
Abstract
The domestication of wild emmer wheat led to the selection of modern durum wheat, grown mainly for pasta production. We describe the 10.45 gigabase (Gb) assembly of the genome of durum wheat cultivar Svevo. The assembly enabled genome-wide genetic diversity analyses revealing the changes imposed by thousands of years of empirical selection and breeding. Regions exhibiting strong signatures of genetic divergence associated with domestication and breeding were widespread in the genome with several major diversity losses in the pericentromeric regions. A locus on chromosome 5B carries a gene encoding a metal transporter (TdHMA3-B1) with a non-functional variant causing high accumulation of cadmium in grain. The high-cadmium allele, widespread among durum cultivars but undetected in wild emmer accessions, increased in frequency from domesticated emmer to modern durum wheat. The rapid cloning of TdHMA3-B1 rescues a wild beneficial allele and demonstrates the practical use of the Svevo genome for wheat improvement.
Similar content being viewed by others

Main
Durum wheat (DW), Triticum turgidum L. ssp. durum (Desf.) Husn., genome BBAA, is a cereal grain mainly used for pasta production and evolved from domesticated emmer wheat (DEW), T. turgidum ssp. dicoccum (Schrank ex Schübl.) Thell. DEW itself derived from wild emmer wheat (WEW), T. turgidum ssp. dicoccoides (Körn. ex Asch. & Graebn.) Thell., in the Fertile Crescent about 10,000 years ago1. Although the first evidence of DW dates to 6,500–7,500 years ago, DW became established as a prominent crop only 1,500–2,000 years ago2. Thus, the human-driven tetraploid wheat evolution process is the result of domestication (WEW to DEW), continued evolution under domestication (DEW to durum wheat landraces, DWL) and breeding improvement from DWL to modern durum wheat cultivars (DWC).
Wild relatives of modern crop plants can serve as sources of valuable genetic diversity for various traits (for example, disease resistance3,4 and nutritional quality5). Comprehensive comparative genomic analyses between cultivated crops and wild progenitors is a key strategy to detect novel beneficial alleles and structural variations that could constrain breeding efforts, as well as to understand the broader genetic consequences of evolution and selection history6,7.
Here we report the fully assembled genome of the modern DW cultivar (cv.) Svevo and provide a genome-wide account of modifications imposed by thousands of years of empirical selection and breeding. This was achieved by comparing the Svevo genome with the assembled genome of WEW accession Zavitan8 and through a survey of the genetic diversity and selection signatures in a Global Tetraploid Wheat Collection consisting of 1,856 accessions. A region bearing a signature of historic selection co-locates with Cdu-B1, a quantitative trait locus (QTL) spanning 0.7 cM on chromosome 5B9 known to control cadmium (Cd) accumulation in the grain. Identification of the gene(s) responsible for Cdu-B1 has been hampered by the large and repetitive nature of the DW genome and the low recombination rate in the region of interest. The efficient, genome-enabled dissection of the Cdu-B1 locus reported here demonstrates the value of the Svevo genome assembly for wheat improvement.
Results
The durum wheat reference genome
The Svevo genome sequence was assembled de novo using protocols previously described8 and its main features are illustrated in Fig. 1. After sequencing (Supplementary Table 1) and assembly, the scaffolds (length of the shortest contig needed to cover 50% of the genome (N50) = 6.0 megabases (Mb); Supplementary Table 2) were ordered and oriented using the Svevo × Zavitan genetic map as previously described10. Thereafter, chromosome conformation capture sequencing (Hi-C)11 resulted in a set of pseudomolecules (9.96 Gb; Supplementary Table 3) corresponding to the 14 chromosomes of DW and one group of unassigned scaffolds (499 Mb). The pseudomolecules encompass 95.3% of the assembled sequences and have 90% of the scaffolds oriented. Alignment of the DW genome with high-density SNP genetic maps12 showed highly recombinogenic distal chromosome regions exhibiting an almost linear relationship between genetic and physical distance (Supplementary Fig. 1). These regions account for about 22% of the genome with an average recombination rate of 1.8 Mb cM−1 (Supplementary Table 4). In contrast, large pericentromeric regions are nearly devoid of recombination and represent about 44% of the genome, with a mean recombination rate of 107 Mb cM−1. Annotation of the Svevo genome led to the identification of 66,559 high confidence (HC) genes, 90.5% of which exhibited detectable evidence of expression in at least one of the 21 RNA-seq datasets listed in Supplementary Table 5. A detailed description of the DW genome is presented in the Supplementary Note (Sections 1.1 and 2.1). Projection onto the DW genome of 2,191 previously reported QTLs resulted in a full meta-QTL analysis (Supplementary Table 6b and Supplementary Dataset 1), revealing a QTL density distribution that closely mirrors the gene density distribution (Supplementary Table 7 and Fig. 1d,e).

Tracks from outside to inside. a, Chromosome name and size (100 Mb tick size, arms differentiated by gray shading). b, Density of WEW HC gene models (HC; 0–25 genes per Mb). c, Links connecting homologous genes between WEW and DW. d, Density of DW HC gene models (0–22 genes per Mb). e, Location of published QTLs. f, k-mer frequencies. g, Long terminal repeat (LTR)-retrotransposon density. h, DNA transposon frequency. i, Mean expression of HC genes calculated as log(FPKM + 1) of the mean expression value of all conditions (range 1.6–8.2). Links in center connect homoeologous genes between subgenomes; blue links between homoeologous chromosomes and green links between large translocated regions.
