Department of Psychology, University of Minnesota Twin Cities, Minneapolis, MN, USA
James J. Lee, Emily A. Willoughby, James J. Lee, Michael B. Miller, William G. Iacono, Matt McGue & Robert F. Krueger
Department of Sociology, University of Colorado Boulder, Boulder, CO, USA
Robbee Wedow & Jason D. Boardman
Institute for Behavioral Genetics, University of Colorado Boulder, Boulder, CO, USA
Robbee Wedow & Jason D. Boardman
Institute of Behavioral Science, University of Colorado Boulder, Boulder, CO, USA
Robbee Wedow & Jason D. Boardman
Department of Complex Trait Genetics, Center for Neurogenomics and Cognitive Research, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands
Aysu Okbay, Richard Karlsson Linnér, Aysu Okbay, S. Fleur W. Meddens, Christiaan de Leeuw, Danielle Posthuma, Philipp D. Koellinger & Philipp D. Koellinger
Department of Economics, School of Business and Economics, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands
Aysu Okbay, Richard Karlsson Linnér, Aysu Okbay, Philipp D. Koellinger & Philipp D. Koellinger
Department of Economics, Harvard University, Cambridge, MA, USA
Edward Kong, Omeed Maghzian, Peter Bowers, Chanwook Lee, Hui Li, Olga Rostapshova, David I. Laibson & David I. Laibson
Department of Sociology, Harvard University, Cambridge, MA, USA
Meghan Zacher
Center for Economic and Social Research, University of Southern California, Los Angeles, CA, USA
Tuan Anh Nguyen-Viet, Mark Alan Fontana, Tushar Kundu, Ruoxi Li, Rebecca Royer, Mark Alan Fontana, Daniel J. Benjamin, Chelsea Watson & Daniel J. Benjamin
Institute for Molecular Bioscience, University of Queensland, Brisbane, Queensland, Australia
Julia Sidorenko, Loïc Yengo, Jian Yang, Peter M. Visscher, Kathryn E. Kemper, Yang Wu, Zhili Zheng, Matthew R. Robinson, Jian Yang & Peter M. Visscher
Estonian Genome Center, University of Tartu, Tartu, Estonia
Julia Sidorenko, Evelin Mihailov, Natalia Pervjakova, Reedik Mägi, Lili Milani, Andres Metspalu, Markus Perola, Tõnu Esko, Maris Alver, Reedik Mägi, Andres Metspalu, Lili Milani & Tõnu Esko
Institute for Behavior and Biology, Erasmus University Rotterdam, Rotterdam, The Netherlands
Richard Karlsson Linnér, Cornelius A. Rietveld, S. Fleur W. Meddens, Ronald de Vlaming, A. Roy Thurik, Philipp D. Koellinger & Philipp D. Koellinger
Center for the Advancement of Value in Musculoskeletal Care, Hospital for Special Surgery, New York, NY, USA
Mark Alan Fontana & Mark Alan Fontana
The Novo Nordisk Foundation Center for Basic Metabolic Research, Section of Metabolic Genetics, University of Copenhagen, Faculty of Health and Medical Sciences, Copenhagen, Denmark
Pascal N. Timshel, Tune H. Pers, Pascal Timshel, Tarunveer S. Ahluwalia, Thorkild I. A. Sørensen & Tune H. Pers
Statens Serum Institut, Department of Epidemiology Research, Copenhagen, Denmark
Pascal N. Timshel, Tune H. Pers, Pascal Timshel & Tune H. Pers
Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
Raymond K. Walters, Patrick Turley, Aarno Palotie & Patrick Turley
Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA
Raymond K. Walters, Patrick Turley, Aarno Palotie & Patrick Turley
Institute for Social and Economic Research, University of Essex, Colchester, UK
Yanchun Bao, Pamela Herd & Meena Kumari
Centre for Global Health Research, Usher Institute of Population Health Sciences and Informatics, University of Edinburgh, Edinburgh, UK
Katharina E. Schraut, Harry Campbell, Peter K. Joshi, Igor Rudan, Ozren Polasek, James F. Wilson, David W. Clark, Peter K. Joshi, Harry Campbell, Melissa C. Smart & James F. Wilson
