
Abstract
Hair color is one of the most recognizable visual traits in European populations and is under strong genetic control. Here we report the results of a genome-wide association study meta-analysis of almost 300,000 participants of European descent. We identified 123 autosomal and one X-chromosome loci significantly associated with hair color; all but 13 are novel. Collectively, single-nucleotide polymorphisms associated with hair color within these loci explain 34.6% of red hair, 24.8% of blond hair, and 26.1% of black hair heritability in the study populations. These results confirm the polygenic nature of complex phenotypes and improve our understanding of melanin pigment metabolism in humans.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
Change history
31 May 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Lin, J. Y. & Fisher, D. E. Melanocyte biology and skin pigmentation. Nature 445, 843–850 (2007).
Randhawa, M. et al. Evidence for the ectopic synthesis of melanin in human adipose tissue. FASEB J. 23, 835–843 (2009).
Sturm, R. A., Teasdale, R. D. & Box, N. F. Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene 277, 49–62 (2001).
Jablonski, N. G. & Chaplin, G. The evolution of human skin coloration. J. Hum. Evol. 39, 57–106 (2000).
Jablonski, N. G. & Chaplin, G. Colloquium paper: human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA 107 Suppl 2, 8962–8968 (2010).
Greaves, M. Was skin cancer a selective force for black pigmentation in early hominin evolution? Proc. Biol. Sci. 281, 20132955 (2014).
Branda, R. F. & Eaton, J. W. Skin color and nutrient photolysis: an evolutionary hypothesis. Science 201, 625–626 (1978).
Norton, H. L. et al. Genetic evidence for the convergent evolution of light skin in Europeans and East Asians. Mol. Biol. Evol. 24, 710–722 (2007).
Wilde, S. et al. Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000y. Proc. Natl. Acad. Sci. USA 111, 4832–4837 (2014).
Field, Y. et al. Detection of human adaptation during the past 2000 years. Science 354, 760–764 (2016).
Aoki, K. Sexual selection as a cause of human skin colour variation: Darwin's hypothesis revisited. Ann. Hum. Biol. 29, 589–608 (2002).
Frost, P. European hair and eye color - a case of frequency-dependent sexual selection? Evol. Hum. Behav. 27, 85–103 (2006).
Madrigal, L. & Kelly, W. Human skin-color sexual dimorphism: a test of the sexual selection hypothesis. Am. J. Phys. Anthropol. 132, 470–482 (2007).
Lin, B. D. et al. Heritability and genome-wide association studies for hair color in a Dutch twin family based sample. Genes (Basel) 6, 559–576 (2015).
Sulem, P. et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 39, 1443–1452 (2007).
Han, J. et al. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 4, e1000074 (2008).
Sulem, P. et al. Two newly identified genetic determinants of pigmentation in Europeans. Nat. Genet. 40, 835–837 (2008).
Eriksson, N. et al. Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet. 6, e1000993 (2010).
Kenny, E. E. et al. Melanesian blond hair is caused by an amino acid change in TYRP1. Science 336, 554 (2012).
Zhang, M. et al. Genome-wide association studies identify several new loci associated with pigmentation traits and skin cancer risk in European Americans. Hum. Mol. Genet. 22, 2948–2959 (2013).
MacArthur, J. et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45(D1), D896–D901 (2017).
Walsh, S. et al. Developmental validation of the HIrisPlex system: DNA-based eye and hair colour prediction for forensic and anthropological usage. Forensic Sci. Int. Genet. 9, 150–161 (2014).
Devlin, B. & Roeder, K. Genomic control for association studies. Biometrics 55, 997–1004 (1999).
Yang, J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011).
Liu, F. et al. Genetics of skin color variation in Europeans: genome-wide association studies with functional follow-up. Hum. Genet. 134, 823–835 (2015).
Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6, e1000888 (2010).
Lee, S. H., Wray, N. R., Goddard, M. E. & Visscher, P. M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 88, 294–305 (2011).