Comparison between Svevo and Zavitan genomes
To gain insights into short-term evolutionary changes, we compared the genome divergence between the modern DW cultivar Svevo and the WEW accession Zavitan8. The comparison revealed strong overall synteny (Fig. 1c) with high similarity in total HC gene number (DW 66,559; WEW 67,182; Supplementary Table 8), chromosome structure and transposable element composition (Supplementary Table 9). We identified syntenic LTR-retrotransposon insertions (Supplementary Fig. 2) not yet subjected to the rapid transposable element turnover of the intergenic space1,13 because of the relatively short separation time between Svevo and Zavitan. To monitor structural variations in the HC gene set, including minor changes that might have been generated during the short evolutionary timespan, a graph-based sequence clustering of all Svevo and Zavitan HC genes (in total 133,741) was undertaken. Stringent clustering (alignment e value < 10−10, overlap > 75% and identity > 75%) grouped only highly similar gene models in the same cluster. This approach produced 36,434 unigene groups, 79% (28,794) of which were clusters with at least two members, while 21% (7,640) contained only singletons (Supplementary Table 10). The main scenarios for conserved and variable genes are summarized in Fig. 2a. The most frequent cluster configuration is made of two homoeologous gene copies per genome (one per A and B subgenomes), which occurs in 35% of all unigene groups. Altogether, the unigene groups with balanced copy numbers for Svevo and Zavitan represent up to 60% of all unigenes and involve 63% of all Svevo genes. The remaining 40% of unigenes (14,660) display asymmetric numbers of intact, full-length genes between Svevo and Zavitan (we named this intact gene number variation). Since the unigene classification is on the basis of HC genes, any mutation leading to a frameshift and/or premature stop codon that rules out a gene from the HC class in Svevo or in Zavitan, results in an asymmetric unigene distribution. For at least two-thirds of the unigenes displaying variation of intact gene number, counterparts for the missing copies can still be found in LC or pseudogene class. The complete gene loss caused by large structural variations was responsible for asymmetric gene distribution in only one-third of the cases. Among the unbalanced gene clusters, there are 6,120 mixed clusters with copies from both genomes, subdivided into more Svevo members or more Zavitan members, as well as 4,313 and 4,227 lineage-specific unigene groups (mostly singleton genes) in Svevo and Zavitan, respectively, which have no close homoeolog in the HC gene set of the other accession. A detailed example of the type of variation leading to intact gene number variation is given for the lineage-specific unigenes (Supplementary Table 10a). In Svevo, this class includes 4,811 genes that represent 7.2% of all HC genes, a value similar to the 5% found after the comparison between two cultivars in a recent pangenome study of hexaploid wheat14. When the Svevo-specific genes were mapped onto the Zavitan genome (Fig. 2a and Supplementary Table 10b), 1,493 genes (31%) were not found on the Zavitan sequence, 1,225 (26%) correspond to shorter counterparts of annotated Zavitan HC genes and 1,095 (23%) were annotated as low confidence (LC) genes or pseudogenes. The remaining 965 (20%, that is, 1.4% of all Svevo HC genes) map to unannotated regions and are candidates for genes missed in the automated annotation.

a, Main unigene group scenarios from co-clustering of Svevo and Zavitan HC genes. The diagram depicts the most common or typical scenarios, only HC intact genes were considered (CNV, copy number variation; CDS, coding sequence). b, Intact gene number variations. Each dot represents a gene cluster consisting of DW (x axis) and WEW (y axis) genes. Dots on the diagonal represent clusters with identical member numbers from both accessions. Functional predictions for some groups of genes with pronounced differences in member numbers are annotated on the diagram. c, Relative distance of DW genes from the centromere separated by gene cluster type. Proportionately more unigenes displaying intact gene number variation than balanced groups are observed towards the ends of the chromosome. HC genes unique to Svevo or Zavitan (not shown) are most highly represented at the ends of the chromosomes, with the median (black line) furthest from the centromere. Shape width represents the relative gene frequency.
Loss and gain events can occur from ancestral four-member unigene clusters with one gene per A and B subgenome of both Svevo and Zavitan. One loss would result in a three-member unigene cluster, with one subgenome location missing. A total of 1,121 clusters with one lost Svevo member were found. The reverse situation with one lost member from Zavitan was found 852 times. The presumed HC gene losses are located predominantly in the more distal chromosomal regions (Supplementary Fig. 3a). A gain event would result in clusters with at least five members with one subgenome carrying two members. This condition was found 472 times with Svevo gains and 503 times with Zavitan gains. Most of the gains are located on the same chromosome indicating tandem gene duplication as the prevailing mechanism (Supplementary Fig. 3b).
Functional categories associated with relevant copy number differences between the two accessions are highlighted in Fig. 2b. A statistical analysis for gene ontology over-representation revealed that specific functions can be classified as: differentially enriched in Svevo, differentially enriched in Zavitan, balanced and Svevo-specific unigene groups (Supplementary Fig. 3c). The balanced gene groups are enriched for a multitude of regulatory functions and indicate a stronger sensitivity to gene dosage effects for the main regulatory networks. Unbalanced unigene groups with more Svevo members and Svevo unique groups are enriched for functions involving protein phosphorylation, for example kinases, which are known to trigger signal transduction cascades in response to environmental cues. The distal highly recombinogenic regions of chromosomes are enriched in unigenes displaying variation of intact gene number (Fig. 2c), contain most of the known QTLs (Fig. 1e) and the HC genes display a reduced expression breadth (that is, average expression value across all tissue/treatment conditions, Fig. 1i). This indicates the presence of an increased number of condition-specific genes. The less dynamic interstitial regions contain a greater number of balanced gene families (Fig. 2c). Here the genes are expressed in nearly all conditions, indicative of an enrichment in housekeeping genes that is consistent with reports from the barley11 and bread wheat15 genomes. The positive correlation between recombination rate and DNA variants supports previous evidence that higher recombination rates and illegitimate recombination are drivers for tandem duplications16.
The balanced copy number groups contain much longer genes (median 1,152 base pairs (bp)) than the groups displaying variation of intact gene number (median 879 bp; Fig. 2a). The highest median gene lengths are found in groups with two copies in each genome (1,242 bp), whereas the lowest are among the unique genes (Svevo, 735 bp; Zavitan, 768 bp) and, surprisingly, in groups with one copy in each genome (756 bp). Such a pronounced shift towards shorter genes indicates an ongoing gene decay by frameshifts and mutations leading to premature stop codons. Collectively, the relatively high number of genes undergoing degeneration could be a consequence of more freedom for gene loss facilitated by the functional redundancy of the tetraploid genome state.
Germplasm structure and phylogenetic relationships
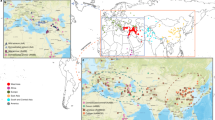
The wheat iSelect 90K SNP Infinium assay17 was used to genotype the Global Tetraploid Wheat Collection consisting of 1,856 accessions representing the four main germplasm groups involved in tetraploid wheat domestication history and breeding: WEW, DEW, DWL and DWC (Supplementary Table 11). A set of 17,340 SNPs (non-redundant, genetically and physically mapped, subgenome-specific Mendelian loci) was used for analysis of genetic diversity, population structure and identification of the selection signatures. Four non-hierarchical clustering analyses (DAPC, sNMF, ADMIXTURE, fineSTRUCTURE18,19,20,21), principal component analysis, and pairwise dissimilarity analysis on the basis of neighbor joining generated global and highly concordant pictures of the genetic relationships among taxa and populations (Fig. 3 and Supplementary Datasets 2 and 3). WEW, DEW, DWL and DWC clearly separated in the neighbor joining tree (Fig. 3a), a result indicative of strong demographic and founder effects and little evidences for polyphyletic origin. Principal component analysis (Fig. 3b) illustrates the broad genetic diversity of DEW, while DWC showed a comparatively limited genetic diversity and a close relationship to a specific DWL population. DEW and DWL from Ethiopia, T. turgidum ssp. turanicum and T. turgidum ssp. carthlicum were genetically isolated.