MRC Epidemiology Unit, Institute of Metabolic Science, University of Cambridge, Cambridge, UK
Felix R. Day, Claudia Langenberg, Jing Hua Zhao, Jian’an Luan, Ken K. Ong, John R. B. Perry & Nicholas J. Wareham
23andMe, Inc., Mountain View, CA, USA
Michelle Agee, Babak Alipanahi, Adam Auton, Robert K. Bell, Katarzyna Bryc, Sarah L. Elson, Pierre Fontanillas, David A. Hinds, Jennifer C. McCreight., Karen E. Huber, Nadia K. Litterman, Matthew H. McIntyre, Joanna L. Mountain, Elizabeth S. Noblin, Carrie A. M. Northover, Steven J. Pitts, J. Fah Sathirapongsasuti, Olga V. Sazonova, Janie F. Shelton, Suyash Shringarpure, Chao Tian, Vladimir Vacic, Catherine H. Wilson, Nicholas A. Furlotte, David A. Hinds, Joyce Y. Tung, Nicholas A. Furlotte, Aaron Kleinman & Joyce Y. Tung
Institute of Social and Preventive Medicine, University Hospital of Lausanne, Lausanne, Switzerland
Peter K. Joshi & Peter K. Joshi
BrainWorkup, LLC, Santa Monica, CA, USA
Joey W. Trampush
Department of Psychiatry and Behavioral Sciences, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Joey W. Trampush
Department of Biomedical and Translational Informatics, Geisinger Health System, Danville, PA, USA
Shefali Setia Verma & Marylyn D. Ritchie
Institute of Mental Health, Singapore, Singapore
Max Lam
Genome Institute, Singapore, Singapore
Max Lam
The Eye Hospital, School of Ophthalmology and Optometry, Wenzhou Medical University, Wenzhou, China
Zhili Zheng
Department of Sociology, Stanford University, Stanford, CA, USA
Jeremy Freese
Department of Sociology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Kathleen Mullan Harris
Carolina Population Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Kathleen Mullan Harris
MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK
Jennifer E. Huffman, Jonathan Marten, Caroline Hayward, Veronique Vitart, James F. Wilson, Alan F. Wright, Caroline Hayward & James F. Wilson
La Follette School of Public Affairs, University of Wisconsin-Madison, Madison, WI, USA
Pamela Herd
Departments of Psychiatry and Molecular Medicine, Hofstra Northwell School of Medicine, Hempstead, NY, USA
Todd Lencz & Anil K. Malhotra
Center for Psychiatric Neuroscience, Feinstein Institute for Medical Research, Manhasset, NY, USA
Todd Lencz & Anil K. Malhotra
Psychiatry Research, The Zucker Hillside Hospital, Glen Oaks, CA, USA
Todd Lencz & Anil K. Malhotra
Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia
Andres Metspalu & Andres Metspalu
Centre for Genomic and Experimental Medicine, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK
Sarah E. Harris, David J. Porteous & David J. Porteous
Division of Population Health Sciences, Ninewells Hospital and Medical School, University of Dundee, Dundee, UK
Blair H. Smith & Blair H. Smith
Medical Research Institute, University of Dundee, Dundee, UK
Blair H. Smith & Blair H. Smith
Department of Economics, University of Toronto, Toronto, Ontario, Canada
Jonathan P. Beauchamp, Riccardo E. Marioni & Jonathan P. Beauchamp
Department of Sociology, Princeton University, Princeton, NJ, USA
Dalton C. Conley & Dalton C. Conley
School of Policy Studies, Queen’s University, Kingston, Ontario, Canada
Steven F. Lehrer & Steven F. Lehrer
Department of Economics, New York University Shanghai, Pudong, Shanghai, China
Steven F. Lehrer & Steven F. Lehrer
National Bureau of Economic Research, Cambridge, MA, USA
Steven F. Lehrer, David Cesarini, Daniel J. Benjamin, Steven F. Lehrer, Daniel J. Benjamin & David Cesarini