Walsh, S. et al. The HIrisPlex system for simultaneous prediction of hair and eye colour fromDNA. Forensic Sci. Int. Genet. 7, 98–115 (2013).
Mengel-From, J., Wong, T. H., Morling, N., Rees, J. L. & Jackson, I. J. Genetic determinants of hair and eye colours in the Scottish and Danish populations. BMC Genet. 10, 88 (2009).
Shekar, S. N. et al. Spectrophotometric methods for quantifying pigmentation in human hair-influence of MC1R genotype and environment. Photochem. Photobiol. 84, 719–726 (2008).
Visser, M., Kayser, M. & Palstra, R. J. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 22, 446–455 (2012).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Henn, B. M. et al. Cryptic distant relatives are common in both isolated and cosmopolitan genetic samples. PLoS One 7, e34267 (2012).
Delaneau, O., Zagury, J. F. & Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. Minimac2: faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Zheng, X. et al. HIBAG-HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014).
Allen, N. et al. UK Biobank: current status and what it means for epidemiology. Health Policy Technol. 1, 123–126 (2012).
Keating, B. et al. First all-in-one diagnostic tool for DNA intelligence: genome-wide inference of biogeographic ancestry, appearance, relatedness, and sex with the Identitas v1 Forensic Chip. Int. J. Legal Med. 127, 559–572 (2013).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012). S1–S3.
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Pybus, M. et al. 1000 Genomes Selection Browser 1.0: a genome browser dedicated to signatures of natural selection in modern humans. Nucleic Acids Res. 42, D903–D909 (2014).
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007).
Consortium, G. T. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Segrè, A. V., Groop, L., Mootha, V. K., Daly, M. J. & Altshuler, D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 6, e1001058 (2010).
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 12052.
The ALSPAC work is supported by a Medical Research Council program grant (MC_UU_12013/4 to D.M.E). The UK Medical Research Council and the Wellcome Trust (grant refs: 092731 and 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. D.M.E. is supported by an Australian Research Council Future Fellowship (FT130101709). This publication is the work of the authors and D.M.E. will serve as guarantor for the contents of this paper. ALSPAC GWAS data was generated by Sample Logistics and Genotyping Facilities at the Wellcome Trust Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe.
The ERF Study was supported by the joint grant from the Netherlands Organization for Scientific Research (NWO, 91203014), the Center of Medical Systems Biology (CMSB), Hersenstichting Nederland, Internationale Stichting Alzheimer Onderzoek (ISAO), Alzheimer Association project number 04516, Hersenstichting Nederland project number 12F04(2).76, and the Interuniversity Attraction Poles (IUAP) program. As a part of EUROSPAN (European Special Populations Research Network), ERF was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the program “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043).
The INGI research was supported by funds from Compagnia di San Paolo, Torino, Italy; Fondazione Cariplo, Italy and Ministry of Health, Ricerca Finalizzata 2008 and CCM 2010 and Telethon, Italy. Additional support was provided by the Italian Ministry of Health (RF 2010 to PG), FVG Region, and Fondo Trieste.
The NTR study was supported by multiple grants from the Netherlands Organization for Scientific Research (NWO: 016-115-035, 463-06-001, 451-04-034), ZonMW (31160008, 911-09-032); from the Institute for Health and Care Research (EMGO + ); and from the Biomolecular Resources Research Infrastructure (BBMRI-NL, 184.021.007), European Research Council (ERC-230374). Genotyping was made possible by grants from NWO/SPI 56-464-14192, Genetic Association Information Network (GAIN) of the Foundation for the National Institutes of Health, Rutgers University Cell and DNA Repository (NIMH U24 MH068457-06), the Avera Institute, Sioux Falls (USA), and the National Institutes of Health (NIH R01 HD042157-01A1, MH081802, Grand Opportunity grants 1RC2 MH089951 and 1RC2 MH089995). B.D.L. is supported by a PhD grant (201206180099) from the China Scholarship Council.