a, Neighbor joining tree from Nei’s genetic distances among the 1,856 accessions of the Global Tetraploid Wheat Collection. Genetic distances were computed from a set of 5,775 whole-genome linkage disquilibrium–pruned (r2 = 0.5) SNPs. Correspondence between branches and main tetraploid wheat taxa/populations on the basis of ADMIXTURE and other population structure analyses are indicated by color code with the exception of T. turgidum ssp. karamychevii and ssp. isphahanicum that are indicated directly on the graph. Significances were estimated through 1,000 bootstrap resampling. b, Principal component analysis plot of the Global Tetraploid Wheat Collection on the basis of genome-wide pairwise distances calculated on the basis of linkage disquilibrium-pruned SNPs. c, ADMIXTURE analyses of the Global Tetraploid Wheat Collection with k (number of populations assumed for the analysis) from 2 to 20.
ADMIXTURE analysis (Fig. 3c) showed that WEW and DEW have highly structured genetic diversity even at high k values, while DWL showed a high rate of admixture already at low k values. WEW germplasm was divided into two main populations from North Eastern Fertile Crescent and Southern Levant (WEW-NE and WEW-SL, respectively). WEW-NE was further divided into several populations from Turkey, Iran and Iraq, while WEW-SL included distinct populations from Israel (3), Jordan, Syria and Lebanon (Supplementary Fig. 4). DEW and DWL germplasm was characterized by a similar though independent radial dispersal pattern: Northern-to-Southern Fertile Crescent and from Fertile Crescent to Mediterranean basin (Western), Greece to Balkans (Western), Iran to Transcaucasia (Eastern) and Oman to India and Ethiopia. The germplasm belonging to DEW and DWL was subdivided in six main populations each, while all DWC clustered to a further distinct group that represents a wide branch of the durum North African and Turkey to Transcaucasian landrace populations (Supplementary Fig. 4).
After removing the accessions with a high level of admixture, the genetic relationships among the main tetraploid germplasm groups were further investigated using hierarchical analysis of variance (ANOVA), by computing the pairwise divergence index (or fixation index) Fst and Nei’s genetic distances (Fig. 4), and by generating population-based whole-genome phylogenetic trees (Supplementary Fig. 5). The results confirm the radial dispersal patterns already reported and indicate the WEW-NE from Turkey as the most probable ancestor of all DEW populations (Fst and genetic distance values consistently lower for all WEW-DEW pairs). Two DEW populations from Southern Levant Fertile Crescent (Fig. 4) showed the closest relationship to all DWL populations (except T. turgidum ssp. turanicum), while the DWC germplasm was mostly related to the two DWL populations from North Africa and Transcaucasia (Fig. 4). The Ethiopian and T. turgidum ssp. turanicum populations were the most differentiated among the DWL germplasm and their contribution to the modern durum varieties was minimal.

Diagonal numbers represent within-population genetic diversity (expected heterozygosity) values. Only low-admixture accessions were used (Q-membership higher than 0.5 for WEW, DEW; Q-membership higher than 0.4 for DWL, DWC). Statistics were estimated with 5,775 linkage disequilibrium-pruned (r2 = 0.5) SNPs. WEW-NE, WEW from the North Eastern Fertile Crescent, Turkey, Iran and Iraq; WEW-SL, WEW from Southern Levant including Lebanon, Syria, Israel and Jordan; DEW-T-TRC-IRN, DEW from Turkey to Transcaucasia and Iran; DEW-T-BLK, DEW from Turkey to the Balkans; DEW-SthEU, DEW spread in Southern Mediterranean areas; DEW-SL-EU1, DEW from Southern Levant Fertile Crescent to Europe (population 1); DEW-SL-EU2, DEW from Southern Levant Fertile Crescent to Europe (population 2); DEW-ETH, DEW from Oman, India and Ethiopia; DWL-SL-NA, DWL from Southern Levant Fertile Crescent to North Africa and Iberia; DWL-GRC-BLK, DWL from Greece to Balkans; DWL-T-TRC, DWL from Turkey to Transcaucasia; DWL-T-FC, DWL diffused in Turkey to the whole Fertile Crescent; DWL-TRN, T. turanicum; DWL-ETH, DWL from Ethiopia; DWC-DRY, DWC from Italian and ICARDA breeding programs adapted to dryland areas; DWC-ITLY, DWC from Italy; DWC-CIM70, DWC from the wide adaptation, temperate-adapted photoperiod insensitive CIMMYT and ICARDA germplasm bred in the 1970s; DWC-CIM80, DWC from the high-yielding CIMMYT germplasm bred in the 1980s; DWC-AMR, DWC from the photoperiod-sensitive North American and French germplasm.
Diversity reduction and signature of selection
The pattern of diversity for each germplasm group was assessed through a SNP-based gene diversity index22 (Fig. 5a). WEW showed the highest average diversity with only two pericentromeric regions (chromosomes 2A and 4A) with a lower than average diversity. Thus, WEW provides a valuable reference for assessing the reduction of diversity associated with domestication and breeding in tetraploid wheat. Compared to WEW, each of the subsequently domesticated/improved germplasm group showed several strong diversity depletions that arose independently and were progressively consolidated through domestication and breeding. With few exceptions, the diversity depletions that occurred in the early transition (WEW to DEW, Fig. 5b or DEW to DWL, Fig. 6a) are confirmed or even reinforced in the subsequent ones (Fig. 6a,b). Consequently, the genome of DWC is characterized by numerous regions showing near-fixation of allelic diversity (Fig. 6b). We applied five different metrics to detect selection signatures: diversity reduction index (DRI23), single site divergence index (Fst; ref. 24), haplotype-based frequency differentiation index (hapFLK25), cross-population extended haplotype homozygosity (XP-EHH26), and spatial pattern of site frequency spectrum (XP-CLR27). Genomic regions supported by one or more indexes were considered as putative signatures of selection. Frequently, two or more indexes occurred in overlapping regions, hereafter referred as selection clusters. In total, 104 pericentromeric (average size 107.7 Mb) and 350 non-pericentromeric (average size 11.4 Mb) clusters were identified in one, two or three transitions.

