed Setd2loxPf/loxP;Zp3-Cre related experiments. M.C.L., W.L., L.Li and W.Xie supervised the project or related experiments. Q.X., Y.X. and W.Xie prepared the manuscript and figures with the help from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Fig. 1 Characterization of H3K36me3 during mouse oogenesis.
a, Scatter plots showing whole genome H3K36me3 enrichment (10kb window) between 500 cells (n = 1), 10k cells (n = 1) and ENCODE dataset in mESCs. Pearson correlation is also shown. b, Pearson correlations are shown for pairwise comparisons of H3K36me3 enrichment between individual sample. Analyses were done using two replicates for GO-P7, GO-P10, GO-P14, FGO, and MII oocytes, and one replicate for 500 and 10k mESCs. ENCODE H3K36me3 data of mESCs is used for comparison. c, Hierarchical clustering analysis of H3K36me3 in individual replicates of mouse early embryos at various stages or mESCs with various cell numbers. d, The dynamics of gene expression and histone modifications for genes activated between GO-P7 to GO-P14.
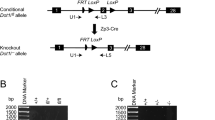
Supplementary Fig. 2 Characterization of Dnmt3L−/− and Setd2mNull FGOs.
a, Strategy of depleting exon 4-7 in the ADD (ATRX-DNMT3-DNMT3L) domain of Dnmt3L (Methods). Inner and outer primers are designed for genotyping. b, Spleen from control and Dnmt3L−/− mice was isolated and western blotting was performed. The asterisk marks possible non-specific band. The arrow marks DNMT3L band. Blot image was cropped (see Supplementary Fig. 13). The experiments were independently performed three times. c, Agarose gel shows the genotyping result of control and Dnmt3L−/− mice by extracting genomic DNA from mouse tail and performing PCR amplification. For mutant mice, the product of inner primer cannot be detected and the outer primer product is truncated. Similar results were obtained from three independent experiments. d, Violin plot showing the levels of DNA methylation in control (n = 2) and Dnmt3L−/− (n = 2) oocytes in FGO. The outer shape indicates all results. The center dot indicates the median average value, and the thick and thin line represent 50% and 95% value ranges, respectively. e, Scatter plot showing gene expression in Dnmt3L+/− and Dnmt3L−/− FGO. Pearson correlation coefficient is shown. Differentially expressed genes are also shown in red (up-regulated) and blue (down-regulated) (2-fold change).
Supplementary Fig. 3 Aberrant DNA methylation in Setd2mNull FGOs.
a, H3K36me3 staining in control (n = 20) and Setd2mNull FGO (n = 20). A representative image from 3 independent experiments is shown. Scale bars: 20 μm. Quantification of H3K36me3 intensity normalized against DNA staining by Hoechst is also shown. Each dot represents the IF intensity of H3K36me3 in nucleus, with mean value indicated by horizontal lines. Error bar represents s.e.m. P-value (t-test with Welch correlation, two sided) is shown. b, Average plot showing H3K36me3 enrichment at gene body of control (n = 2) and Setd2mNull FGO (n = 2). c-d, EU straining in NSN and SN oocytes in control and Setd2mNull FGO. Diagram showing ratios between NSN and SN in control (n = 52) and Setd2mNull FGO (n = 62) (radius between 70-80 μm). e, α-Tubulin staining in control and Setd2mNull MII oocytes. Arrow shows a polar chromosome. Scale bars: 20 μm. These experiments were performed independently three times. f, Gene expression levels (FPKM) of Dnmts in control (n = 2) and Setd2mNull (n = 2) FGO. Centre line indicates average FPKM value between replicates. Error bar represents s.e.m. g, Averaged 5mC enrichment patterns for chromosome 12 in control and Setd2mNull FGO. Gene desert regions with DNA methylation changes are shaded. h, The numbers of 1-kb bins that gain, lose, or show no changes of DNA methylation in Setd2mNull FGOs. i, Percentages of 1-kb bins that gain, lose, or show no changes of DNA methylation in Setd2mNull FGOs that fall into compartment A/B, respectively. Percentages of whole genome in compartment A/B is shown as a control.