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden
Robert Karlsson, Paul Lichtenstein, Nancy L. Pedersen, Patrik K. E. Magnusson & Patrik K. E. Magnusson
Department of Government, Uppsala University, Uppsala, Sweden
Sven Oskarsson, Karl-Oskar Lindgren & Sven Oskarsson
Department of Computational Biology, University of Lausanne, Lausanne, Switzerland
Matthew R. Robinson
Department of Economics, New York University, New York, NY, USA
Kevin Thom, David Cesarini, Kevin Thom & David Cesarini
Autism and Developmental Medicine Institute, Geisinger Health System, Lewisburg, PA, USA
Christopher F. Chabris & Christopher F. Chabris
Center for Translational Bioethics and Health Care Policy, Geisinger Health System, Danville, PA, USA
Michelle N. Meyer & Michelle N. Meyer
Queensland Brain Institute, University of Queensland, Brisbane, Queensland, Australia
Guo-Bo Chen, Zhihong Zhu, Andrew Bakshi, Anna A. E. Vinkhuyzen, Jacob Gratten, Jian Yang, Peter M. Visscher, Jian Yang & Peter M. Visscher
Department of Economics, Stockholm School of Economics, Stockholm, Sweden
Magnus Johannesson & Magnus Johannesson
Department of Economics, University of Southern California, Los Angeles, CA, USA
Daniel J. Benjamin & Daniel J. Benjamin
Center for Experimental Social Science, New York University, New York, NY, USA
David Cesarini & David Cesarini
Department of Applied Economics, Erasmus School of Economics, Erasmus University Rotterdam, Rotterdam, The Netherlands
Cornelius A. Rietveld, Ronald de Vlaming & A. Roy Thurik
Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands
Cornelius A. Rietveld, Ronald de Vlaming, Sven J. van der Lee, Najaf Amin, Frank J. A. van Rooij, Cornelia M. van Duijn, Henning Tiemeier, André G. Uitterlinden & Albert Hofman
Icelandic Heart Association, Kopavogur, Iceland
Valur Emilsson, Albert V. Smith & Vilmundur Gudnason
Faculty of Pharmaceutical Sciences, University of Iceland, Reykjavík, Iceland
Valur Emilsson
Amsterdam Business School, University of Amsterdam, Amsterdam, The Netherlands
S. Fleur W. Meddens & Maël P. Lebreton
New York Genome Center, New York, NY, USA
Joseph K. Pickrell
Department of Biological Psychology, VU University Amsterdam, Amsterdam, The Netherlands
Abdel Abdellaoui, Jouke-Jan Hottenga, Gonneke Willemsen & Dorret I. Boomsma
COPSAC, Copenhagen Prospective Studies on Asthma in Childhood, Herlev and Gentofte Hospital, University of Copenhagen, Copenhagen, Denmark
Tarunveer S. Ahluwalia, Klaus Bønnelykke, Johannes Waage & Hans Bisgaard
Steno Diabetes Center, Gentofte, Denmark
Tarunveer S. Ahluwalia & Johannes Waage
Department of Obstetrics and Gynecology, Institute of Clinical Sciences, Sahlgrenska Academy, Gothenburg, Sweden
Jonas Bacelis & Bo Jacobsson
Research Unit of Molecular Epidemiology, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
Clemens Baumbach & Christian Gieger
Institute of Epidemiology II, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
Clemens Baumbach & Christa Meisinger
deCODE Genetics/Amgen Inc., Reykjavík, Iceland
Gyda Bjornsdottir, Augustine Kong, Gudmar Thorleifsson, Bjarni Gunnarsson, Bjarni V. Halldórsson, Kari Stefansson & Unnur Thorsteinsdottir
Department of Cell Biology, Erasmus Medical Center Rotterdam, Rotterdam, The Netherlands
Johannes H. Brandsma & Raymond A. Poot
Istituto di Ricerca Genetica e Biomedica U.O.S. di Sassari, National Research Council of Italy, Sassari, Italy
Maria Pina Concas, Simona Vaccargiu & Mario Pirastu
Psychology, University of Illinois, Champaign, IL, USA