QIMR funding was provided by the Australian National Health and Medical Research Council (241944, 339462, 389927, 389875, 389891, 389892, 389938, 442915, 442981, 496739, 552485, 552498), the Australian Research Council (A7960034, A79906588, A79801419, DP0770096, DP0212016, DP0343921), the FP-5 GenomEUtwin Project (QLG2-CT-2002-01254), and the US National Institutes of Health (NIH grants AA07535, AA10248, AA13320, AA13321, AA13326, AA14041, MH66206). Statistical analyses were carried out on the Genetic Cluster Computer, which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003). S.E.M. and D.L.D. are supported by the National Health and Medical Research Council (NHMRC) Fellowship Scheme.
The 20-year follow-up of Generation 2 of the Western Australian Pregnancy Cohort (Raine) Study was funded by Australian National Health and Medical Research Council (NHMRC) project grant 1021105, Lions Eye Institute, the Australian Foundation for the Prevention of Blindness, and the Ophthalmic Research Institute of Australia. S.Y. is supported by NHMRC Early Career Fellowship (CJ Martin - Overseas Biomedical Fellowship).
The Rotterdam Study is supported by the Netherlands Organization of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012). This study is funded by the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) project nr. 050-060-810. The Rotterdam Study is supported by the Erasmus MC and Erasmus University Rotterdam; the Netherlands Organization for Scientific Research (NWO); the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE) the Netherlands Genomics Initiative (NGI); the Ministry of Education, Culture and Science; the Ministry of Health Welfare and Sport; the European Commission (DG XII); and the Municipality of Rotterdam. The generation and management of GWAS genotype data for the Rotterdam Study were executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC. F.L. is supported by a Chinese recruiting program, the National Thousand Young Talents Award, and by the National Natural Science Foundation of China (NSFC) (91651507).
The TwinsUK study was funded by the Wellcome Trust (105022/Z/14/Z); European Community's Seventh Framework Programme (FP7/2007-2013). The study also receives support from the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust in partnership with King's College London. SNP genotyping was performed by the Wellcome Trust Sanger Institute and National Eye Institute via NIH/CIDR.
Author information
Authors and Affiliations
Consortia
Contributions
P.G.H., A. Valdes, and F.L. jointly wrote the manuscript, coordinated meta-analyses, and performed prediction modeling. N.A.F., D.M.E., V.B., A. Visconti, G.H., G.M., S.M.R., D.L.D., G.Z., S.D.G., S.E.M., B.D.L., G.W., J.J.H., D.V., G.G., I.G., C.S., M.P.C., M.B., D.T., M.C., A.R., S.Y., A.W.H., Y.C., C.Z., A.G.U., M.A.H., T.N., M.F., and D.A.H. each conducted part of the analyses described in this work. G.D.S., P.G., C.M.v.D., M.A.I., D.A.M., D.I.B., N.G.M., and M.F. contributed populations samples and data used for analyses. M.K. and T.D.S. jointly coordinated the work and participated in manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
N.A.F. and D.A.H. are employees of the 23andMe Inc., a consumer genetics company.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Distribution of self-reported hair color in the UK Biobank and 23andMe cohorts.
Plots show the densities for each hair color category in the UK Biobank (top) and 23andMe cohorts (bottom).
Supplementary Figure 2 Plot of the first five principal components in UK Biobank and 23andMe subjects included in the analyses.
The top-left panel shows the participants of European descent that were included in the analyses (blue) in the backdrop of all multi-ethnic UK Biobank and 23andMe participants. The plots for any two of the first principal components for the subjects of European descent (included in the analyses) are also given.
Supplementary Figure 3 Violin plots of the first principal components for the UK Biobank and 23andMe participants.
Each violin plot shows the distribution of principal components computed in the UK Biobank subjects (top) and the 23andMe cohort (bottom).
Supplementary Figure 4 Expected variation of genomic control inflation factor (λ) as a function of sample size.
