
Abstract
Inference of cell–cell communication from single-cell RNA sequencing data is a powerful technique to uncover intercellular communication pathways, yet existing methods perform this analysis at the level of the cell type or cluster, discarding single-cell-level information. Here we present Scriabin, a flexible and scalable framework for comparative analysis of cell–cell communication at single-cell resolution that is performed without cell aggregation or downsampling. We use multiple published atlas-scale datasets, genetic perturbation screens and direct experimental validation to show that Scriabin accurately recovers expected cell–cell communication edges and identifies communication networks that can be obscured by agglomerative methods. Additionally, we use spatial transcriptomic data to show that Scriabin can uncover spatial features of interaction from dissociated data alone. Finally, we demonstrate applications to longitudinal datasets to follow communication pathways operating between timepoints. Our approach represents a broadly applicable strategy to reveal the full structure of niche–phenotype relationships in health and disease.
Similar content being viewed by others


Main
Complex multicellular organisms rely on coordination within and between their tissue niches to maintain homeostasis and appropriately respond to internal and external perturbations. This coordination is achieved through cell–cell communication (CCC), whereby cells send and receive biochemical and physical signals that influence cell phenotype and function1,2. A fundamental goal of systems biology is to understand the communication pathways that enable tissues to function in a coordinated and flexible manner to maintain health and fight disease3,4.
The advent of single-cell RNA sequencing (scRNA-seq) has made it possible to dissect complex multicellular niches by applying the comprehensive nature of genomics at the ‘atomic’ resolution of the single cell. Concurrently, the assembly of protein–protein interaction databases5 and the rise of pooled genetic perturbation screening6,7 have empowered the development of methods that infer putative axes of cell-to-cell communication from scRNA-seq datasets8,9,10,11,12,13. These techniques generally function by aggregating ligand and receptor expression values for groups of cells to infer which groups of cells are likely to interact with one another14,15,16,17. However, biologically, CCC does not operate at the level of the group; rather, such interactions take place between individual cells. There exists a need for methods of CCC inference that analyze interactions at the level of the single cell, that leverage the full information content contained within scRNA-seq data by looking at upstream and downstream cellular activity, that enable comparative analysis between conditions and that are robust to multiple experimental designs.
Here we introduce single-cell-resolved interaction analysis through binning (Scriabin)—an adaptable and computationally efficient method for CCC analysis. Scriabin dissects complex communicative pathways at single-cell resolution by combining curated ligand–receptor interaction databases13,18,19, models of downstream intracellular signaling20, anchor-based dataset integration21 and gene network analysis22 to recover biologically meaningful CCC edges at single-cell resolution.
Results
A flexible framework for CCC analysis at single-cell resolution
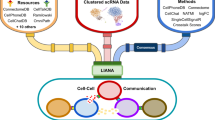
Our goal is to develop a scalable and statistically robust method for the comprehensive analysis of CCC from scRNA-seq data. Scriabin implements three separate workflows depending on dataset size and analytical goals (Fig. 1): (1) the cell–cell interaction matrix (CCIM) workflow, optimal for smaller datasets, analyzes communication for each cell–cell pair in the dataset; (2) the summarized interaction graph workflow, designed for large comparative analyses, identifies cell–cell pairs with different total communicative potential between samples; and (3) the interaction program discovery workflow, suitable for any dataset size, finds modules of co-expressed ligand–receptor pairs.

Scriabin consists of multiple analysis workflows depending on dataset size and the user’s analysis goals. a, At the center of these workflows is the calculation of the CCIM M, which represents all ligand–receptor expression scores for each pair of cells. b, CCIM workflow. In small datasets, M can be calculated directly, active CCC edges predicted using NicheNet20 and the weighted cell–cell interaction matrix used for downstream analysis tasks, such as dimensionality reduction. M is a matrix of N × N cells by P ligand–receptor pairs, where each unique cognate ligand–receptor combination constitutes a unique P. c, Summarized interaction graph workflow. In large comparative analyses, a summarized interaction graph S can be calculated in lieu of a full dataset M. After high-resolution dataset alignment through binning, the most highly variable bins in total communicative potential can be used to construct an intelligently subsetted M. d, Interaction program (IP) discovery workflow. IPs of co-expressed ligand–receptor pairs can be discovered through iterative approximation of the ligand–receptor pair TOM. Single cells can be scored for the expression of each IP, followed by differential expression and modularity analyses.
The fundamental unit of CCC is a sender cell Ni expressing ligands that are received by their cognate receptors expressed by a receiver cell Nj. Scriabin encodes this information in a CCIM M by calculating the geometric mean of expression of each ligand–receptor pair by each pair of cells in a dataset (Fig. 1a). Scriabin currently supports the use of 15 different protein–protein interaction databases for defining potential ligand–receptor interactions and by default uses the OmniPath database, as this database contains robust annotation of gene category, mechanism and literature support for each potential interaction18,19. As ligand–receptor interactions are directional, Scriabin considers each cell separately as a ‘sender’ (ligand expression) and as a ‘receiver’ (receptor expression), thereby preserving the directed nature of the CCC network. M can be treated analogously to a gene expression matrix and used for dimensionality reduction, clustering and differential analyses.
Next, Scriabin identifies biologically meaningful edges, which we define as ligand–receptor pairs that are predicted to affect observed gene expression profiles in the receiving cell (Fig. 1). This requires defining a gene signature for each cell that reflects its relative gene expression patterns and determining which ligands are most likely to drive that observed signature. First, variable genes are identified to immediately focus the analysis on features that distinguish samples of relevance or salient dynamics. When analyzing a single dataset, this set of genes could be the most highly variable genes (HVGs) in the dataset, which would likely reflect cell-type-specific or state-specific modes of gene expression. Alternatively, when analyzing multiple datasets, the genes that are most variable between conditions (or timepoints) could be used. To define the relationship between the selected variable genes and each cell, the single cells and chosen variable genes are placed into a shared low-dimensional space with multiple correspondence analysis (MCA), a weighted generalization of principal component analysis (PCA) that applies to count data, implemented by Cell-ID23. A cell’s gene signature is defined as the set of genes in closest proximity to the variable genes in the MCA embedding (Methods and Supplementary Text). An implementation of NicheNet20 is then used to nominate the ligands that are most likely to result in each cell’s observed gene signature. Ligand–receptor pairs that are recovered from this process are used to weight the CCIM M proportionally to their predicted activity, highlighting the most biologically important interactions (Fig. 1).
Because one dimension of M is N × N cells long, it is impractical to construct M for samples with high cell numbers; this problem will likely be exacerbated as scRNA-seq platforms continue to increase in throughput. Conceptually, solutions to this problem include subsampling and aggregation. Subsampling, however, is statistically inadmissible because it involves omission of available valid data and introduction of sampling noise24; meanwhile, aggregation at any level raises the possibility of obscuring important heterogeneity and/or specificity.
An alternate solution is to first intelligently identify cell–cell pairs of interest and build M using only those sender and receiver cells. We hypothesize that, in the context of a comparative analysis, sender–receiver cell pairs that change substantially in their magnitude of interaction are the most biologically informative. To identify these cells, Scriabin first constructs a summarized interaction graph S, characterized by an N × N matrix containing the sum of all cognate ligand–receptor pair expression scores for each pair of cells. S is much more computationally efficient to generate, store and analyze than a full dataset M (for a 1,000-cell dataset, S is 1,000 × 1,000, whereas M is ~3,000 × 1,000,000). Comparing summarized interaction graphs from multiple samples requires that cells from different samples share a set of labels or annotations of cells representing the same identity. We use recent progress in dataset integration methodology21,25 to develop a high-resolution registration and alignment process that we call ‘binning’, where we assign each cell a bin identity that maximizes the similarity of cells within each bin and maximizes the representation of all samples that we want to compare within each bin while simultaneously minimizing the degree of agglomeration required (Fig. 1 and Supplementary Text). Sender and receiver cells belonging to the bins with the highest communicative variance can then be used to construct M.
Finally, Scriabin implements a workflow for single-cell-resolved CCC analysis that is scalable to any dataset size, enabling discovery of co-expressed ligand–receptor interaction programs. This workflow is motivated by the observation that transcriptionally similar sender–receiver cell pairs will tend to communicate through similar sets of ligand–receptor pairs. To achieve this, we adapted the well-established weighted gene correlation network analysis (WGCNA) pipeline22—designed to find modules of co-expressed genes—to uncover modules of ligand–receptor pairs co-expressed by the same sets of sender–receiver cell pairs, which we call ‘interaction programs’. Scriabin calculates sequences of M subsets that are used to iteratively approximate a topological overlap matrix (TOM), which is then used to discover highly connected interaction programs. Because the dimensionality of the approximated TOM is consistent between datasets, this approach is highly scalable. The connectivity of individual interaction programs is then tested for statistical significance, which can reveal differences in co-expression patterns between samples. Single cells are then scored for the expression of statistically significant interaction programs. Comparative analyses include differential expression analyses on identified interaction programs as well as comparisons of intramodular connectivity between samples.
To illustrate the importance of performing CCC analyses at single-cell resolution, we examined CCC of T cells in the tumor microenvironment. Owing to their low RNA content, it is often difficult to infer the functional states of T cells from their transcriptomes26, yet T cells participate in communicative pathways that are important to clinical and therapeutic outcomes27. Additionally, transcriptional evidence suggests that helper T cells may exist on a phenotypic continuum rather than in traditional discrete functional archetypes28. In a dataset of squamous cell carcinoma (SCC) and matched controls29, we found a high degree of whole-transcriptome phenotypic overlap between intratumoral T cells and those present in normal skin (Fig. 2a). Furthermore, although there were exhausted T cells in this dataset, they did not occupy a discrete cluster but were, rather, distributed across multiple clusters (Fig. 2a and Extended Data Fig. 1), precluding cluster-based CCC approaches from detecting communication modalities unique to exhausted T cells without a priori knowledge. We tested Scriabin’s utility in exposing the heterogeneity of the T cell communicative phenotype by applying the CCIM workflow to pairs of T cells and CD1C+ dendritic cells (DCs), the most abundant antigen-presenting cell (APC) in this dataset. This revealed both a clear distinction between communication profiles between tumor and matched normal as well as distinct populations of cell–cell pairs with exhausted T cells (Fig. 2b). Compared to their non-exhausted counterparts, exhausted T cells communicated with CD1C+ DCs predominantly with exhaustion-associated markers CTLA4 and TIGIT and lost communication pathways involving pro-inflammatory chemokines, such as CCL4 and CCL5 (Fig. 2c)30. This illustrates the communicative heterogeneity that can be missed by agglomerative techniques.