Jaime Derringer
Institute for Computing and Information Sciences, Radboud University Nijmegen, Nijmegen, The Netherlands
Tessel E. Galesloot & Lambertus A. L. M. Kiemeney
Department of Medical, Surgical and Health Sciences, University of Trieste, Trieste, Italy
Giorgia Girotto, Dragana Vuckovic, Ilaria Gandin, Paolo Gasparini & Nicola Pirastu
Department of Public Health, University of Helsinki, Helsinki, Finland
Richa Gupta, Antti Latvala, LifeLines Cohort Study, Anu Loukola & Jaakko Kaprio
Department of Cardiovascular Sciences, University of Leicester, Leicester, UK
Leanne M. Hall, Christopher P. Nelson & Nilesh J. Samani
Centre for Cognitive Ageing and Cognitive Epidemiology, University of Edinburgh, Edinburgh, UK
Leanne M. Hall, Sarah E. Harris, Gail Davies, David C. M. Liewald, Riccardo E. Marioni & Ian J. Deary
Department of Neurology, General Hospital and Medical University Graz, Graz, Austria
Edith Hofer, Katja E. Petrovic, Helena Schmidt & Reinhold Schmidt
Institute for Medical Informatics, Statistics and Documentation, General Hospital and Medical University Graz, Graz, Austria
Edith Hofer
Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Oxford, UK
Momoko Horikoshi
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK
Momoko Horikoshi
Institute of Behavioural Sciences, University of Helsinki, Helsinki, Finland
Kadri Kaasik, Jari Lahti, Liisa Keltigangas-Järvinen & Katri Räikkönen
Nutrition and Dietetics, Health Science and Education, Harokopio University, Athens, Greece
Ioanna P. Kalafati & George V. Dedoussis
Folkhälsan Research Centre, Helsingfors, Finland
Jari Lahti, Katri Räikkönen & Johan G. Eriksson
Institute for Computing and Information Sciences, Radboud University Nijmegen, Nijmegen, The Netherlands
Christiaan de Leeuw
Quantitative Genetics, QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia
Penelope A. Lind & Sarah E. Medland
Lifespan Psychology, Max Planck Institute for Human Development, Berlin, Germany
Tian Liu
Department of Twin Research and Genetic Epidemiology, King’s College London, London, UK
Massimo Mangino, Lydia Quaye, Cristina Venturini & Tim D. Spector
NIHR Biomedical Research Centre, Guy’s and St. Thomas’ Foundation Trust, London, UK
Massimo Mangino & Cristina Venturini
Department of Epidemiology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Peter J. van der Most, Behrooz Z. Alizadeh, Jennifer A. Smith & Judith M. Vonk
Public Health Stream, Hunter Medical Research Institute, New Lambton, New South Wales, Australia
Christopher Oldmeadow, Elizabeth G. Holliday & John R. Attia
Faculty of Health and Medicine, University of Newcastle, Newcastle, New South Wales, Australia
Christopher Oldmeadow, Elizabeth G. Holliday, Rodney J. Scott & John R. Attia
Centre for Integrated Genomic Medical Research, Institute of Population Health, The University of Manchester, Manchester, UK
Antony Payton & William E. R. Ollier
School of Psychological Sciences, The University of Manchester, Manchester, UK
Antony Payton
Department of Health, THL-National Institute for Health and Welfare, Helsinki, Finland
Natalia Pervjakova, Niina Eklund, Seppo Koskinen, Tomi Mäki-Opas, Veikko Salomaa, Jaakko Kaprio & Markus Perola
Psychiatry, VU University Medical Center and GGZ inGeest, Amsterdam, The Netherlands
Wouter J. Peyrot, Yusplitri Milaneschi & Brenda W. J. H. Penninx
Laboratory of Genetics, National Institute on Aging, Baltimore, MD, USA
Yong Qian, Jun Ding & David Schlessinger