The expected λ were calculated using the same number of loci, LD structure and trait heritability observed in the 23andMe cohort (blue) and the UK Biobank cohort (red) in absence of factors artificially inflating association. These genomic inflation factors observed in the discovery cohort are a reflection of the power of these large sample analyzed in presence of polygenicity; they would be merely the equivalent of a λGC = 1.0186 for the 23andMe cohort and λGC = 1.0221 for the UKBB cohort had the effective sample sizes been smaller (n = 20,000) and the underlying genetic effects remained the same.
Supplementary Figure 5 Power to detect genome-wide significant associations.
Plot generated using a sample of n = 140,000 subjects at α = 10-7. Power is shown for the most representative range of allele frequencies detected in GWAS (MAF = 0.10, red line; MAF = 0.30, blue line). The value on the y-axis denotes the probability that a true association would be detected at the pre-defined α; for example, for a locus with a MAF = 0.10 with an effect size of 0.035 s.d. per each copy of the risk allele over the trait, there is a 60% probability to replicate the same association at a genome-wide level of significance.
Supplementary Figure 6 Correlation of effect sizes between men and women participating in the UK Biobank and the 23andMe cohorts.
The black line denotes parity of effects, and each point represents one of the GWAS associated SNPs shown in Supplementary Table 2.
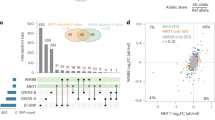
Supplementary Figure 7 Evidence of natural selection for the SNPs significantly associated with hair color.
Three different natural selection tests were selected (see Online Methods), and the values plotted here represent the centile rank of the natural selection score within European populations (iHS test, red), compared to YRI Africans (XP-EHH CEU vs. YRI, green) and to CHB Chinese populations (blue) using 1000 Genomes data. The plot shows an enrichment for higher ranks (lower centiles) of selection for some SNPs within European populations (two tailed Wilcoxon test P = 0.04), evidence for stronger selection for these SNPs in Europeans compared to Africans (two-tailed Wilcoxon P = 0.014), but less significant compared to Chinese (two tailed Wilcoxon P = 0.056).
Supplementary Figure 8 Hair color prediction in participants of the Rotterdam and QIMR studies.
The prediction model is based on multinomial logistic regression including 20 HIrisPlex SNPs, a polygenic score for blond-brown-black (233 SNPs), and a polygenic score for red (25 SNPs); see Supplementary Note for marker and model specifications.
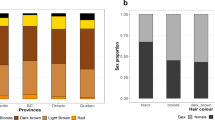
Supplementary Figure 9 Sex-specific prevalence of hair color categories in the UK Biobank and 23andMe cohorts.
In both cohorts, there is a higher prevalence of blond, red and light brown hair colors among women and a higher prevalence of black and dark brown hair colors among men.
Supplementary information
Supplementary Figures
Supplementary Figures 1–9
Supplementary Note and Supplementary Tables
Supplementary Note 1 and Supplementary Tables 1–10
Rights and permissions
About this article

Cite this article
Hysi, P.G., Valdes, A., Liu, F. et al. Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritability. Nat Genet 50, 652–656 (2018). https://doi.org/10.1038/s41588-018-0100-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0100-5
This article is cited by
-
Comprehensive in Silico Analyses of Single Nucleotide Variants of the Human Orthologues of 171 Murine Loci to Seek Novel Insights into the Genetics of Human Pigmentation
Proceedings of the Zoological Society (2022)
-
A large Canadian cohort provides insights into the genetic architecture of human hair colour
Communications Biology (2021)
-
GenNet framework: interpretable deep learning for predicting phenotypes from genetic data
Communications Biology (2021)
-
Searching for improvements in predicting human eye colour from DNA
International Journal of Legal Medicine (2021)
-
Mapping of cis-acting expression quantitative trait loci in human scalp hair follicles
BMC Dermatology (2020)