a, UMAP projections of 1,624 intratumoral T cells from the SCC dataset from Ji et al.29, colored by cluster identity (top left), sample of origin (tumor or matched normal; bottom left) and T cell exhaustion score (middle) (Methods). The dot plot at right depicts the percent and average expression of the T cell exhaustion score in each cluster. b, UMAP projections of 202,708 T cell–CD1C+ DC cell–cell pairs from Scriabin’s CCIM workflow. Points are colored by sample of origin (left) and the T cell exhaustion score of the T cell in the cell–cell pair (right). c, Bar plot depicting differentially expressed ligand–receptor pairs among T cell–CD1C+ DC cell–cell pairs between exhausted and non-exhausted T cell senders. Individual bars are colored by the power from Seurat’s implementation of a ROC-based differential expression (DE) test. d, Schematic illustrating the workflow to evaluate the impact of technical noise on the robustness of cell–cell communication analyses with Scriabin. e, Left: box plot depicting the ability of downsampled CCIMs to recapitulate the GT CCIM. The y axis depicts the proportion of GT cell–cell pairs that are recapitulated by a query cell–cell pair (LISI score >1), and points are colored by the mean LISI score for GT cell–cell pairs. Each experimental condition was repeated on 12 different random subsamples of 300 cells from three independent datasets. Right: bar plot depicting the degree of downsampling required for each dataset to reach inDrop coverage.
Scriabin is robust and efficient for single-cell CCC analysis
One potential concern of performing single-cell-resolution CCC analysis is that scRNA-seq measurements are inherently sparse and noisy. Aggregative techniques, although frequently obscuring biological heterogeneity, do carry the advantage of using less sparse and, therefore, more robust expression values. Additionally, using single-cell resolution versus aggregated pseudobulk measurements for CCC analysis is not a binary option but, rather, the ends of an entire spectrum of resolution. Probabilistic denoising techniques for scRNA-seq data31,32 use information from transcriptionally similar cells to smooth noise created by putative technical zeroes and represent a mild form of aggregation by smoothing measured expression values. Furthermore, cluster-based agglomerative CCC techniques can operate at a wide range of potential clustering resolutions. We sought to quantitatively examine the impact of technical noise on single-cell-resolution CCC analysis and identify if there is an optimal degree of aggregation that avoids issues with data sparsity without agglomerating over distinct communication phenotypes.
To do this, we simulated technical noise by randomly downsampling a deeply sequenced scRNA-seq dataset (Fig. 2d). We used as ground truth (GT) three datasets generated by the Fluidigm C1 or Smart-Seq2 platforms8,13,21,33, which profile cells approximately one to two orders of magnitude more deeply than droplet-based methods. We then randomly downsampled these datasets to the sequencing depth of inDrop, between two-fold and 270-fold depending on the sequencing depth of the original dataset (Fig. 2e, right)34,35. We performed Scriabin’s CCIM workflow directly on the downsampled datasets, on the downsampled datasets denoised by adaptively thresholded low-rank approximation (ALRA)32, on datasets created by aggregating cells over similarity neighborhoods of nine different sizes or on pseudobulk expression values from clustering at four different resolutions. Next, we integrated the CCIM generated from the GT datasets with the CCIMs generated from the randomly downsampled datasets. To quantify the degree to which the CCIMs from the downsampled datasets recapitulated the GT CCIMs, we calculated the local inverse Simpson’s index (LISI; Fig. 2d)36. This value defines the number of datasets in the neighborhood of each GT cell–cell pair and ranges between 1, denoting that only GT cell–cell pairs are present in the neighborhood, and 2, denoting an equal mixture of GT and downsampled cell–cell pairs.
We found that CCIMs generated either from raw downsampled data or from ALRA-denoised data best recapitulated GT data (Fig. 2e). Downsampling introduced technical noise only for the most highly downsampled dataset, but this technical noise was almost completely rescued via data denoising. When defining each cell’s transcriptome as the mean transcriptome of that cell and its k-nearest neighbors, increasing k worsened the recapitulation of the GT dataset. ALRA-denoised data outperformed all nine k tested. Furthermore, at all cluster resolutions tested, at least 50% of GT CCC states are not captured by using pseudobulk expression values. These data indicate that agglomeration at nearly any level results in loss of unique CCC states. Additionally, in datasets from platforms with a high degree of sparsity, denoising methods may represent an optimal degree of data smoothing that decreases the impacts of technical noise while preserving data structure and heterogeneity.
We next explored Scriabin’s performance in comparison to other published CCC methods. Scriabin was faster than five agglomerative CCC methods15,16,17,37,38 in analyzing a single dataset at all the dataset sizes tested (Extended Data Fig. 2a). Of these five agglomerative CCC methods, only Connectome38 supports a full comparative workflow and was slower than Scriabin in a comparative CCC analysis of two datasets (Extended Data Fig. 2b). We also compared the top CCC edges predicted by these methods39 to a pseudobulk version of Scriabin. Applying these methods to four scRNA-seq datasets, we found that the top results returned by Scriabin overlapped with three of the five published methods analyzed (Connectome, CellChat and NATMI; Extended Data Fig. 2c). The remaining two methods (iTALK and SCA) did not have overlapping results with each other or any of the other tested methods for any of the datasets tested (Extended Data Fig. 2c).
Although the pseudobulk version of Scriabin’s results agreed with several published methods, we also sought to demonstrate more directly that these results were biologically correct. We hypothesized that spatial transcriptomic datasets could be leveraged for this purpose, as cells that Scriabin predicts to be highly interacting should be, on average, in closer proximity. We ran Scriabin on 11 spatial transcriptomic datasets, removing secreted ligand–receptor interactions that could operate over a distance from the ligand–receptor database (Fig. 3a). Cells that Scriabin predicted were the most highly interacting were in significantly closer proximity relative to randomly permuted distances (Fig. 3b and Extended Data Fig. 2d,e), indicating that Scriabin can detect spatial features from dissociated data alone.

a, Left: description of workflow to validate Scriabin using spatial transcriptomic datasets; right: density plots showing the distribution of cell–cell distances within the top 1% of highly interacting cell–cell pairs predicted by Scriabin. The vertical black lines denote the median distance of all cell–cell pairs. b, The procedure depicted in a was repeated for 11 biologically independent datasets, and the median distance quantile of the top 1% interacting cell–cell pairs was calculated using real cell distances relative to randomly permuted cell distances. Shown is an exact two-sided P value from the Wilcoxon rank-sum test. c, ROC plots depicting Scriabin’s ability to correctly predict the gRNA with which a single cell was transduced based on its communicative profile. Each of the n = 15 lines represents a different gene target by gRNAs in a CRISPRa dataset of stimulated T cells40. d, Experimental scheme to validate Scriabin through transfection of exogenous CCC edges. In total, 21,538 cells from NK cell–B cell co-cultures were profiled by scRNA-seq. Specific sample sizes for the four transfection conditions are as follows: 4,934 (GFP–GFP), 5,665 (GFP–CD40), 4,908 (CD40L–GFP) and 6,031 (CD40L–CD40). e, CCIMs were generated by Scriabin for each co-culture condition with or without ligand activity ranking. The bar plot depicts the top differentially expressed ligand–receptor pairs between cell–cell pairs from control (GFP/GFP) versus transfected (CD40L/CD40) samples. f, Box plot depicting CD40LG–CD40 cell–cell pair interaction scores in each co-culture condition. The CD40LG–CD40 interaction score is derived from CCIMs generated with ligand activity ranking. The interaction scores are calculated from the sample sizes for each condition noted in Fig. 3d. g, Scatter plot depicting the relationship between the CD40LG–CD40 interaction score and the CCC perturbation Dunn z-test statistic for each of 311 bin–bin pairs (Methods). Pearson correlation coefficient, exact two-sided P value and a 95% confidence interval are shown. h, Bar plot depicting the Pearson correlation coefficient between bin perturbation and CD40LG–CD40 interaction score using a full-transcriptome SNN graph for binning compared to a reference-based weighted SNN (WSNN) that does not contain structure related to transfection.
We next hypothesized that we could leverage a single-cell-resolution pooled genetic perturbation screen to validate Scriabin’s ability to identify biologically relevant shifts in cellular communication phenotypes. In an analysis of a CRISPRa Perturb-seq screen of activated human T cells that included guide RNAs (gRNAs) targeting 15 different cell surface ligands or receptors40, we found that Scriabin could accurately predict the gRNA with which a cell was transduced by analyzing cellular CCC profiles (average area under the curve (AUC): 0.93; Fig. 3c).
To provide direct experimental evidence of Scriabin’s ability to detect changes in CCC, we devised an experiment where we transfected isolated natural killer (NK) cells with mRNA encoding CD40L and isolated B cells with mRNA encoding its cognate receptor CD40 (Supplementary Text and Fig. 3d). After co-culture of the transfected cells, we performed scRNA-seq to assess how the forced expression of exogenous CD40 or CD40L impacted CCC. As NK cells do not normally express CD40L, but B cells can express low levels of CD40 at baseline (Extended Data Fig. 3), we hypothesized that we would observe enhanced communication along the CD40L–CD40 edge only when CD40LG was transfected and that this would be enhanced when both CD40LG and CD40 were transfected. Using Scriabin’s CCIM workflow, we found that the CD40LG–CD40 communication edge was the only ligand–receptor pair that was substantially changed in the transfected conditions (Fig. 3e). This difference was enhanced by incorporating ligand activity weighting into construction of the CCIM (Fig. 3e). In line with our predictions, we also found that communication along the CD40LG–CD40 axis was strongest when NK cells were transfected with CD40LG and further increased by transfecting B cells with CD40 (Fig. 3f).
Although the aggregative method Connectome38 returned CD40LG–CD40 as a differential communication edge, it also returned 25 other ligand–receptor pairs as statistically significant (Extended Data Fig. 4). These additional unexpected differential results appeared to be driven by small shifts in expression of very lowly expressed ligands and receptors (Extended Data Fig. 4). We also used NicheNet alone to identify differentially active ligands between the transfected and untransfected conditions. Although CD40L was returned among the top 20 predicted active ligands, NicheNet predicted that FASLG and PTPRC were more differentially active despite there being little appreciable difference in the expression of these ligands (Extended Data Fig. 4). This underlines the utility of using information on both relative ligand and receptor expression as well as downstream gene expression changes in performing comparative CCC analyses.
Finally, we used Scriabin’s summarized interaction graph workflow to bin cells from the four transfection conditions and found a significant correlation between the bin perturbation score and the degree to which the cells in each bin were transfected (Fig. 3g), demonstrating the utility of this workflow in identifying single cells that have the highest degree of communicative perturbation. This correlation was completely abrogated when binning was performed on data structures not related to transfection, such as proximity in a reference neighbor graph (Fig. 3h). These data provide empirical evidence that Scriabin accurately identifies meaningful changes in CCC.
Scriabin reveals known CCC concealed by aggregative methods
We further evaluated if Scriabin’s single-cell-resolution CCC results returned communicating edges that are obscured by agglomerative CCC methods. To this end, we analyzed a publicly available dataset of a well-characterized tissue niche: the granulomatous response to Mycobacterium leprae infection (Fig. 4a). Granulomas are histologically characterized by infected macrophages and other myeloid cells surrounded by a ring of Th1 T cells41,42,43. These T cells produce interferon (IFN)-γ that is sensed by myeloid cells; this communication edge between T cells and myeloid cells is widely regarded as the most important interaction in controlling mycobacterial spread44,45,46. Ma et al.41 performed scRNA-seq on skin granulomas from patients infected with Mycobacterium leprae, the causative agent of leprosy. This dataset includes granulomas from five patients with disseminated lepromatous leprosy (LL) and four patients undergoing a reversal reaction (RR) to tuberculoid leprosy, which is characterized by more limited disease and a lower pathogen burden (Fig. 4a). Analysis of CCC with Scriabin revealed IFNG as the most important ligand sensed by myeloid cells in all analyzed granulomas, matching biological expectations (Fig. 4b). Baseline NicheNet also returned IFNG as the most differentially active ligand in RR granulomas, although with a lesser degree of specificity than Scriabin (Extended Data Fig. 5).