a, SNP-based diversity index (DI) for the main germplasm groups identified in the Global Tetraploid Wheat Collection: WEW, DEW, DWL and DWC. DI is reported as a centered 25 SNP-based average sliding window (single SNP step). Top and bottom 2.5% DI quantile distributions are highlighted as red- and blue-filled dots, respectively. b, Cross-population selection index metrics for the comparison between WEW and DEW. Selection metrics are provided for: diversity reduction index (DRI), divergence index (Fst), cross-population extended haplotype homozygosity (XP-EHH), multilocus test for allele frequency differentiation (XP-CLR) and haplotype-based differentiation test (hapFLK). For DRI, top and bottom 2.5% DI quantile distributions are highlighted as red- and blue-filled dots, respectively, while for the other selection metrics top 5% quantile distributions are highlighted as red-filled dots. The physical location of genes (Supplementary Table 12) and QTL confidence intervals relevant to domestication and breeding is reported.

Genome-wide cross-population selection signatures in DEW to DWL and DWL to DWC on the basis of 17,340 informative SNPs. a, Cross-population selection index metrics for the DEW to DWL. b, Cross-population selection index metrics for the DWL to DWC. For both panels, selection metrics are provided for: DRI, Fst, XP-EHH, XP-CLR and hapFLK. For DRI, top and bottom 2.5% DI quantile distributions are highlighted as red- and blue-filled dots, respectively, while for the other selection metrics top 5% quantile distributions are highlighted as red-filled dots. The physical location of genes (Supplementary Table 12) and QTL confidence intervals relevant to domestication and breeding is reported.
When 41 loci known to be under selection during emmer domestication and durum wheat evolution or breeding (Supplementary Table 12) were projected on the genome, many of them overlapped with selection clusters (Figs. 5 and 6; Supplementary Dataset 4). Most of the strongest pericentromeric diversity depletions (DRI > 4) occurred during emmer domestication (chromosomes 2A, 4A, 4B, 5A, 5B, 6A and 6B). Furthermore, one of the two brittle rachis regions marking the early domestication process (BRT-3B8) showed a localized sharp reduction in diversity confirmed by Fst and XP-CLR indexes. The same region, then, underwent an extreme diversity reduction in the DEW-to-DWL transition (DRI 3.4). Additional 14 pericentromeric and 90 non-pericentromeric (DRI > 2) diversity depletions, including one harboring the major tough glume QTL governing threshability (Tg-2B28), occurred during the DEW-to-DWL transition. Finally, several reductions in diversity (75 with DRI > 2) were specifically associated with breeding of modern durum cultivars, including some associated with disease resistance (for example, Sr13; ref. 29 and Lr14; ref. 30) and grain yellow pigment content loci (for example, Psy-B1; ref. 31). A detailed description of the selections signatures is presented in Supplementary Note.
Variation for cadmium grain content in tetraploid wheat
Cdu-B1 is a QTL located on the long arm of chromosome 5B, which accounts for >80% of the phenotypic variation in cadmium (Cd) concentration in grain9,32,33. The Cdu-B1 region corresponds to a physical interval of 4.27 Mb. A detailed comparison of the Zavitan and Svevo (low and high Cd, respectively) genomes, coupled with exome sequencing, revealed a segment of increased nucleotide variation in this refined region (Supplementary Fig. 6). Furthermore, the region contains 192 gene models, 48 of which have informative functional annotations (Supplementary Table 13). One of these genes, TRITD5Bv1G197370, encodes a P1B-type heavy-metal ATPase 3 (HMA3) that is orthologous to rice HMA3 (OsHMA3) (Supplementary Table 13 and Supplementary Fig. 7). OsHMA3 is a tonoplast-localized transporter that transports Cd into vacuoles, thereby limiting its translocation from roots to shoots and grain34,35,36. Closer inspection of TRITD5Bv1G197370 (TdHMA3-B1 herein) revealed a 17-bp duplication in the first exon that creates two alternative alleles, namely TdHMA3-B1a in Zavitan (functional; low grain Cd) and TdHMA3-B1b in Svevo (non-functional; high grain Cd; Supplementary Fig. 7). The alternative alleles are clearly identified by Xusw59, a diagnostic polymerase chain reaction (PCR) marker that amplifies the region including the 17-bp duplication (Supplementary Fig. 7) and perfectly discriminated low- and high-Cd accumulators in a global collection of DW lines (Supplementary Fig. 8). Heterologous expression of TdHMA3-B1a/b alleles in Cd- and zinc (Zn)-sensitive yeast confirmed that TdHMA3-B1a is a tonoplast-localized Cd and Zn transporter (Fig. 7 and Supplementary Figs. 9–12). Furthermore, disruption of P-ATPase ion transport activity (that is, TdHMA3-B1a-D411A knockout) reduced both TdHMA3-B1a-mediated yeast complementation (Fig. 7a,b and Supplementary Fig. 11) and cellular Cd and Zn accumulation (Fig. 7d and Supplementary Fig. 13). These findings demonstrate that TdHMA3-B1a transports Cd and Zn into vacuoles. Functional analysis of the TdHMA3-B1 homoeolog, TdHMA3-A1 (Supplementary Figs. 7b,c and 9) and the longest alternative open reading frame for allele TdHMA3-B1b, TdHMA3-B1b-ORF2 (Supplementary Fig. 7b), are provided in the Supplementary Note. Consistent with the predicted function of TdHMA3-B1, the primary in planta effect of the non-functional allele, TdHMA3-B1b, is the reduced Cd retention in roots and a two- to threefold increase in Cd transport to shoots and grain (Supplementary Figs. 14 and 15 and Supplementary Table 14)37. Although other genes in the Cdu-B1 physical interval could potentially contribute to Cdu-B1, only TdHMA3-B1 is functionally consistent with the Cdu-B1 phenotype and is supported by these results as a candidate for the Cd phenotypic differences in DW.