Supplementary Fig. 4 Aberrant H3K4me3 and H3K27me3 in Setd2mNull FGOs.
a, Pie charts demonstrating proportions of the genome covered by H3K4me3, H3K27me3, bivalent marks (H3K4me3 and H3K27me3), H3K36me3, or none of these marks in control and Setd2mNull FGO (Methods). b, A snapshot of UCSC browser showing H3K4me3, H3K27me3 and H3K36me3 enrichment in 2-3 replicates of control and Setd2mNull FGO. Regions that acquire/lost H3K4me3 or acquire H3K27me3 are shaded. c, Box plots showing enrichment of H3K4me3 and H3K27me3 for regions defined as ‘K4me3-gain domains’, ‘K4me3-loss domains’, ‘K27me3-gain domains’, and ‘K27me3-loss domains’ in individual replicates of control (H3K4me3, n = 3; H3K27me3, n = 2) and Setd2mNull FGO (H3K4me3, n = 3; H3K27me3, n = 2). The median of each dataset is shown by center line. The bottom, top edges and whiskers represent the 25th, 75th percentiles and 1.5 times the interquartile range, respectively. d, Box plots showing enrichment of H3K4me3 and H3K27me3 in control (H3K4me3, n = 3; H3K27me3, n = 2) and Setd2mNull FGO (H3K4me3, n = 3; H3K27me3, n = 2) for regions defined as ‘K4me3-gain domains’, ‘K4me3-loss domains’, ‘K27me3-gain domains’, and ‘K27me3-loss domains’. Enrichment of whole genome is shown as controls. The median of each dataset is shown by center line. The bottom, top edges and whiskers represent the 25th, 75th percentiles and 1.5 times the interquartile range, respectively. P-values (t-test, two sided) are also shown.
Supplementary Fig. 5 Setd2mNull GO-P7 oocytes share a similar epigenome with Setd2mNull FGOs.
a, Snapshots from UCSC genome browser showing H3K4me3 and H3K27me3 enrichment in control and Setd2mNull GO-P7 and FGO oocytes. b, Box plots showing enrichment of H3K4me3 and H3K27me3 in control (Zhang et al., 2016) and Setd2mNull P7 oocytes (H3K4me3, n = 1; H3K27me3, n = 1) for regions defined as ‘K4me3-gain domains’, ‘K4me3-loss domains’, ‘K27me3-gain domains’, and ‘K27me3-loss domains’ in control and Setd2mNull FGO. Similar analysis was performed for transcription levels in control (n = 2) and Setd2mNull P7 oocytes (n = 2) for misregulated (up, upregulated; down, downregulated) genes in Setd2mNull FGO. The median of each dataset is shown by center line. The bottom, the top edges and whiskers represent the 25th, 75th percentiles and 1.5 times the interquartile range, respectively. P-values (t-test, two sided) are also shown. c, Hierarchical clustering analysis of global H3K4me3 and H3K27me3 distribution in control (Zhang et al., 2016) and Setd2mNull GO-P7 and FGO. d, Violin plot showing the levels of DNA methylation in control (GO-P7, n = 1; FGO, n = 2) and Setd2mNull oocytes (GO-P7, n = 1; FGO, n = 2) in GO-P7 and FGO. The outer shape indicates all results. The center dot indicates the median average value, and the thick and thin line represent the 50% and 95% value ranges, respectively.
Supplementary Fig. 6 Ectopic H3K27me3 correlates with the downregulation of gene expression in Setd2mNull FGOs.
a, Heat maps showing differentially expressed genes (2-fold change) that also show altered H3K4me3 in gene bodies in Setd2mNull FGO. Promoter H3K4me3 levels are also mapped and shown. Genes showing changes in both gene expression and H3K4me3 were selected for this analysis. b, Heat maps showing differentially expressed genes (2-fold change) that also show alteration of H3K27me3 enrichment at gene bodies in Setd2mNull FGO. Promoter H3K27me3 levels are also mapped and shown. Genes showing changes in both gene expression and H3K27me3 were selected for this analysis. Gene ontology analysis and example genes are also shown on the right side. c, Scatter plot showing changes of gene expression and changes of H3K4me3 (n = 3) or H3K27me3 (n = 2) at promoters in Setd2mNull FGO, compared to control FGO (n = 2 for both H3K4me3 and H3K27me3). Pearson correlation coefficient is shown. d, A snapshot from UCSC browser showing H3K4me3, H3K27me3 and H3K36me3 enrichment in control and Setd2mNull FGO. Gene expression is also shown.