a, Schematic of the scRNA-seq dataset of leprosy granulomas published by Ma et al.41. Sample sizes for each profiled granuloma are shown in Supplementary Table 1. b, Ligands prioritized by Scriabin’s implementation of NicheNet as predicting target gene signatures in granuloma myeloid cells. Points are colored and sized by the number of granulomas in which the ligand is predicted to result in the downstream gene signature. c, Circos plot summarizing RR versus LL differential CCC edges between T cells (senders) and myeloid cells (receivers) generated by Connectome. Blue: edges upregulated in RR; red: edges upregulated in LL. The two black arrows mark T-cell-expressed ligands IL1B and CCL21, which are further analyzed in d and e. d, Percentage and average of expression of IL1B by T cells per granuloma (left) and total number of T cells per granuloma (right). e, Percentage and average expression of CCL21 by T cells per granuloma (left); percentage and average expression of CCR7- and CCL21-stimulated genes by myeloid cells per granuloma. f, RR versus LL differential interaction heat map between T cell bins (senders; rows) and myeloid cell bins (receivers; columns) generated by Scriabin, colored by the t-statistic between the mean summarized interaction scores of n = 4 RR granulomas relative to n = 5 LL granulomas. In blue are the bins more highly interacting in RR; in red are the bins more highly interacting in LL. The black box indicates groups of bins predicted to be highly interacting in RR granulomas relative to LL. g, UMAP projection of 74,437 perturbed T cell–myeloid cell sender–receiver pairs indicating changes in ligand–receptor pairs used for T cell–myeloid communication in LL versus RR granulomas. h, Scatter plot depicting differential gene expression by T cells. The average log(fold change) of expression by cluster 2 bins is plotted on the x axis; the average log(fold change) of expression by RR granulomas is plotted on the y axis. i, Target genes predicted to be upregulated by IFNG in RR granuloma myeloid cells in cluster 2 bins. Points are sized and colored by the number of cells in which the target gene is predicted to be IFNG responsive. j, Alluvial plot depicting the RR granuloma cell types that are predicted to receive the IFNG-responsive target genes from cluster 2 myeloid cells. DEG, differentially expressed gene.
To assess if Scriabin was capable of avoiding pitfalls associated with agglomerative methods in comparative CCC analyses, we analyzed differential CCC pathways from T cells to myeloid cells between LL and RR granulomas using an agglomerative method (Connectome, which implements a full comparative workflow38) and Scriabin. We first assessed if it would be possible to analyze higher levels of granularity by using author-provided subclustering annotations. However, as Connectome performs differential CCC analyses by aggregating data at the level of cell type or cluster, this requires that each subcluster have representatives from the conditions being compared. In the Ma et al.41 dataset, satisfying this condition meant decreasing clustering resolution from 1 to 0.1 so that all subclusters are present in all profiled granulomas and comparing all aggregated LL granulomas to all aggregated RR granulomas (Extended Data Fig. 5). This requirement moves analysis further from single-cell resolution, and we, therefore, elected to use author-annotated T cells and myeloid cells for analysis without subclustering.
Comparative CCC analysis with Connectome revealed IL1B and CCL21 as the two most upregulated T-cell-expressed ligands received by myeloid cells in RR granulomas (Fig. 4c). However, there was no clear evidence of IL1B upregulation among RR granulomas (Fig. 4d); rather, the RR granuloma that contributed the most T cells expressed the highest level of IL1B, and the LL granuloma that contributed the most T cells expressed the lowest level of IL1B (Fig. 4d). Additionally, CCL21 was expressed by T cells of a single RR granuloma, and the myeloid cells of a different RR granuloma expressed the highest levels of the CCL21 receptor CCR7 and three CCL21 target genes (Fig. 4e). This indicates that the most highly scored differential CCC edges may be due to agglomeration of RR and LL granulomas required by Connectome (Extended Data Fig. 5) rather than conserved biological changes between these two groups.
To compare differential CCC between LL and RR granulomas with Scriabin, we aligned data from the nine granulomas together using Scriabin’s binning procedure (Fig. 1); generated single-cell summarized interaction graphs for each granuloma; and calculated a t-statistic to quantify the difference in interaction for each pair of bins between LL and RR granulomas (Fig. 4f). This analysis revealed a group of T cell and myeloid bins whose interaction was strongly increased in RR granulomas relative to LL (Fig. 4f, black box). We visualized the cells in these perturbed bins by generating cell–cell interaction matrices for these cells in each sample and embedding them in shared low-dimensional space (Fig. 4g). The T cells in these bins were defined by expression of CRTAM, a marker of cytotoxic CD4 T cells, and upregulated IFNG in the RR granulomas (Fig. 4h). These perturbed T cells were enriched in ‘RR CTL’ and ‘amCTL’ subclusters described by Ma et al.41 that correspond to IFNG-expressing cytotoxic T cells (Extended Data Fig. 5). Perturbed myeloid cells were enriched in transitional macrophage and type I IFNhigh macrophage subclusters (Extended Data Fig. 5). Myeloid cells in these bins upregulated several pro-inflammatory cytokines in RR granulomas, including IL1B, CCL3 and TNF, in response to IFNG from this T cell subset (Fig. 4i). IFNG-responsive IL1B and TNF were also predicted to be RR-specific ligands received by myeloid cells, fibroblasts and endothelial cells in RR granulomas (Fig. 4j). Collectively, Scriabin identified a subset of CRTAM+ T cells that upregulated IFNG in RR granulomas that is predicted to act on myeloid cells to upregulate additional pro-inflammatory cytokines. These CCC results match previous results demonstrating that enhanced production of IFNG can drive RRs47,48 and implicate cytotoxic CD4 T cells as initiators of this reaction.
Scalable discovery of co-expressed interaction programs
We next assessed Scriabin’s interaction program discovery workflow. To illustrate the scalability of this process, we chose to analyze a large single-cell atlas of developing fetal gut49 composed of 76,592 cells sampled from four anatomical locations (Fig. 5a). Scriabin discovered a total of 75 significantly correlated interaction programs across all anatomical locations. Scoring all single cells on the expression of the ligands and receptors in these interaction programs revealed strong cell-type-specific expression patterns for many programs (Fig. 5b) as well as subtle within-cell-type differences in sender or receiver potential, highlighting the importance of maintaining single-cell resolution (Extended Data Fig. 6 and Supplementary Text).

a, UMAP projections of the dataset of Fawkner-Corbett et al.49 with 76,592 individual cells colored by author-provided cell type annotations (left) or by anatomical sampling location (right). b, Heat map depicting average expression of interaction program (IP) ligands (left) or IP receptors (right) by each cell type. c, UMAP projections of 25,969 intestinal epithelial cells, colored by expression of stem cell markers LGR5 and SOX9 as well as by the receptor expression score for IP1. d, UMAP projection of all cells colored by ligand (shades of blue) or receptor (shades of red) expression of IP1. e, Intramodular connectivity scores for each ligand–receptor pair in each anatomical location for IP1. The black arrows mark ligand–receptor pairs that include RSPO3. f, Heat map of two-dimensional bin counts depicting the correlation between IP1 sender score and the sender score for the IP module that contains the ligand GREM1. Shown are Pearson r and a two-sided P value. g, UMAP projection of all cells colored by ligand (shades of blue) or receptor (shades of red) expression of the GREM1 IP. h, UMAP projections of 4,447 gut endothelial cells colored by expression of LEC markers LYVE1 (top) and PROX1 (bottom). i, Bar plot depicting predicted active ligands for intestinal epithelial cells and correlation of predicted ligand activity with expression of ISC markers LGR5 and SOX9. Bars are colored by the average log(fold change) in expression of each ligand by LECs relative to other gut endothelial cells. j, Alluvial plot depicting target genes predicted to be upregulated in ISCs in response to CCL21 and NTS.
We next examined ways in which our identified interaction programs reflected known biological networks of intestinal development. Recently, several important interactions were shown to be critical in maintaining the intestinal stem cell (ISC) niche50,51,52. We were able to identify ISCs, defined by expression of LGR5 and SOX9, within the intestinal epithelial cells of this dataset, and we discovered a single interaction program (hereafter referred to as IP1) whose receptors were co-expressed with these ISC markers (Fig. 5c). IP1 represents a program of fibroblast-specific ligand and intestinal epithelial cell receptor expression (Fig. 5d). Among IP1 ligands were the ephrins EPHB3, whose expression gradient is known to control ISC differentiation53, and RSPO3 (Fig. 5e). Two recent studies each reported that RSPO3 production by lymphatic endothelial cells (LECs) and GREM1+ fibroblasts is critical for maintaining the ISC niche in mice51,52. In this human dataset, we did not observe expression of RSPO3 in LECs (Extended Data Fig. 6), and, although Fawkner-Corbett et al.49 identified RSPO3 as a potential communication ligand for ISCs, they did not examine the precise source of this ligand. In our application of Scriabin’s interaction program workflow, we found that GREM1+ fibroblasts expressed RSPO3 as a part of IP1 that was predicted to be sensed primarily by ISCs, thus demonstrating that this interaction pathway may communicate between different cell types in mouse than in human (Fig. 5d–f). We also found a separate interaction program containing the ligand GREM1; the ligands of this interaction program were co-expressed with IP1 ligands (Fig. 5f) and predicted to communicate to a different receiver cell type, namely gut endothelial cells (Fig. 5g).
Despite the absence of RSPO3 expression in LECs, it remains possible that LECs maintain the ISC niche in human intestinal development, particularly as these cells can reside in close spatial proximity to ISCs51,52. Although Fawkner-Corbett et al.49 included several CCC analyses on endothelial cells, these analyses were performed on aggregated endothelial cells and not specifically on LECs. We were able to identify a small population of LECs (Fig. 5h) and used Scriabin’s single-cell-resolution ligand activity ranking workflow to examine communication between LECs and ISCs. We found that two LEC-specific markers, CCL21 and NTS, were predicted to be active ligands for ISCs (Fig. 5i). CCL21 and NTS were both predicted to result in upregulation of target genes that notably included MYC and ID1 (Fig. 5j), which are known to participate in intestinal crypt formation and ISC maintenance54,55. None of these ligand–receptor CCC edges was returned by an agglomerative CCC analysis by Connectome (Extended Data Fig. 6). Our results suggest that, unlike in mice, in humans, LECs may contribute to ISC maintenance through production of CCL21 and NTS. Taken together, our results demonstrate the utility of interaction programs both in identifying known CCC edges and in providing new biological insights.