a, Growth (OD600) of Cd-sensitive ycf1 yeast expressing empty vector (EV, p413TEF), YCF1, TdHMA3-B1a, TdHMA3-B1a-D411A and TdHMA3-B1b in the presence of 40 and 55 μM Cd. Plotted growth curves are means of three (D411A), five (YCF1, B1a, B1b), or six (EV) experiments ± 95% confidence intervals shown as shaded backgrounds. b, Growth (OD600) of Zn-sensitive zrc1cot1 yeast expressing empty vector (EV, p413TEF), ZRC1, TdHMA3-B1a, TdHMA3-B1a-D411A and TdHMA3-B1b in the presence of 125 and 225 μM Zn. Plotted growth curves are means of three (B1b) or five (EV, ZRC1, B1a, D411A) experiments ± 95% confidence intervals shown as shaded backgrounds. c, Localization of TdHMA3-B1a-GFP and TdHMA3-B1b-GFP expressed in ycf1 (differential interference contrast (DIC) and merged images provide spatial references). Scale bars, 2 μm. d, Cd accumulation in ycf1 and Zn accumulation in zrc1cot1 expressing TdHMA3-B1a and transport activity knockout, TdHMA3-B1a-D411A, after exposure to 5 μM Cd or 50 μM Zn for 4 h. Data are shown as means ± 95% confidence intervals for n = 4 independent cultures (circles). Experiments repeated with similar results. P values were calculated by two-tailed, unpaired t-tests (d.f. = 6). The dashed lines indicate the Zn concentrations of the inoculating yeast cultures.
The distribution of the TdHMA3-B1a and B1b alleles in the Global Tetraploid Wheat Collection revealed a clear association with domestication status and geographical provenance (Fig. 8, Supplementary Fig. 16, Supplementary Tables 15 and 16 and Supplementary Dataset 2). The functional TdHMA3-B1a allele was genetically fixed in all 115 WEW accessions, while the TdHMA3-B1b allele showed a trend of increasing frequency in DEW (13%), DWL (26%) and DWC (68%). Among DEW accessions, the non-functional, high-Cd-accumulating TdHMA3-B1b allele was enriched in the subgroup from Turkey38,39 (38%), indicating Turkey to be the region of origin and location from which the allele spread. Among DWL accessions, TdHMA3-B1b showed a maximum of 41% occurrence in North African landraces (Fig. 8b). The steady increase in the frequency of the TdHMA3-B1b allele from DEW to DWC indicates a process of systematic selection or divergence during durum wheat evolution and breeding as demonstrated by the local Fst data (Supplementary Dataset 5). This result might indicate either a positive effect of the high-Cd allele per se or the presence of a selective sweep for another gene in linkage disequilibrium with TdHMA3-B1b. To assess the presence of genes which could have been targeted by selection in this locus, we identified the boundaries of high linkage disequilibrium with TdHMA3-B1 (squared correlation coefficient r2 ≥ 0.5) in both DWL and DWC. The region contains 219 HC and LC genes in both Svevo and Zavitan as well as 73 and 58 genes present only in WEW and DW, respectively (Supplementary Dataset 6). Although the vernalization responsive gene VRN-B1 (ref. 40) lies in this region (6.94 Mb distal from TdHMA3-B1), inspection of allelic diversity revealed VRN-B1 as a low-variant gene, with the ancestral wild-type vrn-B1 dominating across the entire Global Tetraploid Wheat Collection, thus excluding VRN-B1 as a contributor to a possible selective sweep around Cdu-B1.

a, FineSTRUCTURE clustering and bar plots of individual ADMIXTURE membership coefficients at critical k = 7 for the tetraploid diversity panel. b, Bar plot of individual TdHMA3-B1a/b allelic score in WEW, DEW, DWL and DWC. Pie charts of TdHMA3-B1a/b relative ratio in main tetraploid germplasm groups. Blue bars, accessions carrying TdHMA3-B1a; yellow bars, accessions carrying TdHMA3-B1b.
Discussion
The genome assembly of the modern DW cv. Svevo, with a quality level consistent with those recently obtained for other species8,11,15, represents an essential tool to study durum wheat domestication, evolution and breeding as well as to gain new insights into gene function and the genome-wide organization of QTLs for relevant agronomic traits. This study presents an inclusive analysis of a large panel of tetraploid wheat representing all known taxa and provides a global picture of genetic relationship and population structure. The process leading to modern durum wheat was revealed by the four main germplasm groups of the Global Tetraploid Wheat Collection. The combination of genetic diversity and selection signature analysis revealed a dynamic description of the modifications imposed on the genome by domestication and breeding. The strongest reductions in diversity occurred in well-defined pericentromeric regions during the domestication of WEW. Then, the reduction of diversity continued more moderately, but spread over the genome, during the evolution of DWL and, more recently, as a consequence of the breeding activity23,41. Multiple divergence and haplotype metrics identified several regions coincident with known domestication loci, as well as others that might indicate new putative loci under domestication or selection.
Identification of TdHMA3-B1 as the gene most likely responsible for phenotypic variation in grain Cd accumulation, a result supported by genetic and functional evidence, and the recovery of the TdHMA3-B1a allele for low Cd accumulation, provides an example of the relevance of the genomic tools presented here. The increase in frequency of TdHMA3-B1b during DW breeding could be due to the presence of a selective sweep for another gene in the linkage disequilibrium region, although no evidence has been found and further studies are required to support this hypothesis. Alternatively, the non-functional TdHMA3-B1b allele could exert some beneficial effects on plant fitness. Zinc assimilation by plants results in the co-transportation of Cd, and like other P1B-2-type ATPase transporters42, TdHMA3-B1 can transport both metals. Although Cdu-B1 has no effect on agronomic performance under Zn-sufficient conditions, the low-Cd line from a pair of Cdu-B1 near-isogenic lines showed reduced biomass compared to the high-Cd line when grown under Zn-deficiency43. Therefore, TdHMA3-B1b could provide a growth benefit in Zn-deficient soils, such as those that widely occur in wheat-growing regions of Turkey44 where TdHMA3-B1b originated. A reduction in root vacuolar sequestration of Cd and Zn in high-Cd genotypes (non-functional TdHMA3-B1b allele) under Zn-limiting conditions may increase the pool of Zn available for transport to the shoot, thereby sustaining shoot growth.