Research Centre of Applied and Preventive Cardiovascular Medicine, University of Turku, Turku, Finland
Olli Raitakari
Department of Medical Genetics, University of Lausanne, Lausanne, Switzerland
Rico Rueedi & Zoltan Kutalik
Swiss Institute of Bioinformatics, Lausanne, Switzerland
Rico Rueedi & Zoltan Kutalik
Department Of Health Sciences, University of Milan, Milan, Italy
Erika Salvi & Daniele Cusi
Institute for Medical Informatics, Biometry and Epidemiology, University Hospital of Essen, Essen, Germany
Börge Schmidt, Lewin Eisele & Karl-Heinz Jöckel
Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, MD, USA
Jianxin Shi
Faculty of Medicine, University of Iceland, Reykjavík, Iceland
Albert V. Smith, Vilmundur Gudnason, Kari Stefansson & Unnur Thorsteinsdottir
MRC Integrative Epidemiology Unit, University of Bristol, Bristol, UK
Beate St Pourcain, David M. Evans, George McMahon, Lavinia Paternoster, Susan M. Ring, Thorkild I. A. Sørensen, Nicholas J. Timpson & George Davey Smith
School of Oral and Dental Sciences, University of Bristol, Bristol, UK
Beate St Pourcain
Institute for Community Medicine, University Medicine Greifswald, Greifswald, Germany
Alexander Teumer, Sebastian E. Baumeister, Henry Völzke & Wolfgang Hoffmann
Department of Cardiology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Niek Verweij, Klaus Berger & Pim van der Harst
Institute of Epidemiology and Social Medicine, University of Münster, Münster, Germany
Juergen Wellmann
Divisions of Genetics and Rheumatology, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Harm-Jan Westra
Partners Center for Personalized Genetic Medicine, Boston, MA, USA
Harm-Jan Westra
Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
Harm-Jan Westra, Philip L. de Jager & Aarno Palotie
Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, IL, USA
Jingyun Yang, Patricia A. Boyle & David A. Bennett
Department of Neurological Sciences, Rush University Medical Center, Chicago, IL, USA
Jingyun Yang & David A. Bennett
Department of Epidemiology, University of Michigan, Ann Arbor, MI, USA
Wei Zhao, Erin B. Ware & Sharon L. R. Kardia
Department of Gastroenterology and Hepatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Behrooz Z. Alizadeh
Institute of Epidemiology and Preventive Medicine, University of Regensburg, Regensburg, Germany
Sebastian E. Baumeister
Institute of Molecular Genetics, National Research Council of Italy, Pavia, Italy
Ginevra Biino
Department of Behavioral Sciences, Rush University Medical Center, Chicago, IL, USA
Patricia A. Boyle
Warwick Medical School, University of Warwick, Coventry, UK
Francesco P. Cappuccio
Department of Psychology, University of Edinburgh, Edinburgh, UK
Gail Davies, David C. M. Liewald & Ian J. Deary
Saïd Business School, University of Oxford, Oxford, UK
Jan-Emmanuel De Neve
William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, UK
Panos Deloukas & Stavroula Kanoni
Princess Al-Jawhara Al-Brahim Centre of Excellence in Research of Hereditary Disorders (PACER-HD), King Abdulaziz University, Jeddah, Saudi Arabia
Panos Deloukas
The Berlin Aging Study II; Research Group on Geriatrics, Charité-Universitätsmedizin Berlin, Berlin, Germany
Ilja Demuth & Elisabeth Steinhagen-Thiessen
Institute of Medical and Human Genetics, Charité-Universitätsmedizin Berlin, Berlin, Germany
Ilja Demuth
German Socio-Economic Panel Study, DIW Berlin, Berlin, Germany
Peter Eibich & Martin Kroh
Health Economics Research Centre, Nuffield Department of Population Health, University of Oxford, Oxford, UK
Peter Eibich