Assembly of longitudinal communicative circuits
A frequent analytical question in longitudinal analyses concerns how events at one timepoint influence cellular phenotype in the following timepoint56,57. We hypothesized, in datasets with close spacing between timepoints, that Scriabin’s high-resolution bin identities would allow us to assemble ‘longitudinal communicative circuits’—chains of sender–receiver pairs across consecutive timepoints. A communicative circuit consists of at least four cells across at least two timepoints: sender cell at timepoint 1 (S1), receiver cell at timepoint 1 (R1), sender cell at timepoint 2 (S2) and receiver cell at timepoint 2 (R2). If the interaction between S1 and R1 is predicted to result in the upregulation of ligand LA by R1, S1–R1–S2–R2 participates in a longitudinal circuit if R1 and S2 share the same bin (that is, S2 represents the counterpart of R1 at timepoint 2) and if LA is predicted to be an active ligand in the S2–R2 interaction (Fig. 6a). This process enables the stitching together of multiple sequential timepoints to identify communicative edges that are downstream in time and mechanism.

a, Schematic representing a longitudinal communicative circuit. Four cells participate in a longitudinal circuit if an interaction between S1 and R1 is predicted to result in the upregulation of ligand LA by R1, if R1 and S2 share a bin and if expression of LA by S2 participates in an active communication edge with R2. b, Alluvial plot depicting longitudinal communicative circuits. Stratum width corresponds to the number of cells in each cell grouping participating in the circuit corrected for the total number of cells in that group. Red strata are infected with SARS-CoV-2; blue strata are composed of uninfected cells. c, Dot plot depicting percent and scaled average expression of IL1B by club, basal and ciliated cells at 1 dpi. d, Volcano plot depicting log(fold change) (x axis) and −log(P value) (y axis) of IL1B+ basal cells relative to IL1B− basal cells at 1 dpi. Positive log(fold change) indicates that the gene is more highly expressed by IL1B+ basal cells. P values are calculated from Seurat’s implementation of the Wilcoxon rank-sum test (two-sided, adjusted for multiple comparisons using Bonferroni correction). e, Each point represents a ligand predicted to be active and participating in circuits at 1 dpi. The size of each dot represents the number of unique circuits in which that ligand participates. The y axis represents the log(fold change) of expression of each ligand between 1 dpi (positive y axis) and mock (negative y axis). The color represents the log(fold change) of expression of each ligand at 1 dpi between infected (red) and uninfected (blue) cells. f, Target genes predicted by Scriabin’s implementation of NicheNet20 to be upregulated in the receiver cells at the ends of the longitudinal communicative circuits at 3 dpi. Points are colored by the active ligand and sized by the number of cells in which the target is predicted to be upregulated by the active ligand. DEG, differentially expressed gene; NS, not significant.
To illustrate this process, we analyzed a published dataset of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in human bronchial epithelial cells (HBECs) in air–liquid interface (ALI) that was sampled daily for 3 d58. This dataset contains all canonical epithelial cell types of the human airway and indicates that ciliated and club cells are the preferentially infected cell types in this model system, with some cells having more than 50% of unique molecular identifiers (UMIs) from SARS-CoV-2 (Extended Data Fig. 7). We first defined a per-cell gene signature of genes variable across time and used this gene signature to predict active ligands expected to result in the observed cellular gene signatures20,23. Next, we used Scriabin’s high-resolution binning workflow to align the datasets from the three post-infection timepoints, which we then used to assemble longitudinal communicative circuits.
Scriabin identified circuits at the level of individual cells that spanned all three post-infection timepoints. We summarized these circuits by author-annotated cell type and whether SARS-CoV-2 reads were detected in the cell (Fig. 6b). Interestingly, we found that uninfected cells were more frequently the initiators of longitudinal circuits operating over all three timepoints (Fig. 6b). The most frequent circuit-initiating ligand was IL1B produced by basal, ciliated and club cells; in these cell types at 1 day post-infection (dpi), IL1B was more strongly expressed in bystander cells relative to infected cells (Fig. 6c). Uninfected basal cells at 1 dpi displayed the highest expression of IL1B (Fig. 6c), and these IL1B+ cells were also characterized by higher expression of other pro-inflammatory cytokines, including CCL20 and CXCL8 (Fig. 6d). Among the other ligands active at 1 dpi, acute phase reactant-encoding genes, including SAA1 and CTGF59,60, were strongly upregulated at 1 dpi relative to the mock condition and were both more highly expressed by uninfected cells (Fig. 6e); these genes are known to be induced in the setting of SARS-CoV-2 infection and are hypothesized to be involved in downstream tissue remodeling processes61. Thus, the unique ability of Scribain to elucidate longitudinal signaling circuits between cells implicates the activity of uninfected bystander cells as potentially important mediators of downstream responses to infection. This may reflect described processes in other viral infections where non-productively infected cells may be key drivers of downstream inflammatory activity62,63,64.
When we assessed the predicted downstream targets at the ends of the longitudinal circuits in both infected and bystander cells, we found that TGFB1 produced by infected basal cells was predicted to result in the upregulation of TNFSF10 (encoding TRAIL) and the alarmin S100A8 predominantly by other infected cells (Fig. 6b,f). Additionally, TGFB1 was predicted to upregulate both NOTCH1 and the NOTCH1 ligand JAG1, which indicates that these circuits may induce downstream Notch signaling. In sum, these data illustrate how the single-cell resolution of Scriabin’s CCC analysis workflow can perform integrated longitudinal analyses, nominating hypotheses for experimental validation.
Discussion
Most existing CCC methodologies aggregate ligand and receptor expression values at the level of the cell type or cluster, potentially obscuring biologically valuable information. Here we introduce a framework to perform comparative analyses of CCC at the level of the individual cell. Scriabin maximally leverages the single-cell resolution of the data to maintain the full structure of both communicative heterogeneity and specificity. We used this framework to find rare communication pathways in the developing intestine known to be required for stem cell maintenance as well as to define the kinetics of early dynamic communication events in response to SARS-CoV-2 infection through assembly of longitudinal communicative circuits.
A major challenge of single-cell-resolved CCC analysis is data inflation: because CCC analysis fundamentally involves performing pairwise calculations on cells or cell groups, it is frequently computationally prohibitive to analyze every sender–receiver cell pair. Some existing tools, such as NICHES65, support single-cell resolution CCC analysis but involve subsampling strategies when applied at scale. Scriabin implements two complementary workflows to address the issue of data inflation while avoiding subsampling and aggregation. Subsampling and aggregation preclude a truly comprehensive view of CCC structure and risk obscuring important biology; either can be particularly problematic in situations where a small subset of cells disproportionately drives intercellular communication, with agglomeration potentially concealing the full activity of those cells and subsampling potentially removing those cells altogether. One biological situation in which the preservation of single-cell-resolution data could be particularly important is in the setting of activation-induced T cell exhaustion66. Although exhausted T cells exert divergent effects on their communication targets relative to their activated counterparts, we show that exhausted T cells can often be difficult to distinguish from activated cells by clustering or subclustering. By avoiding aggregation and subsampling, Scriabin increases the likelihood of detecting these potentially meaningful differences in CCC pathways.
We observe that aggregation obscures potentially biologically meaningful subsets of T cells in SCC as well as in RRs in leprosy granulomas. Owing to the degree of transcriptional perturbation in T cells during RRs, subclustering is not always a tenable approach to increasing the resolution of CCC analyses because it, in turn, can preclude analysis at a per-sample level. We also show that aggregating across samples, which is a common practice in existing CCC tools, can return putatively differential CCC edges that are driven disproportionately by individual samples, potentially leading to inaccurate conclusions that are not generalizable.
As the throughput of scRNA-seq workflows continues to increase, it will be important that single-cell-resolution CCC methods are scalable to any dataset size. We introduce two complementary workflows to address this challenge. First, for large comparative analyses, the summarized interaction graph workflow saves computational resources by summarizing the total magnitude of communication between cell–cell pairs, and a dataset alignment strategy called ‘binning’ enables identification of cells of the greatest biological interest between samples. We provide empirical evidence that this strategy identifies subspaces with the greatest degree of communication perturbation. However, this approach is not robust to situations where ligand–receptor pair mechanisms of CCC change between cell–cell pairs without changing the overall magnitude of CCC.
As an alternative, we also introduce a second single-cell-resolution CCC workflow that is scalable to datasets of any size. The interaction program discovery workflow of Scriabin accomplishes this by focusing first on common patterns of ligand–receptor pair co-expression rather than individual cell–cell pairs. Individual cells can be scored for expression of these interaction programs post hoc, enabling downstream comparative analyses with a comprehensive view of CCC structure. We apply this workflow to an atlas-scale dataset of human fetal gut development, where we validate a mode of communication between a fibroblast subset and ISCs that has recently been shown to be important for maintaining the ISC niche51,52. Owing to the relative scarcity of these cells, we show that agglomerative methods fail to discover these important interactions for downstream mechanistic validation.
Longitudinal datasets pose an additional opportunity and challenge for comparative analyses because there is a priori knowledge about the sequential relationship between different samples. The single-cell nature of Scriabin’s workflows permits us to analyze how pathways of CCC operate both within and between timepoints in longitudinal datasets. By identifying circuits of CCC that function over multiple timepoints in a longitudinal infection dataset, we can observe how uninfected bystander cells may initiate important inflammatory pathways first, which are later amplified by infected cells. A fundamental assumption of the circuit assembly workflow is that ligands upregulated at one timepoint can be observed to exert their biological activity at the following timepoint. This assumption is highly dependent on a priori biological knowledge of the communication pathways of interest as well as on the spacing between timepoints. Assembly of longitudinal communication circuits may represent a valuable strategy to elucidate complex dynamic and temporal signaling events, particularly when longitudinal sampling is performed at frequencies on the same scale as signaling and transcriptional response pathways.
The cell–cell interaction matrix M is more highly enriched for zero values than gene expression matrices. This is because genes encoding molecules involved in CCC tend to be more lowly expressed than other genes (as the most highly expressed genes tend to encode intracellular proteins involved in cell housekeeping) and because a zero value in either the ligand or the receptor of a cell–cell pair will result in a zero value in the interaction vector. Consequently, these zero values can make it difficult for Scriabin to determine if putatively downregulated or ‘missing’ CCC edges are biological or due to dropout. We show that data denoising algorithms for scRNA-seq are capable of removing technical noise caused by data sparsity, substantially improving the yield of bona fide single-cell CCC states. This process can make the presence and absence of CCC edges more interpretable. We recommend the use of denoising algorithms when analyzing datasets generated by low-coverage platforms and particularly for non-UMI methods, which are more likely to be zero-inflated67,68.
Another complementary set of techniques for CCC inference are computational methods that infer which cells are communicating by identifying putative multiplets in the dataset or by directly sequencing interacting cells. The central premise of these techniques, which include Neighbor-seq69 and PIC-seq70, is that physically interacting cells are likely to be more difficult to dissociate when preparing single-cell suspensions and, therefore, that multiplets may be more likely to represent cells that are genuinely interacting. Although this provides an additional layer of evidence for biologically meaningful interactions, there are some communication edges that cannot be detected with these methods. For example, CCC involving secreted ligands will not be adequately modeled with these techniques. Additionally, as each scRNA-seq dataset represents a single snapshot of a sample, cells that have previously interacted but are no longer associated will not be detected. This latter problem has been addressed by techniques such as LIPSTIC71 that permanently label cells that have interacted using particular ligands or receptors. However, these methods remain poorly scalable and require prior cell engineering. We anticipate that future technological developments will enable synergy of these complementary approaches toward more comprehensive solutions for CCC analysis.