Access to the fully annotated genome sequence in combination with the wealth of genotypic, genetic mapping12 and gene expression data provides great potential for future innovation for the wheat scientific community and the breeding sector. Gene discovery, QTL cloning and the precision of genomics-assisted breeding to enhance grain quality and quantity of pasta wheat will benefit from the resources presented here. Furthermore, the durum sequence provides a fundamental tool to more effectively bridge and harness the allelic diversity present in wheat ancestors most of which remains largely untapped.
Methods
DW genomic sequencing and assembly
DNA extraction and sequencing
DW genomic DNA was isolated from fresh leaf tissue (~10 g) of DW cv. Svevo using a phenol/chloroform large-scale nucleus extraction protocol45. Five size-selected genomic DNA libraries ranging from 450 bp to 10 kb were sequenced to generate a total of 3.35 terabases (Tb) of data (equivalent to 279× coverage, on the basis of an estimated genome size of 12 Gb) using standard protocols. The libraries were constructed at the University of Illinois Roy J. Carver Biotechnology Center and sequenced at University of Salerno and Genomix4life by using an Illumina HiSeq2500 platform and HiseqX instrument, according to the manufacturer’s instructions (Supplementary Table 1).
Scaffold assembly
The DW genome was assembled from the 3.35 Tb of data using the proprietary software package DenovoMAGIC2 (NRGene) as previously described8. PCR duplicates, adapters and linkers were removed, and paired-end reads found to contain probable sequencing errors (that is, sub-sequences of ≥23 bp not found in at least one other independent read) were culled. From the paired-end libraries, only pairs with at least 10 bp sequence overlap were merged to create stitched reads. The stitched reads were then used to build an initial De Bruijn graph of contigs (k-mer, 191 bp). By exploring graph structure, the software identifies non-repetitive contigs and uses stitched reads information to resolve repeats and extend non-repetitive sequences of the contigs, where possible (Supplementary Table 2). Scaffolding was completed using a directed graph containing contigs as nodes and edges were on the basis of the paired-end and mate-pair links as vertices. Erroneous connections were identified and filtered out to generate unconnected sub-graphs that were ordered into scaffolds. Paired-end reads were used to find reliable paths in the graph for additional repeat resolving. This was accomplished by searching the graph for a unique path of contigs connecting pairs of reads mapping to two different non-repetitive contigs. The scaffolds were then further ordered and linked using the mate-pair libraries, estimating gaps between the contigs according to the distance of mate-pair links. Linking scaffolds with mate-pair reads required confirmation of at least three filtered mate-pairs or at least one filtered mate-pair with supporting confirmation from two or more filter failed mate-pairs where the Nextera adapter was not found. Scaffolds shorter than 380 bp were masked and links between non-repetitive contigs mapping to the same scaffolds were merged, generating a directed scaffold graph. The scaffolding procedure identified the non-branched components in the scaffolds graph, filtered out the rare connections between them and generated topological sorting based ordering of the initial scaffolds into the final scaffolds (Supplementary Table 2).
Chromosome conformation capture sequencing (Hi-C) and pseudomolecule construction
Scaffolds were assembled into pseudomolecules using data from two chromosomes conformation capture sequencing (Hi-C) libraries generated using the TCC protocol46 as previously described11. The libraries were sequenced using an Illumina HiSeq2500 instrument following the manufacturer’s instructions (each TCC library on two lanes, paired-end, 2 × 100). In silico HindIII digestion of the Svevo sequence assembly by NRGene and assignment of the pre-processed reads to the restriction fragments was done following the methods described47.
To identify possible misjoins in the Svevo NRGene assembly, we aligned the IWGSC chromosome survey sequencing hexaploid wheat contigs48 to the assembly and lifted the POPSEQ map positions and flow-sorted chromosome assignment from the chromosome survey sequencing contigs to the durum scaffolds as described8. A total of 18 putative chimeric scaffolds (an example is reported in Supplementary Fig. 17) were detected and split. All the identified misjoins had also evidence from flow-sorting sequencing data and Hi-C links from different chromosomes. The improved durum wheat assembly (10.45 Gb, scaffold N50 of 5,972,063 bp; Supplementary Table 2) was used to construct the final pseudomolecules (Supplementary Table 3).
The construction of a chromosome-scale Hi-C map of the NRGene v.2 assembly comprised two steps: (1) chromosome assignment and genetic anchoring of the Svevo scaffolds, (2) ordering and orienting scaffolds by Hi-C link information. Scaffolds were assigned to chromosomes using POPSEQ and flow-sorting data as previously described10. We used the same methods for Hi-C map construction as for the barley and WEW genomes8,47. We considered only intrachromosomal Hi-C links between pairs of scaffolds less than 20 cM apart. In the case of chromosome 3B, we used a threshold of 5 cM to avoid erroneous joins. Moreover, we did not consider scaffolds with fewer than 100 HindIII restriction sites for Hi-C mapping. Scaffolds were oriented as previously described47 using a bin size of 1 Mb. After the manual removal of four outlier scaffolds on chromosome 5A, the Hi-C map was highly collinear to the Svevo × Zavitan genetic map (Hi-C, Supplementary Fig. 18).
The FASTA sequence assemblies representing the 14 durum wheat chromosomes (that is, pseudomolecules, 2,938 scaffolds and 9.96 Gb in total) were constructed on the basis of the final Hi-C map. One hundred N characters were inserted as gap sequence between adjacent scaffolds. In addition, 126,526 scaffolds not assigned to chromosomes were placed in a sequence named ‘chrUn’ (499 Mb). The inclusion of gap sequences increased the total size of the assembly from 10.45 Gb to 10.46 Gb.
Additional methods
Additional methods are detailed in the Supplementary Note.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
A dedicated DW genome browser with all genome information presented in this manuscript (sequences, annotations, markers, passport data, etc.) is available at http://www.interomics.eu/durum-wheat-genome. Svevo genome and gene model were submitted to EBI-ENA under the study PRJEB22687. Svevo genome, gene model, SNP and QTL data and passport information of the Global Tetraploid Wheat Collection are available at GrainGenes (https://wheat.pw.usda.gov/GG3/jbrowse_Durum_Svevo). RNA-seq and microRNA (miRNA) datasets can be downloaded at the SRA database under accessions SRP149116 (study: PRJNA473404). Durum wheat cv. Svevo GFF3 and VCF annotation files can be downloaded at figshare, http://doi.org/10.6084/m9.figshare.6984035. The WEW acc. Zavitan gene model version 2 is available at https://doi.org/10.5447/ipk/2019/0. The accessions used in this study are available on request and on the basis of a Material Transfer Agreement from the corresponding author (L.C.).