The University of Queensland Diamantina Institute, The Translational Research Institute, Brisbane, Queensland, Australia
David M. Evans
Survey Research Center, Institute for Social Research, University of Michigan, Ann Arbor, MI, USA
Jessica D. Faul & David R. Weir
Department of Genetics, Division of Statistical Genomics, Washington University School of Medicine, St Louis, MO, USA
Mary F. Feitosa, Aldi T. Kraja, Ingrid B. Borecki & Michael A. Province
Institute of Human Genetics, University of Bonn, Bonn, Germany
Andreas J. Forstner
Department of Genomics, Life and Brain Center, University of Bonn, Bonn, Germany
Andreas J. Forstner
Institute of Biomedical and Neural Engineering, School of Science and Engineering, Reykjavík University, Reykjavík, Iceland
Bjarni V. Halldórsson
Laboratory of Epidemiology, Demography, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA
Tamara B. Harris
Department of Psychiatry, Washington University School of Medicine, St Louis, MO, USA
Andrew C. Heath & Pamela A. Madden
Division of Applied Health Sciences, University of Aberdeen, Aberdeen, UK
Lynne J. Hocking
Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald, Germany
Georg Homuth & Uwe Völker
Manchester Medical School, The University of Manchester, Manchester, UK
Michael A. Horan
Program in Translational NeuroPsychiatric Genomics, Departments of Neurology and Psychiatry, Brigham and Women’s Hospital, Boston, MA, USA
Philip L. de Jager
Harvard Medical School, Boston, MA, USA
Philip L. de Jager
Department of Genes and Environment, Norwegian Institute of Public Health, Oslo, Norway
Astanand Jugessur, Ronny Myhre & Bo Jacobsson
Department of Genomics of Common Disease, Imperial College London, London, UK
Marika A. Kaakinen
Department of Clinical Physiology, Tampere University Hospital, Tampere, Finland
Mika Kähönen
Department of Clinical Physiology, University of Tampere, School of Medicine, Tampere, Finland
Mika Kähönen
Public Health, Medical School, University of Split, Split, Croatia
Ivana Kolcic
Institute of Social and Preventive Medicine, Lausanne University Hospital (CHUV), Lausanne, Switzerland
Zoltan Kutalik
Neuroepidemiology Section, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA
Lenore J. Launer
Amsterdam Brain and Cognition Center, University of Amsterdam, Amsterdam, The Netherlands
Maël P. Lebreton
Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, USA
Douglas F. Levinson
Institute of Human Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
Peter Lichtner & Thomas Meitinger
Medical Genetics Section, Centre for Genomic and Experimental Medicine, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK
Riccardo E. Marioni
Department of Internal Medicine, Lausanne University Hospital (CHUV), Lausanne, Switzerland
Pedro Marques-Vidal & Peter Vollenweider
Tema BV, Hoofddorp, The Netherlands
Gerardus A. Meddens
Molecular Epidemiology, QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia
Grant W. Montgomery & Dale R. Nyholt
NIHR Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, UK
Christopher P. Nelson & Nilesh J. Samani
Institute of Health and Biomedical Innovation, Queensland Institute of Technology, Brisbane, Queensland, Australia
Dale R. Nyholt
Psychiatric and Neurodevelopmental Genetics Unit, Department of Psychiatry, Massachusetts General Hospital, Boston, MA, USA
Aarno Palotie
Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Helsinki, Finland
Aarno Palotie, Antti-Pekka Sarin & Jaakko Kaprio
Department of Neurology, Massachusetts General Hospital, Boston, MA, USA