One current limitation of Scriabin is that it does not take into account situations where multiple receptor subunits encoded by different genes are required in combination to respond to a ligand or where receptor subunits are known to differentially contribute to collective ligand–receptor avidity. An additional limitation is that Scriabin assumes uniform validity of ligand–receptor interactions in curated protein–protein interaction databases and treats all ligand–receptor pairs as equally important. In situations where it is known a priori which ligand–receptor pairs have a higher level of literature support, this information could be used to prioritize downstream analysis of particular ligand–receptor pairs. Additionally, Scriabin assumes the interaction directionality that is presented by the user-selected ligand–receptor database; however, not all interactions are unidirectional, and biologically important receptor–receptor interactions are also possible72. Scriabin supports the use of custom ligand–receptor pair databases for users who a priori have specific analytical questions involving non-traditional interaction directionality.
Similarly, all downstream signaling analyses in Scriabin rely on NicheNet’s ligand–target activity matrix, which may be biased by the cell types and stimulation conditions used to generate it. The NicheNet database also does not allow for analysis of inhibitory signaling, and, thus, Scriabin will only return CCC edges predicted to result in activated signaling. Although Scriabin uses NicheNet to predict active CCC edges by examining downstream gene expression changes, an additional analysis goal includes identifying the upstream signaling machinery that results in the upregulation of a ligand or denotes successful signaling, as additional power could be gained by using sets of genes to infer upstream signaling rather than relying on ligand expression alone (which could be impacted by dropout or differences between mRNA and protein expression). More generally, Scriabin assumes that gene expression values for ligands and receptors correlate well with their protein expression. A future point of improvement would be to support analysis of multimodal datasets where cell surface proteins that contribute to CCC are measured directly or to enable analysis of protein measurements that are imputed from integration with multimodal references73. Future iterations of Scriabin will seek to address these issues as well as further improve computational efficiency.
Collectively, our work provides a toolkit for comprehensive comparative analysis of CCC in scRNA-seq data, which should empower discovery of information-rich communicative circuitry and niche–phenotype relationships.
Methods
CCIM workflow
Generation of CCIM
We define the cell–cell interaction vector between a pair of cells as the geometric mean of expression values of each cognate ligand–receptor pair. Formally, the interaction vector V between sender cell Ni and receiver cell Nj is given by
where ln,rn represent a cognate ligand–receptor pair. We chose to multiply ligand and receptor expression values so that zero values of either ligand or receptor expression would result in a zero value for the corresponding index of the interaction vector. Additionally, we chose to take the square root of the product of ligand–receptor expression values so that highly expressed ligand–receptor pairs do not disproportionately drive downstream analysis. This definition is equivalent to the geometric mean. The cell–cell interaction matrix M is constructed by concatenating the cell–cell interaction vectors. M is used as input to low-dimensional embeddings for visualization and nearest neighbor graphs for graph-based clustering.
Weighting CCIM by upstream regulome
The CCIM M can be weighted by ligand–receptor edges that are predicted to be active based on observed downstream gene expression changes. First, we identify genes in the dataset that are variable across some axis of interest. For analyses of single datasets, variable genes can be defined as the set of genes with the highest residual variance in the dataset—for example, by calling FindVariableFeatures as implemented by Seurat. For comparative analyses, Scriabin provides several utility functions to aid in the identification of variable genes between samples or between timepoints, depending on the user’s analytical questions.
Next, the package CelliD23, which provides a convenient and scalable workflow to define single-cell gene signatures, is used to define per-cell gene signatures. In brief, user-defined variable genes are used to embed the dataset into low-dimensional space by MCA. A cell’s gene signature is then defined as the set of genes to which that cell is nearest in the MCA bi-plot. A quantile cutoff is used to threshold gene proximity, by default the 5% of nearest genes.
NicheNet’s20 ligand–target matrix, which denotes the regulatory potential scores between ligands and target genes, is then used to rank ligands based on their predicted ability to result in the per-cell gene signature. First, expressed genes are defined by the percentage of cells in which they are detected (by default, 2.5%). Next, a set of potential ligands is defined as those ligands that are expressed genes and for which at least one receptor is also an expressed gene. Next, the ligand–target matrix is filtered to contain only the set of potential ligands and targets in the set of expressed genes. The authors of NicheNet showed that the Pearson correlation coefficient between a ligand’s target prediction and observed transcriptional response is the most informative metric of ligand activity20. Therefore, the activity of a single ligand for a single cell is defined as the Pearson correlation coefficient between the vector of that cell’s gene signature and the target gene scores for that ligand. For each active ligand, target gene weights for each cell are defined as the ligand–target matrix regulatory score for the top 250 targets for each ligand that appear in a given cell’s gene signature. We selected a Pearson coefficient threshold (by default, 0.075) to define active ligands in each cell.
Finally, we weight individual values of \(V_{N_iN_j}\). Scriabin supports two methods for weighting the CCIM by predicted ligand activities. Method ‘product’ (default) weights interaction vectors proportionally to predicted ligand activities. The vector of ligand activities for receiver cell Nj, Aj, is scaled so that values above the Pearson threshold lie between two scaling factors (by default, 1.5 and 3), and values below the Pearson threshold are set to 1. The interaction vector is then given by:
Method ‘sum’ treats a ligand activity as orthogonal evidence of receptor expression. Pearson coefficients in the vector of ligand activities for receiver cell Nj, Aj, that are below the Pearson threshold are set to 0. The interaction vector is then given by:
Use cases for ligand activity weighting methods, as well as other parameters involved in calculating ligand activities, are described in the Supplementary Text.
Downstream analysis of weighted CCIMs
M can be treated analogously to the gene expression matrix and used for downstream analysis tasks, such as dimensionality reduction. After generation and (optional) weighting of M by active ligands, M is placed into an assay of a Seurat object for downstream analysis. M is scaled by ScaleData; latent variables are found via PCA; and the top principal components (PCs) (identified by ElbowPlot for each dataset) are used to embed the dataset in two dimensions using uniform manifold approximation and projection (UMAP)74. Neighbor graphs are constructed by FindNeighbors, which can then be clustered via modularity optimization graph-based clustering75 as implemented by Seurat’s FindClusters73. Differential ligand–receptor edges among clusters, cell types or samples can be identified using FindMarkers. Scriabin provides several utility functions to facilitate visualization of gene expression profiles or other metadata on Seurat objects built from cell–cell interaction matrices.
Summarized interaction graph and binning workflow
Generation of summarized interaction graph
Because M scales exponentially with dataset size, it is frequently impractical to calculate M for all cell–cell pairs Ni,Nj. In this situation, Scriabin supports two workflows that do not require aggregation or subsampling. In the first workflow, a summarized cell–cell interaction graph S is built in lieu of M where \(S_{i,\,{j}} = \Sigma V_{N_iN_j}\). S thus represents the magnitude of predicted interaction across all cognate ligand–receptor pairs expressed by all sender–receiver cell pairs. S is then corrected for associations with sequencing depth by linear regression. The sequencing depth of cell–cell pair Ni,Nj is defined as \(nUMI_{N_i} + nUMI_{N_{j}}\). A linear model is fit to describe the relationship between the summarized interaction score (\(S_{i,j} = \Sigma V_{N_iN_j}\), where S is the summarized interaction matrix and \(V_{N_iN_j}\) is the interaction vector for cell–cell pair Ni,Nj) and the total sequencing depth of each cell–cell pair. The residuals of this model are used as a sequencing depth-corrected S. S may optionally be weighted through prediction of ligand activity as described above. The second workflow is described below in the ‘Interaction program discovery workflow’ subsection.
Dataset binning for comparative CCC analyses
Once summarized interaction graphs are built for multiple samples, alignment of these graphs requires knowledge about which cells between samples represent a shared molecular state. The goal of binning is to assign each cell a bin identity so that S from multiple samples can be summarized into equidimensional matrices based on shared bin identities.
The binning process begins by constructing a shared nearest neighbor (SNN) graph using FindNeighbors, defining connectivity between all cells to be compared. Alternate neighbor graphs—for example, those produced using Seurat’s weighted nearest neighbor (WNN) workflow, which leverages information from multimodal references—can also be used. Next, mutual nearest neighbors (MNNs) are identified between all sub-datasets to be compared using Seurat’s integration workflow (FindIntegrationAnchors)21. In brief, two sub-datasets to be compared are placed into a shared low-dimensional space via diagonalized canonical correlation analysis (CCA), and the canonical correlation vectors are log-normalized. Normalized canonical correlation vectors are then used to identify k-nearest neighbors for each cell in its paired dataset, and the resulting MNN pairings are scored as described21. Low-scoring MNN pairings are then removed, as they have a higher tendency to represent incorrect cell–cell correspondences when orthogonal data are available (Extended Data Fig. 10).
For each cell that participates in an MNN pair, Scriabin defines a bin as that cell and all cells with which it participates in an MNN pair. Considering a dataset of n cells i of which a subset i′ participates in an MNN pair, for each cell i′n we define a bin jn that contains i′n and all MNNs of i′n. Next, Scriabin constructs a connectivity matrix G where Gi,j is the mean connectivity in the SNN graph between cell i and the cells within bin j. Each cell in is assigned a bin identity of the bin jn with which it shares the highest connectivity in G. Thus, at the end of this process, each cell has a single bin identity, which reflects its SNN similarity to a group of cells with cross-dataset MNN connectivity.
However, at this stage, each bin jn may not contain cells from all the samples being compared. Thus, we next optimize for the set of bins that results in the best representation of all samples. Bins j with the lowest total connectivity and lowest multi-sample representation in G are iteratively removed, and cell bin identities are re-scored until the mean sample representation of each bin plateaus. Within-bin connectivity and sample representation are further improved by reassigning cells that result in better sample representation of an incompletely represented bin while maintaining equal or greater SNN connectivity with the cells in that bin. Finally, remaining incompletely represented bins are merged with the nearest completely represented bin with which it shares the highest SNN connectivity. At the end of this process, each cell will, thus, have a single assigned bin identity, where each bin contains cells from all samples to be compared.
Statistical analysis of bin significance
Bins are then tested for the statistical significance of their connectivity structure using a permutation test. For each bin, random bins of the same size and number of cells per sample are generated iteratively (by default, 10,000 times). The connectivity vector of the real bins is tested against each of the random bins by a one-sided Mann–Whitney U-test. If the bin fails 500 or more of these tests (P > 0.05), it is considered non-significant.
Because bin SNN connectivity is generally non-zero, but randomly sampled cells generally have an SNN connectivity of zero, this strategy will tend to return most bins as statistically significantly connected. Thus, we recommend passing high-resolution cell type labels to the binning significance testing. In this situation, randomly generated bins are generated by randomly selecting cells from the same sample and cell type annotation, and the permutation test proceeds as described above. Bins where more than a threshold (by default, 95%) of cells belong to the same cell type annotation are automatically considered significant. This avoids rare cell types that may only form a single bin from being discarded. Cells that were assigned to bins that failed the significance testing are reassigned to the bin with which they share the highest SNN connectivity.
Identification of variable bins
For each bin, a Kruskal–Wallis test is used to assess differences in the magnitude of CCC between cell–cell pairs from different samples. The Kruskal–Wallis P value and test statistic can be used to identify which bins contain cells that exhibit the highest change in prediction interaction scores. Specific samples that contribute to each significantly variable bin’s perturbation are then identified through the Dunn post hoc test. This set of sender and receiver cells can then be used to construct M as described above.