References
Dubcovsky, J. & Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316, 1862–1866 (2007).
Faris, J. Genomics of Plant Genetic Resources Ch. 18 (Springer, 2014).
Steffenson, B. J. et al. A walk on the wild side: mining wild wheat and barley collections for rust resistance genes. Aust. J. Agric. Res. 58, 532–544 (2007).
Ellis, J. G., Lagudah, E. S., Spielmeyer, W. & Dodds, P. N. The past, present and future of breeding rust resistant wheat. Front. Plant Sci. 5, 641 (2014).
Uauy, C., Distelfeld, A., Fahima, T., Blechl, A. & Dubcovsky, J. A. NAC gene regulating senescence improves grain protein, zinc and iron content in wheat. Science 314, 1298–1300 (2006).
Gaut, B. S. Evolution is an experiment: assessing parallelism in crop domestication and experimental evolution. Mol. Biol. Evol. 32, 1661–1671 (2015).
Brozynska, M., Furtado, A. & Henry, R. J. Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotech. J. 14, 1070–1085 (2016).
Avni, R. et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357, 93–97 (2017).
Wiebe, K. et al. Targeted mapping of Cdu1, a major locus regulating grain cadmium concentration in durum wheat (Triticum turgidum L. var durum). Theor. Appl. Genet. 121, 1047–1058 (2010).
Avni, R. et al. Ultra-dense genetic map of durum wheat×wild emmer wheat developed using the 90K iSelect SNP genotyping assay. Mol. Breed. 34, 1549–1562 (2014).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Maccaferri, M. et al. A high-density, SNP-based consensus map of tetraploid wheat as a bridge to integrate durum and bread wheat genomics and breeding. Plant Biotech. J. 13, 648–663 (2015).
Wicker, T. et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 19, 103 (2018).
Montenegro, J. D. et al. The pangenome of hexaploid bread wheat. Plant J. 90, 1007–1013 (2017).
The International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding through a fully annotated and anchored reference genome sequence. Science 361, eaar7191 (2018).
Saintenac, C., Jiang, D. & Akhunov, E. D. Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biol. 12, R88 (2011).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotech. J. 12, 787–796 (2014).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Frichot, E., Mathieu, F., Trouillon, T., Bouchard, G. & François, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 196, 973–983 (2014).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Lawson, D. J., Hellenthal, G., Myers, S. & Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 8, e1002453 (2012).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl Acad. Sci. USA 12, 3321–3323 (1973).
Pankin, A., Altmüller, J., Becker, C. & von Korff, M. Targeted resequencing reveals genomic signatures of barley domestication. New Phytol. 218, 1247–1259 (2018).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet. 10, 639–650 (2009).
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M. & Servin, B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193, 929–941 (2013).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Faris, J. D., Zhang, Z. & Chao, S. Map-based analysis of the tenacious glume gene Tg-B1 of wild emmer and its role in wheat domestication. Gene 542, 198–208 (2014).
Zhang, W. et al. Identification and characterization of Sr13, a tetraploid wheat gene that confers resistance to the Ug99 stem rust race group. Proc. Natl Acad. Sci. USA 114, E9483–E9492 (2017).
Maccaferri, M. et al. A major QTL for durable leaf rust resistance widely exploited in durum wheat breeding programs maps on the distal region of chromosome arm 7BL. Theor. Appl. Genet. 117, 1225–1240 (2008).
He, X. Y., He, Z. H., Ma, W., Appels, R. & Xia, X. C. Allelic variants of phytoene synthase 1 (Psy1) genes in Chinese and CIMMYT wheat cultivars and development of functional markers for flour colour. Mol. Breed. 23, 553–563 (2009).
Penner, G. A., Clarke, J., Bezte, L. J. & Leisle, D. Identification of RAPD markers linked to a gene governing cadmium uptake in durum wheat. Genome 38, 543–547 (1995).
Knox, R. E. et al. Chromosomal location of the cadmium uptake gene (Cdu1) in durum wheat. Genome 52, 741–747 (2009).
Ueno, D. et al. Gene limiting cadmium accumulation in rice. Proc. Natl Acad. Sci. USA 107, 16500–16505 (2010).
Miyadate, H. et al. OsHMA3, a P1B-type of ATPase affects root-to-shoot cadmium translocation in rice by mediating efflux into vacuoles. New Phytol. 189, 190–199 (2011).
Yan, J. et al. A loss-of-function allele of OsHMA3 associated with high cadmium accumulation in shoots and grain of Japonica rice cultivars. Plant Cell Environ. 39, 1941–1954 (2016).
Harris, N. S. & Taylor, G. J. Cadmium uptake and partitioning in durum wheat during grain filling. BMC Plant Biol. 13, 103 (2013).
Özkan, H., Brandolini, A., Schäfer-Pregl, R. & Salamini, F. AFLP analysis of a collection of tetraploid wheats indicates the origin of emmer and hard wheat domestication in southeast Turkey. Mol. Biol. Evol. 19, 1797–1801 (2002).
Luo, M. C. et al. The structure of wild and domesticated emmer wheat populations, gene flow between them, and the site of emmer domestication. Theor. Appl. Genet. 114, 947–959 (2007).
Yan, L. et al. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl Acad. Sci. USA 100, 6263–6268 (2003).
Varshney, R. K. et al. Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 49, 1082–1088 (2017).
Mills, R. F. et al. The plant P1B-type ATPase AtHMA4 transports Zn and Cd and plays a role in detoxification of transition metals supplied at elevated levels. FEBS Lett. 579, 783–791 (2005).
Hart, J. J., Welch, R. M., Norvell, W. A., Clarke, J. M. & Kochian, L. V. Zinc effects on cadmium accumulation and partitioning in near-isogenic lines of durum wheat that differ in grain cadmium concentration. New Phytol. 167, 391–401 (2005).
Cakmak, I. et al. Zinc deficiency as a critical problem in wheat production in Central Anatolia. Plant Soil 180, 165–172 (1996).
Dvorak, J., McGuire, P. E. & Cassidy, B. Apparent sources of the A genomes of wheats inferred from polymorphism in abundance and restriction fragment length of repeated nucleotide sequences. Genome 30, 680–689 (1988).
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F. & Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98 (2011).
Beier, S. et al. Construction of a map-based reference genome sequence for barley, Hordeum vulgare L. Sci. Data 4, 170044 (2017).