Aarno Palotie
Medical Genetics, Institute for Maternal and Child Health IRCCS “Burlo Garofolo”, Trieste, Italy
Antonietta Robino, Sheila Ulivi, Diego Vozzi & Paolo Gasparini
Social Impact, Arlington, VA, USA
Olga Rostapshova
Department of Economics, University of Minnesota Twin Cities, Minneapolis, MN, USA
Aldo Rustichini
Department of Psychiatry and Behavioral Sciences, NorthShore University HealthSystem, Evanston, IL, USA
Alan R. Sanders & Pablo V. Gejman
Department of Psychiatry and Behavioral Neuroscience, University of Chicago, Chicago, IL, USA
Alan R. Sanders & Pablo V. Gejman
Public Health Genomics Unit, National Institute for Health and Welfare, Helsinki, Finland
Antti-Pekka Sarin
Research Unit for Genetic Epidemiology, Institute of Molecular Biology and Biochemistry, Center of Molecular Medicine, General Hospital and Medical University, Graz, Graz, Austria
Helena Schmidt
Information Based Medicine Stream, Hunter Medical Research Institute, New Lambton, New South Wales, Australia
Rodney J. Scott
Research Unit Hypertension and Cardiovascular Epidemiology, Department of Cardiovascular Science, University of Leuven, Leuven, Belgium
Jan A. Staessen
R&D VitaK Group, Maastricht University, Maastricht, The Netherlands
Jan A. Staessen
Institute of Genetic Epidemiology, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
Konstantin Strauch
Institute of Medical Informatics, Biometry and Epidemiology, Chair of Genetic Epidemiology, Ludwig Maximilians-Universität, Munich, Germany
Konstantin Strauch
Department of Geriatrics, Florida State University College of Medicine, Tallahassee, FL, USA
Antonio Terracciano
Department of Health Sciences and Genetics, University of Leicester, Leicester, UK
Martin D. Tobin
Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands
Frank J. A. van Rooij
Research Center for Group Dynamics, Institute for Social Research, University of Michigan, Ann Arbor, MI, USA
Erin B. Ware
Platform for Genome Analytics, Institutes of Neurogenetics and Integrative and Experimental Genomics, University of Lübeck, Lübeck, Germany
Lars Bertram
Neuroepidemiology and Ageing Research Unit, School of Public Health, Faculty of Medicine, Imperial College London, London, UK
Lars Bertram
Department of Health Sciences, Community and Occupational Medicine, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Ute Bültmann
Istituto di Ricerca Genetica e Biomedica (IRGB), Consiglio Nazionale delle Ricerche, c/o Cittadella Universitaria di Monserrato, Monserrato, Cagliari, Italy
Francesco Cucca
Institute of Biomedical Technologies, Italian National Research Council, Segrate, Milan, Italy
Daniele Cusi
Department of General Practice and Primary Health Care, University of Helsinki, Helsinki, Finland
Johan G. Eriksson
Departments of Human Genetics and Psychiatry, Donders Centre for Neuroscience, Nijmegen, The Netherlands
Barbara Franke
Department of Genetics, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Lude Franke & Pim van der Harst
Sidra, Experimental Genetics Division, Sidra, Doha, Qatar
Paolo Gasparini
Department of Psychiatry and Psychotherapy, University Medicine Greifswald, Greifswald, Germany
Hans-Jörgen Grabe
Department of Psychiatry and Psychotherapy, HELIOS-Hospital Stralsund, Stralsund, Germany
Hans-Jörgen Grabe
Econometric Institute, Erasmus School of Economics, Erasmus University Rotterdam, Rotterdam, The Netherlands
Patrick J. F. Groenen
Durrer Center for Cardiogenetic Research, ICIN-Netherlands Heart Institute, Utrecht, The Netherlands