Interaction program discovery workflow
Iterative approximation of a ligand–receptor pair TOM
An alternative to the summarized interaction graph workflow is to instead identify co-expressed ligand–receptor pairs, which we refer to as ‘interaction programs’. This approach represents an adaptation of the well-established WGCNA22 and is scalable to any dataset size and still permits analysis of CCC at single-cell resolution. The first step in this workflow is to generate a signed covariance matrix of ligand–receptor pairs for each sample, defined as
where \(lr_i,lr_j\) are individual ligand–receptor pair vectors of M. In large datasets, \(s_{ij}^{signed}\) is approximated by iteratively generating subsets of M. \(s_{ij}^{signed}\) is next converted into an adjacency matrix via soft thresholding
where β is the soft power. Soft power is a user-defined parameter that is recommended to be the lowest value that results in a scale-free topology model fit of >0.6. Next, this adjacency matrix is converted into a TOM as described76. This process proceeds separately for each sample to be analyzed in a multi-sample dataset.
Identification and significance testing of interaction programs
The TOM is hierarchically clustered, and interaction programs are identified through adaptive branch pruning of the hierarchical clustering dendrogram. Intramodular connectivity for each ligand–receptor pair in each interaction program is then calculated as described77. If interaction programs are being discovered in a multi-sample dataset, similar modules (defined by Jaccard overlap index above a user-defined threshold) are merged. Next, interaction programs are then tested for statistically significant co-expression structure via a permutation test where random interaction programs are generated 10,000 times. The correlation vector of the real module is tested against each of the random modules by a one-sided Mann–Whitney U-test. If the module fails 500 or more of these tests (P > 0.05), it is considered non-significant. Each sample is tested for significant correlation of each module.
Downstream analysis of interaction programs
Single cells are scored separately for the expression of the ligands and receptors of each significant module with Seurat’s AddModuleScore. This function calculates a module score by comparing the expression level of an individual query gene to other randomly selected control genes expressed at similar levels to the query genes and is, therefore, robust to scoring modules containing both lowly and highly expressed genes as well as to scoring cells with different sequencing depth. Scriabin includes several utility functions to conveniently visualize interaction program expression for sender and receiver cells.
Identification of longitudinal CCC circuits
A longitudinal CCC circuit is composed of S1–L1–R1–S2–L2–R2, where S are sender cells and R are receiver cells at timepoints 1 and 2 and where L1 is expressed by/sensed by S1/R1, and L2 is expressed by/sensed by S2/R2. For computational efficiency, construction of longitudinal CCC circuits starts at the end of the circuit and proceeds upstream. First, ligands L2 predicted by NicheNet to be active in receiver cells at timepoint 2 are identified. Next, sender cells that express L2 and have the L2 in its per-cell gene signature are identified. Among the bins occupied by these S2 candidates, Scriabin then searches for receiver cells at timepoint 1 that occupy the same bin and have the corresponding timepoint 2 ligand L2 within its list of upregulated target genes and identifies the ligand(s) L1 predicted by NicheNet to result in upregulation of that target. Finally, Scriabin identifies S1 candidates that express the timepoint 1 ligands L1 and have L1 in its per-cell gene signature. S1–R1–S2–R2 cell groups that meet these criteria are retained for further analysis. This process repeats for every pair of timepoints. Finally, Scriabin searches for overlap between circuits of sequential timepoint pairs to identify circuits that operate over more than two timepoints.
Ligand–receptor pair databases for analysis
Scriabin supports the use of 15 ligand–receptor interaction databases for all analytical functions; these resources were collected from LIANA78. By default, Scriabin uses the OmniPath database18,19 filtered for curation strength of >7 to ensure that ligand–receptor interactions with strong experimental evidence are included in downstream analysis. Scriabin also supports the use of custom ligand–receptor pair lists for users with specific analytical questions.
Transfection and co-culture of primary NK and B cells
Peripheral blood mononuclear cells (PBMCs) were acquired from a healthy blood donor who was consented for release of genetic data by the Stanford Blood Center. PBMCs were isolated by Ficoll-Paque (GE Healthcare) density gradient centrifugation and cryopreserved in 90% FBS + 10% DMSO (v/v). PBMCs were thawed at 37 °C in complete RPMI 1640 media (supplemented with 10% FBS, L-glutamine and penicillin–streptomycin–amphotericin; RP10) containing benzonase (EMD Millipore). NK cells and B cells were purified from thawed PBMCs by magnetic bead isolation via negative selection according to the manufacturer’s specifications (Miltenyi Biotec, 130-092-657 and 130-101-638, respectively). NK and B cells were maintained in complete RP10 media without additional cytokines to ensure a resting state. All cell culture was performed at 37 °C/5% CO2 in a humidified environment.
eGFP-encoding, CD40-encoding and CD40L-encoding mRNAs were purchased from TriLink BioTechnologies and used without further purification. Notably, open reading frame (ORF) sequences for mRNAs encoding CD40 and CD40L were codon optimized using the codon optimization tool developed by Integrated DNA Technologies: this serves both to improve translational efficacy as well as to enable distinguishing endogenous versus exogenous CD40 and CD40LG mRNAs through sequencing.
mRNAs were delivered to isolated NK and B cells via transfection by charge-altering releasable transporters (CARTs) as previously described79. In brief, CART/mRNA polyplexes were prepared by diluting 0.84 of mRNA (1 μg μl−1) into 14.52 μl of PBS (pH 5.5). To this solution was added 1.44 μl of CART BDK-O7:N7:A13 (2 mM DMSO) to achieve a charge ratio of 10:1 (±, assuming all ionizable cationic groups are protonated). After mixing by finger vortex for 15 s, 2.5 μl of the polyplexes was added to cells and incubated for 6 h in serum-free media. After this incubation, an aliquot was taken from each transfection condition for flow cytometric analysis; FBS was added to a final concentration of 10%; the cells were counted; and NK cells and B cells from the respective transfection conditions were mixed together in a 1:1 ratio for co-culture. Cells were co-cultured for 12 h before analysis by flow cytometry and scRNA-seq.
Flow cytometry
Antibodies used for flow cytometric analyses are listed in Supplementary Table 2. eBioscience Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific) was used as a viability stain. After application of viability stain, cells were surface stained for 20 min at room temperature before acquisition on an Aurora flow cytometer (Cytek Biosciences) and analysis by FlowJo version 10.6.1 software.
scRNA-seq by Seq-Well
The Seq-Well platform for scRNA-seq was used as described previously56,80,81,82,83. Immediately after co-culture, cells were counted and diluted in RP10 to a concentration of 75,000 cells per milliliter. Then, 200 μl of this cell suspension (15,000 cells) was loaded onto Seq-Well arrays pre-loaded with mRNA capture beads (ChemGenes). After four washes with DPBS to remove serum, the arrays were sealed with a polycarbonate membrane (pore size, 0.01 µm) for 30 min at 37 °C. Next, arrays were placed in lysis buffer; transcripts were hybridized to the mRNA capture beads; beads were recovered from the arrays and pooled for downstream processing. Immediately after bead recovery, mRNA transcripts were reverse transcribed using Maxima H-RT (Thermo Fisher Scientific, EPO0753) in a template-switching-based RACE reaction; excess unhybridized bead-conjugated oligonucleotides were removed with Exonuclease I (New England Biolabs (NEB), M0293L); second-strand synthesis was performed with Klenow fragment (NEB, M0212L) to enhance transcript recovery in the event of failed template switching81. Whole-transcriptome amplification (WTA) was performed with KAPA HiFi PCR Mastermix (Kapa Biosystems, KK2602) using approximately 6,000 beads per 50-μl reaction volume. Resulting libraries were then pooled in sets of six (approximately 36,000 beads per pool), and products were purified by Agencourt AMPure XP beads (Beckman Coulter, A63881) with a 0.6× volume wash followed by a 0.8× volume wash. Quality and concentration of WTA products were determined using an Agilent TapeStation, with a mean product size of more than 800 base pairs (bp) and a non-existent primer peak indicating successful preparation. Library preparation was performed with a Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1096) with 1 ng of pooled library using single-index primers. Tagmented and amplified libraries were again purified by Agencourt AMPure XP beads with a 0.6× volume wash followed by a 1.0× volume wash, and quality and concentration were determined by TapeStation analysis. Libraries between 400 bp and 1,000 bp with no primer peaks were considered successful and pooled for sequencing. Sequencing was performed on a NovaSeq 6000 instrument (Illumina; Chan Zuckerberg Biohub). The read structure was paired-end with read 1 beginning from a custom read 1 primer80 containing a 12-bp cell barcode and an 8-bp UMI and with read 2 containing 50 bp of mRNA sequence.
Alignment and quality control of scRNA-seq data
Sequencing reads were aligned and count matrices assembled using STAR84 and dropEst85, respectively. In brief, the mRNA reads in read 2 demultiplexed FASTQ files were tagged with the cell barcode and UMI for the corresponding read in the read 1 FASTQ file using the dropTag function of dropEst. Next, reads were aligned with STAR using the GRCh38.p13 (hg38) human reference genome from Ensembl. This reference also included sequences and annotations for the codon-optimized ORFs for GFP-encoding, CD40-encoding and CD40L-encoding mRNAs so that both endogenous and exogenous mRNAs could be quantified. Count matrices were built from resulting BAM files using dropEst85. Cells that had fewer than 750 UMIs or more than 15,000 UMIs, as well as cells that contained more than 20% of reads from mitochondrial genes or rRNA genes (RNA18S5 or RNA28S5), were considered low quality and removed from further analysis. To remove putative multiplets, cells that expressed more than 75 genes per 100 UMIs were also filtered out.
Pre-processing of scRNA-seq data
The R package Seurat21,73,86 was used for data scaling, transformation, clustering, dimensionality reduction, differential expression analysis and most visualizations. Unless otherwise noted, data were scaled and transformed and variable genes identified using the SCTransform() function, and linear regression was performed to remove unwanted variation due to cell quality (% mitochondrial reads and % rRNA reads). PCA was performed using the 3,000 most highly variable genes, and the first 50 PCs were used to perform UMAP to embed the dataset into two dimensions74,87. Next, the first 50 PCs were used to construct an SNN graph (FindNeighbors()), and this SNN was used to cluster the dataset (FindClusters()). Although upstream quality control removed many dead or low-quality cells, if any clusters were identified that were defined by few canonical cell lineage markers and enriched for genes of mitochondrial or ribosomal origin, these clusters were removed from further analysis88,89.
Annotation of transfected NK and B cells in scRNA-seq data
Because of the strong degree of transcriptional perturbation caused by transfection (Extended Data Fig. 3), we elected to annotate NK and B cells in this dataset by integration with a multimodal reference rather than by graph-based clustering. First, we noted two clusters with high expression of CD3-encoding genes and monocyte-specific genes (including LYZ and CD14), respectively; we considered these clusters contaminating T cells and monocytes and removed them from further analysis. Next, we used the multimodal (whole transcriptome plus 228 cell surface proteins) PBMC dataset published by Hao et al.73 as a reference. We subsetted the reference to contain only NK and B cells, scaled both the transcriptome and protein assays and ran PCA on both modalities. Next, we found multimodal neighbors between the modalities using WNN analysis, which learns the relative utility of each data modality in each cell. Supervised PCA (SPCA) was then run on the WNN SNN graph, which seeks to capture a linear transformation that maximizes its dependency to the WNN SNN graph. These SPCA-reduced dimensions were then used for identification of anchors between the reference and query datasets as previously described21. Finally, these anchors were used to transfer reference cell type annotations to the query dataset.