Chapman, J. A. et al. A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 16, 26 (2015).
Acknowledgements
We acknowledge the funding support of: the Italian Ministry of Education and Research with projects CNR Flagship InterOmics PB05 (L.M., A.C., G.S.), PON ELIXIR CNR-BiOmics PIR01_00017 (L.M., M.G., M.Mo.) and PON ISCOCEM (P.D.); CREA project Interomics (L.C.); Fondazione in rete per la ricerca agroalimentare AGER project From Seed to Pasta (R.T.); FP7-KBBE Project DROPS ID244347 (R.T.); Genome Canada (A.G.S., C.P.); the Western Grain Research Foundation (A.G.S., C.P.); the Manitoba Wheat and Barley Commission (A.G.S., C.P.); the Saskatchewan Wheat Development Commission (A.G.S., C.P.); the Alberta Wheat Development Commission (A.G.S., C.P.); the Saskatchewan Ministry of Agriculture (A.G.S., C.P.); the administrative support of Genome Prairie (A.G.S., C.P.); Canadian Triticum Applied Genomics -CTAG2- (A.G.S., C.P.); Binational Science Foundation grant no. 2015409 (I.H., A.D.); Israel Science Foundation grant no. 1137/17 (A.D.); USDA-Agricultural Research Service Current Research Information System project 3060-21000-038-00-D (J.D.F., S.S.X.); German Federal Ministry of Food and Agriculture grant no. 2819103915 (N.S., K.F.X.M.); German Ministry of Education and Research grant no. 031A536 (K.F.X.M.); and Natural Sciences and Engineering Council of Canada grant nos. SPG 336119-06 and RGPIN 92787 (G.J.T., C.P.). The authors are grateful to E. Elias (North Dakota State University) for providing nine DW cultivars, included in the Global Tetraploid Wheat Collection and E. Scarpella (University of Alberta, Edmonton, Canada) for assistance with confocal microscopy.
Author information
Authors and Affiliations
Contributions
L.C., C.J.P., K.F.X.M., A.C. and RT conceived the study. A.S., G.S., P.D.V., N.P., L.M., H.B., A.D., A.C., C.J.P. and L.C. planned, and organized the sequencing steps. S.C.M., A.H., M.Mas. and N.S. carried out chromosome conformation capture and curated the final genome assembly. S.O.T., H.G., M.S., D.O., T.L., V.P., P.B., P.F., P.C., M.L., B.L., A.S., S.C., M.Mac., S.B., F.D., C.M., C.C., E.M., D.M., E.M., D.N., A.G., H.B. and A.M.M. contributed to genome annotations. A.M., M.G., M.Mo. and L.M. curated the genome visualization. S.O.T., H.G., M.S., D.O., T.L., V.P., R.A., J.D., A.D. and K.F.X.M. conducted the comparison between Svevo and Zavitan genomes. A.T.O.M. and I.H. carried out the identification of loss of functional mutations. M.Mac., R.K.P., D.O., E.F., S.C., S.S., F.D., H.O., B.K., D.M., R.J., E.M., S.C., J.D.F., M.P., A.M.M., S.S.X., A.D. and M.J.H. planned and carried out the analysis of genetic diversity and the identification of selective sweeps. N.S.H., K.W., J.E., R.P.M.L., J.M.C., A.G.S., S.C.K., K.Y.H.L., G.J.T., R.K., S.W. and C.J.P. planned and carried out the isolation and functional analysis of TdHMA3-B1. L.C., M.Mac., A.M.M., S.W., K.F.X.M. and C.J.P. organized and managed the contributions of others to this publication and produced the first draft of it. L.C. served as Senior Project Coordinator. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note, Supplementary Figures 1–39 and Supplementary Tables 1–24
Supplementary Dataset 1
Inventory of linkage mapping and GWAS-QTLs mapped on tetraploid wheat and projected on the Svevo genome assembly.
Supplementary Dataset 2
Population structure of the Global Tetraploid wheat Collection (GTC, 1,856 accessions) assessed by ADMIXTURE/fineSTRUCTURE analysis and their allelic score at HMA3-B1 assessed by Xusw59 perfect marker.
Supplementary Dataset 3
Population structure of the Global Tetraploid wheat Collection (GTC) assessed with four model- and non model- based quantitative clustering methods: DAPC-K means, DAPC-Ward’s, sNMF, ADMIXTURE.
Supplementary Dataset 4
Whole-genome diversity reduction and selection signal analysis in tetraploid wheat.
Supplementary Dataset 5
Detailed Fst analysis of the tetraploid diversity panel for chromosome 5B.
Supplementary Dataset 6
Gene content and functional analysis of the Cdu-B1 region in chromosome 5B as defined by the linkage disequilibrium interval in the durum wheat landrace and cultivar panels ± 2 Mb.
Supplementary Dataset 7
Experimental conditions and tissue used for the RNA-seq and small-RNA-seq expression analysis.
Supplementary Dataset 8
Sequencing data on 216 experimentally validated wheat genes.
Supplementary Dataset 9
Unigene clusters made with both wild emmer wheat Zavitan and durum wheat Svevo HC genes.
Supplementary Dataset 10
Genome-wide atlas of 597 putative high impact variants differentiating between wild emmer wheat accession Zavitan and durum wheat cultivar Svevo by chromosome.
Supplementary Dataset 11
Detailed genetic maps used to project iSelect Illumina 9K, 15K, 90K SNP, DArT, SSR, EST-SSR, and STS markers on the Svevo genome assembly.
Supplementary Dataset 12
Locus identifiers and protein sequences used for the HMA phylogenetic analysis.
Supplementary Dataset 13
List of miRNAs detected in all or in one unique sequencing pool.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maccaferri, M., Harris, N.S., Twardziok, S.O. et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51, 885–895 (2019). https://doi.org/10.1038/s41588-019-0381-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0381-3
This article is cited by
-
Genetic mapping of deoxynivalenol and fusarium damaged kernel resistance in an adapted durum wheat population
BMC Plant Biology (2024)
-
A membrane associated tandem kinase from wild emmer wheat confers broad-spectrum resistance to powdery mildew
Nature Communications (2024)
-
Genetic diversity, linkage disequilibrium, and population structure of tetraploid wheat landraces originating from Europe and Asia
BMC Genomics (2023)
-
Gene expression profile of the developing endosperm in durum wheat provides insight into starch biosynthesis
BMC Plant Biology (2023)