Supplementary Fig. 7 Abnormal imprints in Setd2mNull FGOs.
a, Snapshots from UCSC browser showing H3K4me3, H3K27me3, H3K36me3, and DNA methylation levels at maternal imprinted loci in control and Setd2mNull FGO, as well as WT and Dnmt3a/b-/- oocytes. Imprinting control regions are shaded. b, Box plot showing the DNA methylation level in regions with H3K4me3, H3K27me3, or neither in Setd2mNull FGO (n = 2). The median of each dataset is shown by center line. The bottom, top edges and whiskers represent the 25th, 75th percentiles and 1.5 times the interquartile range, respectively. P-values (t-test, two sided) are also shown.
Supplementary Fig. 8 Dynamics of H3K36me3 in mouse early embryos.
a, Immunostaining of H3K36me3 in oocytes and early embryos. Scale bars: 20 μm. One representative result out of three independent experiments is shown. PB, polar body. b, A snapshot of UCSC browser showing H3K36me3 enrichment in two replicates of sperm, 1C, late 2C, 8C and ICM from blastocyst. c, Hierarchical clustering analysis of H3K36me3 in individual replicates of mouse early embryos at various stages. d, Pearson correlations for pairwise comparisons of H3K36me3 enrichment at individual stages (n = 2). Two replicates were done for each sample. e, A bar chart showing the percentages of reads assigned to the maternal or paternal allele in mouse embryos at individual stage.
Supplementary Fig. 9 Allelic H3K36me3 in early embryos.
a, Hierarchical clustering analysis of H3K36me3 at individual stage of mouse early embryos. b, Heat maps showing allelic H3K36me3 enrichment and related allelic gene expression in mouse early embryos. c, Snapshots showing allelic H3K36me3 and H3K27me3 at Sfmbt2, Eva1a, Adam19 and Bzw2 loci.
Supplementary Fig. 10
a, Bright fields of mouse embryos isolated at E2.5 for control (morula stage) and Setd2mNull embryos (arrested at 1-cell stage). One representative result of three independent experiments is shown. Scale bars: 20 μm. b, Hoechest staining of control (2-cell) and Setd2mNull embryos (arrested at 1-cell prior to pronuclei fusion) obtained at E1.5. One representative result of two independent experiments is shown. Scale bars: 20 μm. c, Bright fields of mouse embryos isolated at E3.5 for control (blastocyst stage) and Setd2f/f;Zp3-Cre embryos. One representative result of two independent experiments is shown. Scale bars: 20 μm. d, EU staining in control and Setd2mNull zygotes at E0.5 (1-cell stage). Dashed lines marked maternal and paternal pronuclei. One representative result of three independent experiments is shown. e, EU signal intensity was calculated by immunofluorescence intensity of maternal/paternal pronuclei against background in control (n = 9) and Setd2mNull zygotes (n = 10) . IF intensity of individual pronuclei from three independent experiments are analyzed. Dots represent single pronuclei, with medians indicated by horizontal lines. Error bar represents s.e.m.. f, Hierarchical clustering analysis of global gene expression in oocytes and early embryos for control (blue) and Setd2mNull mutants (red). g, Heat map showing gene expression in control and Setd2mNull oocytes and embryos.
Supplementary Fig. 11 Aberrant epigenome in Setd2mNull 1-cell embryos.
a, Gene ontology of up- or down-regulated genes between Setd2mNull (n = 2) and control 1C (n = 2). P-values (t-test, two sided, not adjusted by multiple comparisons) are transformed by log10. b, Gene ontology of genes that are both up- or down-regulated between Setd2mNull and control in FGO (n = 2) and 1C embryos (n = 2). P-values (t-test, two sided, not adjusted by multiple comparisons) are transformed by log10. c, Heat map showing expression and H3K27me3 levels for genes that are both downregulated in Setd2mNull FGO and 1C embryos, and acquire ectopic H3K27me3 in Setd2mNull FGO or 1C embryos. Example genes are also shown. d, A heat map showing H3K4me3 enrichment in FGO and 1C for ‘H3K4me3-gain domains’ or ‘H3K4me3-loss domains’ defined in FGO (left). A similar analysis result was shown for H3K27me3 (right). e, Snapshots from UCSC genome browser showing allelic H3K4me3 (top) or H3K27me3 (bottom) enrichment in control and Setd2mNull zygotes. H3K4me3 and H3K27me3 in 1C embryos from a previous study are also shown as control. f, A bar chart showing the percentages of H3K4me3 or H3K27me3 reads assigned to the maternal (129 or C57BL/6) or paternal (PWK) allele in control and Setd2mNull 1C embryos.