Processing, analysis and visualization of public scRNA-seq datasets
Published scRNA-seq datasets were acquired as described in the ‘Data availability’ section. In each case, we acquired raw count matrices or processed Seurat objects containing raw count matrices. Any upstream processing was performed as described in the respective manuscripts.
Raw count matrices from Ravindra et al.58 required filtering before downstream analysis; cells meeting the following criteria were kept: >1,000 UMIs, <20,000 UMIs, >500 unique features, <0.85 UMI-to-unique feature ratio, <20% UMIs of mitochondrial origin and <35% reads from ribosomal protein-encoding genes. Pbmc5k and pbmc10k datasets from 10x Genomics were filtered to enforce a minimum number of features per cell of 200 and to remove genes not expressed in at least three cells.
Cell type annotations were provided for the Ji et al.29, Ma et al.41 and Fawkner-Corbett et al.49 datasets, which were used for downstream analytical tasks. For the Ravindra et al.58 dataset, manual annotation of cellular identity was performed by finding differentially expressed genes for each cluster using Seurat’s implementation of the Wilcoxon rank-sum test (FindMarkers()) and comparing those markers to known cell-type-specific genes listed in Ravindra et al.58. PBMC datasets were annotated by WNN projection and label transfer from a multimodal PBMC reference as described73,83.
For analysis of T cell exhaustion in the SCC dataset from Ji et al.29, an exhaustion signature was defined by PDCD1, TOX, CXCL13, CTLA4, TNFRSF9, HAVCR2, LAG3, CD160 and CD244. This signature incorporates several markers of exhausted T cell reported in the literature66,90,91,92. Individual T cells were scored for expression of this signature using Seurat’s AddModuleScore.
Analysis of the impact of sparsity on single-cell-resolution CCC analysis
We collected three scRNA-seq datasets generated from methods with high coverage: Fluidigm C1 pancreas islets21,33, Smart-seq2 uterine decidua13 and Smart-seq2 HNSCC8. We used Scriabin’s CCIM workflow to generate a CCIM for each of these datasets, which we consider to be the GT of CCC for that dataset. Next, we randomly downsampled the datasets to inDrop coverage using downsampleMatrix() from scuttle93,94,95,96,97,98,99,100. For these analyses, inDrop coverage was defined as the mean UMIs per cell of the inDrop dataset included in the pancreas islet dataset available through SeuratData21, which is 5,828 UMIs per cell.
From these downsampled datasets, we generated several interaction matrices:
-
1.
Raw. We calculated an unweighted CCIM through Scriabin’s standard CCIM workflow.
-
2.
ALRA-denoised. We used ALRA32, a denoising algorithm for scRNA-seq, to denoise the downsampled dataset and then built an unweighted CCIM from the denoised dataset.
-
3.
Neighborhood-aggregated. We generated nearest neighbor graphs from each downsampled dataset using Seurat’s FindNeighbors() using the first 15 PCs. Next, we defined the neighborhood for each cell as that cell and its nearest k neighbors. Finally, we defined the transcriptome of each cell as the mean of that cell and its nearest k neighbors. We used values of k between 5 and 100. We used this matrix of neighborhood-aggregated expression values to generate CCIMs.
-
4.
Cluster-aggregated. We used Seurat’s graph-based clustering algorithm with resolutions between 2.5 and 5 to cluster each downsampled dataset. We then generated a matrix of pseudobulk expression vectors for each cluster and used this matrix to generate a cluster–cluster interaction matrix.
Next, we used Seurat’s dataset integration pipeline to integrate each CCIM from the downsampled dataset with the GT CCIM21. The first 30 PCs were used for CCIM integration. Finally, we used the LISI36 to quantify the degree to which the CCIMs from the downsampled datasets recapitulated the GT CCIMs. This value defines the number of datasets in the neighborhood of each GT cell–cell pair and ranges between 1, denoting that only GT cell–cell pairs are present in the neighborhood, and 2, denoting an equal mixture of GT and downsampled cell–cell pairs.
Comparative analyses between Scriabin and published CCC analysis methods
Pbmc5k and pbmc10k datasets from 10x Genomics were used to benchmark the computational efficiency of Scriabin. For single dataset analyses, pbmc5k was randomly subsetted to multiple dataset sizes. Cell type annotations were passed to Connectome38, NATMI17, CellChat15, iTALK16 and SingleCellSignalR (SCA)37, which were run using default parameters defined by LIANA39. The time for these methods to return results was compared to a version of Scriabin that generated and visualized a full dataset summarized interaction graph and returned pseudobulk ligand–receptor pair scores for each cell type annotation. Connectome38 is the only of these packages that supports a full comparative workflow. For comparative analysis, we analyzed differences in CCC between the pbmc5k and pbmc10k datasets. We compared Connectome’s total runtime to the runtime of Scriabin to generate full dataset summarized interaction graphs, perform dataset binning and visualize the most perturbed bins.
Multiple ligand–receptor resources compiled by LIANA39 were used to compare results returned by published CCC analysis methods and Scriabin. This analysis was performed on four datasets: 10x PBMC 5k, Fluidigm C1 pancreas islets21,33, Smart-seq2 uterine decidua13 and Smart-seq2 HNSCC8. The following results parameters were used from each method: prob (CellChat), LRscore (SingleCellSignalR), weight_norm (Connectome), weight_comb (iTALK) and edge_avg_expr (NATMI). To visualize the overlap in results between the methods and resources, we extracted the top 1,000 results from each method–resource pair and calculated the Jaccard index between these top results (as described by ref. 39).
Analysis of spatial transcriptomic datasets with Scriabin
To evaluate if Scriabin returns biologically meaningful CCC edges, we downloaded spatial coordinates and gene expression count matrices from 10 spatial transcriptomic datasets from the 10x Visium platform available at https://www.10xgenomics.com/resources/datasets. We also analyzed a spatial transcriptomic dataset published by Ma et al.41 of a human leprosy granuloma. We treated each count matrix analogously to scRNA-seq data, performing data transformation and dimensionality reduction as described above. We calculated per-cell gene signatures for each dataset based on variable genes across the dataset, which we then used to rank ligands based on their predicted ability to result in the observed gene expression profile using NicheNet20. Next, we constructed a summarized interaction graph using a ligand–receptor pair database that was restricted to membrane-bound ligands and receptors, which we weighted according to the predicted ligand activities. Finally, we compared the distance quantile of the top 1% of interacting cell–cell pairs compared to randomly permuted distances.
Analysis of CRISPRa Perturb-seq data
To quantify Scriabin’s ability to detect changes in CCC at single-cell resolution, we analyzed data from a pooled genetic perturbation screen. We elected to analyze the Perturb-seq dataset published by Schmidt et al.40 as this dataset was collected on primary cells and contained a high number of gRNAs (15) targeting cell surface ligands or receptors used in CCC. We collected a processed and publicly available h5Seurat object of the anti-human CD3/CD28 re-stimulated T cells from the Schmidt et al.40 dataset from https://zenodo.org/record/5784651. The authors’ gRNA calls were used for all downstream analysis; we identified gRNAs g in this dataset that targeted cell surface ligands or receptors that were present in OmniPath’s ligand–receptor interaction database. The dataset was then subsetted to include only cells transduced with a gRNA targeting one of these cell surface ligands or receptors or cells transduced with a non-targeting gRNA. Untransduced T cells were removed from further analysis. We repeated the following process for each gRNA gi. Given a gRNA, gA, targeting a ligand-encoding gene A: we isolated cells transduced with gA and cells transduced with a non-targeting gRNA. From this subsetted dataset, we generated a CCIM without ligand activity ranking using Scriabin’s CCIM workflow. We next isolated interaction vectors V for ligand A and all receptors of A, RA. For each interaction vector VAR, we constructed a receiver operating characteristic (ROC) curve using VAR as the predictor variable and the gRNA assignment (either gA or non-targeting) as the response variable to quantify and visualize the sensitivity and specificity of the prediction.
Visualization
For all box plot features: minimum whisker, 25th percentile −1.5 × interquartile range (IQR) or the lowest value within; minimum box, 25th percentile; center, median; maximum box, 75th percentile; maximum whisker, 75th percentile +1.5 × IQR or greatest value within.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw and processed scRNA-seq data generated in this manuscript are available on the Gene Expression Omnibus (GEO) as accession GSE228415 (ref. 101). Spatial transcriptomic datasets and datasets of PBMCs (pbmc5k and pbmc10k) were downloaded from 10x Genomics; for comparison of mouse and human PBMCs, datasets from 10x Genomics’ cell multiplexing oligo demonstration were used (https://www.10xgenomics.com/resources/datasets). Processed scRNA-seq data of SCC and matched normals29 were provided directly by the study authors. Processed count matrices from the Smart-Seq2 human HNSCC dataset were downloaded from GEO accession GSE103322 (ref. 8). Processed count matrices from the Smart-Seq2 human uterine decidua dataset were downloaded from European Bioinformatics Institute accession E-MTAB-6678 (ref. 13). Processed Seurat objects of the Fluidigm C1 pancreas islet dataset are available through the R package SeuratData21,33. Processed CRISPRa Perturb-seq data were downloaded from Zenodo record 5784651 (ref. 40). scRNA-seq data of human leprosy granulomas41 were downloaded from https://github.com/mafeiyang/leprosy_amg_network. Data from developing fetal intestine49 were acquired from the CELLxGENE portal: https://cellxgene.cziscience.com/collections/60358420-6055-411d-ba4f-e8ac80682a2e. Data of longitudinal responses to SARS-CoV-2 infection in HBECs58 were downloaded from GEO accession GSE166766. The GRCh38.p13 reference genome is available from the National Center for Biotechnology Information.
Code availability
Scriabin is available for download and use as an R package at https://github.com/BlishLab/scriabin (ref. 102).
References
Almet, A. A., Cang, Z., Jin, S. & Nie, Q. The landscape of cell–cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 26, 12–23 (2021).
Armingol, E., Officer, A., Harismendy, O. & Lewis, N. E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88 (2020).
Tanay, A. & Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017).
Yosef, N. & Regev, A. Writ large: genomic dissection of the effect of cellular environment on immune response. Science 354, 64–68 (2016).
Ramilowski, J. A. et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun. 6, 7866 (2015).
Dixit, A. et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866 (2016).
Schraivogel, D. et al. Targeted Perturb-seq enables genome-scale genetic screens in single cells. Nat. Methods 17, 629–635 (2020).
Puram, S. V. et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624 (2017).
Camp, J. G. et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 546, 533–538 (2017).
Pavličev, M. et al. Single-cell transcriptomics of the human placenta: inferring the cell communication network of the maternal–fetal interface. Genome Res. 27, 349–361 (2017).
Zepp, J. A. et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell 170, 1134–1148 (2017).
Cohen, M. et al. Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell 175, 1031–1044 (2018).