Pim van der Harst
Centre for Population Health Research, School of Health Sciences and Sansom Institute, University of South Australia, Adelaide, South Australia, Australia
Elina Hyppönen
South Australian Health and Medical Research Institute, Adelaide, South Australia, Australia
Elina Hyppönen & Christine Power
Population, Policy and Practice, UCL Institute of Child Health, University College London, London, UK
Elina Hyppönen
Department of Epidemiology and Biostatistics, MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, London, UK
Marjo-Riitta Järvelin
Center for Life Course Epidemiology, Faculty of Medicine, University of Oulu, Oulu, Finland
Marjo-Riitta Järvelin
Unit of Primary Care, Oulu University Hospital, Oulu, Finland
Marjo-Riitta Järvelin
Biocenter Oulu, University of Oulu, Oulu, Finland
Marjo-Riitta Järvelin
Fimlab Laboratories, Tampere, Finland
Terho Lehtimäki
Department of Clinical Chemistry, University of Tampere, School of Medicine, Tampere, Finland
Terho Lehtimäki
Genetic Epidemiology, QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia
Nicholas G. Martin
Centre for Clinical and Cognitive Neuroscience, Institute Brain Behaviour and Mental Health, Salford Royal Hospital, Manchester, UK
Neil Pendleton
Manchester Institute Collaborative Research in Ageing, University of Manchester, Manchester, UK
Neil Pendleton
Faculty of Medicine, University of Split, Split, Croatia
Ozren Polasek
Department of Clinical Genetics, VU Medical Centre, Amsterdam, The Netherlands
Danielle Posthuma
Institute of Preventive Medicine, Bispebjerg and Frederiksberg Hospitals, The Capital Region, Frederiksberg, Denmark
Thorkild I. A. Sørensen
Montpellier Business School, Montpellier, France
A. Roy Thurik
Panteia, Zoetermeer, The Netherlands
A. Roy Thurik
Department of Psychiatry, Erasmus Medical Center, Rotterdam, The Netherlands
Henning Tiemeier
Department of Child and Adolescent Psychiatry, Erasmus Medical Center, Rotterdam, The Netherlands
Henning Tiemeier
Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands
André G. Uitterlinden
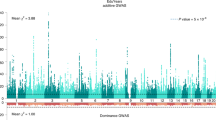
D.J.B., D.C., P.T. and P.M.V. designed and oversaw the study. A.O. was the lead analyst of the study, responsible for quality control and meta-analyses. Analysts who assisted A.O. in major ways include: E.K. (quality control), O.M. (COJO, MTAG, quality control), T.A.N.-V. (figure preparation), H.L. (quality control), C.L. (quality control), J.S. (UKB association analyses) and R.K.L. (UKB association analyses). P.B. and E.K. conducted the within-family association analyses. The cross-cohort heritability and genetic-correlation analyses were conducted by R.W. and M.Z. The analyses of the X chromosome in UK Biobank were conducted by J.S.; A.O. ran the meta-analysis. J.J.L. organized and oversaw the bioinformatic analyses, with assistance from T.E., E.K., K.T., T.H.P. and P.N.T. Polygenic-prediction analyses were designed and conducted by A.O., K.T. and R.W. Besides the contributions explicitly listed above, T.K., R.L. and R.R. conducted additional analyses for several subsections. C.W. helped with the coordination of the participating cohorts. J.P.B., D.C.C., T.E., M.J., J.J.L., P.D.K., D.I.L., S.F.L., S.O., M.R.R., K.T. and J.Y. provided helpful advice and feedback on various aspects of the study design. All authors contributed to and critically reviewed the manuscript. E.K., J.J.L. and R.W. made especially large contributions to the writing and editing.