Supplementary Fig. 12 Development of reconstructed embryos.
a, Reconstructed embryos (Cwt+Mwt+Pwt, n = 78; Cwt+Mmut+Pwt, n = 74; Cmut+Mwt+Pwt, n = 22) were cultured to 24h, 48h and 96h after spindle transfer, respectively. One representative result of five independent experiments is shown. Scale bars: 20 μm. b, Morphology of Cwt+Mwt+Pwt (n = 44) and Cwt+Mmut+Pwt (n = 38) embryos dissected from E10.5. One representative image from 2-3 independent experiments is shown. Pl, placenta; Em, embryo; YS, yolk sac; EPC, ectoplacental cone. c, Box plots showing enrichment of H3K4me3 and H3K27me3 for regions defined as ‘K4me3-gain/loss domains’, ‘K27me3-gain/loss domains’, in control and Cwt+Mmut+Pwt embryos at early 2C (H3K4me3, n = 1; H3K27me3 n = 1) and 8C (H3K4me3, n = 1; H3K27me3 n = 1). The median of each dataset is shown by center line. The bottom, top edges and whiskers represent the 25th, 75th percentiles and 1.5 times the interquartile range, respectively. P-values (t-test, two sided) are shown. d, Snapshot from UCSC browser showing H3K4me3 enrichment at a maternally imprinted locus in control, Setd2mNull FGO, ST embryos, with gene expression at 8C. e, A model illustrating epigenetic reprogramming in control and mutant FGOs or embryos. In Setd2mNull FGO, H3K27me3 and H3K4me3 ectopically spread to former H3K36me3 territories (Gene1 and Gene2). The aberrant epigenome persists to early 2-cell embryos even in reconstructed embryos with WT cytosol (Cwt+Mmut+Pwt). At 8-cell stage, the aberrant H3K4me3 is largely globally reset to canonical H3K4me3, except for ectopic H3K4me3 at ICR of maternal imprinting genes which persists to 8-cell embryos and is associated with increased expression from maternal allele in Cwt+Mmut+Pwt embryos. Aberrant H3K27me3 is still retained in these Cwt+Mmut+Pwt embryos.
Supplementary Fig. 13 Original uncropped gel of Supplementary Fig. 2b.
Boxes indicate the cropped regions in Supplementary Fig. 2b.
Supplementary information
Supplementary Information
Supplementary Figs. 1–13
Supplementary Table 1
Sequencing information of STAR ChIP-seq.
Supplementary Table 2
Genes with allele specific H3K36me3 in mouse early embryos.
Supplementary Table 3
Differential expressed genes in Dnmt3L control and knock out fullgrown oocyte.
Supplementary Table 4
Differentially expressed genes in control and Setd2mNull fullgrown oocyte.
Supplementary Table 5
Primers and sgRNAs used in this study.
Rights and permissions
About this article

Cite this article
Xu, Q., Xiang, Y., Wang, Q. et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51, 844–856 (2019). https://doi.org/10.1038/s41588-019-0398-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0398-7
This article is cited by
-
The impact of selective HDAC inhibitors on the transcriptome of early mouse embryos
BMC Genomics (2024)
-
RNA m6A methylation and regulatory proteins in pulmonary arterial hypertension
Hypertension Research (2024)
-
Emergence of replication timing during early mammalian development
Nature (2024)
-
CRL4DCAF13 E3 ubiquitin ligase targets MeCP2 for degradation to prevent DNA hypermethylation and ensure normal transcription in growing oocytes
Cellular and Molecular Life Sciences (2024)
-
Imprinting at the KBTBD6 locus involves species-specific maternal methylation and monoallelic expression in livestock animals
Journal of Animal Science and Biotechnology (2023)