Vento-Tormo, R. et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 563, 347–353 (2018).
Raredon, M. S. B. et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 5, eaaw3851 (2019).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Wang, Y. et al. iTALK: an R package to characterize and illustrate intercellular communication. Preprint at bioRxiv https://doi.org/10.1101/507871 (2019).
Hou, R., Denisenko, E., Ong, H. T., Ramilowski, J. A. & Forrest, A. R. R. Predicting cell-to-cell communication networks using NATMI. Nat. Commun. 11, 5011 (2020).
Türei, D., Korcsmáros, T. & Saez-Rodriguez, J. OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13, 966–967 (2016).
Türei, D. et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 17, e9923 (2021).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Cortal, A., Martignetti, L., Six, E. & Rausell, A. Gene signature extraction and cell identity recognition at the single-cell level with Cell-ID. Nat. Biotechnol. 39, 1095–1102 (2021).
McMurdie, P. J. & Holmes, S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10, e1003531 (2014).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Wang, J. et al. Evaluation of ultra-low input RNA sequencing for the study of human T cell transcriptome. Sci Rep. 9, 8445 (2019).
Wu, L. et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol. Res. 7, 1700–1713 (2019).
Kiner, E. et al. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. 22, 216–228 (2021).
Ji, A. L. et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182, 497–514 (2020).
Joller, N. & Kuchroo, V. K. Tim-3, Lag-3, and TIGIT. Curr. Top. Microbiol. Immunol. 410, 127–156 (2017).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Linderman, G. C. et al. Zero-preserving imputation of single-cell RNA-seq data. Nat. Commun. 13, 192 (2022).
Lawlor, N. et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 27, 208–222 (2017).
Baron, M. et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360 (2016).
Zilionis, R. et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 12, 44–73 (2016).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Cabello-Aguilar, S. et al. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 48, e55–e55 (2020).
Raredon, M. S. B. et al. Computation and visualization of cell–cell signaling topologies in single-cell systems data using Connectome. Sci. Rep. 12, 1–12 (2022).
Dimitrov, D. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 13, 1–13 (2022).
Schmidt, R. et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science 375, eabj4008 (2022).
Ma, F. et al. The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol. 22, 839–850 (2021).
Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003).
Ridley, D. S. & Jopling, W. H. Classification of leprosy according to immunity. A five-group system. Int. J. Lepr. Other Mycobact. Dis. 34, 255–273 (1966).
Flynn, J. L. et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254 (1993).
Herbst, S., Schaible, U. E. & Schneider, B. E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 6, e19105 (2011).
Ní Cheallaigh, C. et al. A common variant in the adaptor mal regulates interferon gamma signaling. Immunity 44, 368–379 (2016).
Verhagen, C. E. et al. Reversal reaction in borderline leprosy is associated with a polarized shift to type 1-like Mycobacterium leprae T cell reactivity in lesional skin: a follow-up study. J. Immunol. 159, 4474–4483 (1997).
Teles, R. M. B. et al. Identification of a systemic interferon-γ inducible antimicrobial gene signature in leprosy patients undergoing reversal reaction. PLoS Negl. Trop. Dis. 13, e0007764 (2019).
Fawkner-Corbett, D. et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 184, 810–826 (2021).
Biton, M. et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175, 1307–1320 (2018).
Goto, N. et al. Lymphatics and fibroblasts support intestinal stem cells in homeostasis and injury. Cell Stem Cell 29, 1246–1261.e6 (2022).
Niec, R. E. et al. Lymphatics act as a signaling hub to regulate intestinal stem cell activity. Cell Stem Cell 29, 1067–1082.e18 (2022).
Darling, T. K. & Lamb, T. J. Emerging roles for Eph receptors and ephrin ligands in immunity. Front. Immunol. 10, 1473 (2019).
Kim, M. J. et al. PAF-Myc-controlled cell stemness is required for intestinal regeneration and tumorigenesis. Dev. Cell 44, 582–596 (2018).
Zhang, N. et al. ID1 is a functional marker for intestinal stem and progenitor cells required for normal response to injury. Stem Cell Rep. 3, 716–724 (2014).
Kazer, S. W. et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat. Med. 26, 511–518 (2020).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Ravindra, N. G. et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 19, e3001143 (2021).
Gressner, O. A., Peredniene, I. & Gressner, A. M. Connective tissue growth factor reacts as an IL-6/STAT3-regulated hepatic negative acute phase protein. World J. Gastroenterol. 17, 151–163 (2011).
Sack, G. H. Jr. Serum amyloid A—a review. Mol. Med. 24, 46 (2018).
Xu, J. et al. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 21, 182 (2020).
Doitsh, G. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514 (2013).
Vignuzzi, M. & López, C. B. Defective viral genomes are key drivers of the virus–host interaction. Nat. Microbiol. 4, 1075–1087 (2019).
López, C. B. Defective viral genomes: critical danger signals of viral infections. J. Virol. 88, 8720–8723 (2014).
Raredon, M. S. B. et al. Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHES. Bioinform. 39, btac775 (2023).
Andreatta, M. et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 2965 (2021).
Svensson, V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 38, 147–150 (2020).
Cao, Y., Kitanovski, S., Küppers, R. & Hoffmann, D. UMI or not UMI, that is the question for scRNA-seq zero-inflation. Nat. Biotechnol. 39, 158–159 (2021).
Ghaddar, B. & De, S. Reconstructing physical cell interaction networks from single-cell data using Neighbor-seq. Nucleic Acids Res. 50, e82 (2022).
Giladi, A. et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 38, 629–637 (2020).
Pasqual, G. et al. Monitoring T cell–dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500 (2018).
Guidolin, D., Marcoli, M., Tortorella, C., Maura, G. & Agnati, L. F. Receptor–receptor interactions as a widespread phenomenon: novel targets for drug development? Front. Endocrinol. 10, 53 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. https://doi.org/10.1038/nbt.4314 (2018).
Waltman, L. & van Eck, N. J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471 (2013).
Yip, A. M. & Horvath, S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinformatics 8, 22 (2007).
Dong, J. & Horvath, S. Understanding network concepts in modules. BMC Syst. Biol. 1, 24 (2007).
Dimitrov, D. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 13, 3224 (2022).
Wilk, A. J. et al. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Adv. 4, 4244–4255 (2020).
Gierahn, T. M. et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 (2017).
Hughes, T. K. et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immun. 53, 878–894.e7 (2020).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
Wilk, A. J. et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 218, e20210582 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Petukhov, V. et al. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at arXiv https://doi.org/10.48550/arXiv.1802.03426 (2018).
Carter, R. A. et al. A single-cell transcriptional atlas of the developing murine cerebellum. Curr. Biol. 28, 2910–2920 (2018).
Freytag, S., Tian, L., Lönnstedt, I., Ng, M. & Bahlo, M. Comparison of clustering tools in R for medium-sized 10x Genomics single-cell RNA-sequencing data. F1000Res. 7, 1297 (2018).
Miller, B. C. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Chibueze, C. E., Yoshimitsu, M. & Arima, N. CD160 expression defines a uniquely exhausted subset of T lymphocytes in HTLV-1 infection. Biochem. Biophys. Res. Commun. 453, 379–384 (2014).
Agresta, L., Hoebe, K. H. N. & Janssen, E. M. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front. Immunol. 9, 2809 (2018).
McCarthy, D. J., Campbell, K. R., Lun, A. T. L. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Polo, J. M., Ci, W., Licht, J. D. & Melnick, A. Reversible disruption of BCL6 repression complexes by CD40 signaling in normal and malignant B cells. Blood 112, 644–651 (2008).
McKinlay, C. J. et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl Acad. Sci. USA 114, E448–E456 (2017).
McKinlay, C. J., Benner, N. L., Haabeth, O. A., Waymouth, R. M. & Wender, P. A. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl Acad. Sci. USA 115, E5859–E5866 (2018).
Molfetta, R., Quatrini, L., Santoni, A. & Paolini, R. Regulation of NKG2D-dependent NK cell functions: the yin and the yang of receptor endocytosis. Int. J. Mol. Sci. 18, 1677 (2017).
Carlsten, M. et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 183, 4921–4930 (2009).
Wilk, A. J., Shalek, A. K., Holmes, S. & Blish, C. A. Comparative analysis of cell–cell communication at single-cell resolution. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE228415 (2023).
Wilk, A. J., Shalek, A. K., Holmes, S. & Blish, C. A. Comparative analysis of cell–cell communication at single-cell resolution. GitHub. https://github.com/BlishLab/scriabin (2023).
Acknowledgements
We thank W. J. Greenleaf and S. W. Kazer for helpful conversations in the conceptualization of Scriabin’s workflow. We thank P. A. Wender and N. L.-B. Weidenbacher for synthesis of CARTs. We thank C. Tzouanas and J. Ordovas-Montanes for insights on intestinal cell–cell communication pathways. We thank A. Ji for providing processed scRNA-seq data of SCC and matched normals. We also thank all current and former members of the Blish laboratory for helpful discussions of this work. A.J.W. is supported by the Stanford Medical Scientist Training Program (T32 GM007365-44) and the Stanford Bio-X Interdisciplinary Graduate Fellowship. Work with CARTs was supported by NIH/NIAID R01 AI161803 to C.A.B. This work was also supported by NIH/NIDA DP1 DA04508902 to C.A.B.; R01HD103571 to C.A.B.; NIH/NCI 1U54CA217377, U01 28020510 and 1U2CCA23319501 to A.K.S.; NIH/NIDA 1DP1DA053731 to A.K.S.; the Bill & Melinda Gates Foundation INV-027498 and OPP1202327 to A.K.S.; the MIT Stem Cell Initiative through Foundation MIT to A.K.S.; and a 2019 Sentinel Pilot Project from the Bill & Melinda Gates Foundation to C.A.B. and A.K.S.
Author information
Authors and Affiliations
Contributions
A.J.W., A.K.S., S.H. and C.A.B. conceived of the work. A.J.W. built Scriabin and performed computational analyses, cell culture work, flow cytometric analysis and scRNA-seq profiling. A.J.W. wrote the manuscript, with input from all authors. S.H. and C.A.B. jointly supervised the work.
Corresponding author
Ethics declarations
Competing interests
A.K.S. reports compensation for consulting and/or scientific advisory board (SAB) membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Ochre Bio, Third Rock Ventures, Hovione, Relation Therapeutics, FL82, Empress Therapeutics, IntrECate Biotherapeutics, Senda Biosciences and Dahlia Biosciences. C.A.B. reports compensation for consulting and/or SAB membership from Catamaran Bio, DeepCell, Immunebridge and Revelation Biosciences. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Yvan Saeys and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Additional analyses of exhausted intratumoral SCC T cells.
a) UMAP projection of all T cells from the dataset published by Ji, et al.29, colored by author-annotated T cell subtype. b) Dot plot depicting average and percent expression of the exhaustion signature score by author-annotated T cell subtypes. c) ROC curves depicting the ability of each cluster from the single-cell T cell object (left) or Scriabin generated T cell-CD1C+ DC CCIM (right) to be classified as exhausted or non-exhausted. Each line corresponds to a single cluster. The diagonal black line corresponds to an AUC = 0.5, where there is no predictive power of classification. AUC = 0, the cluster can be perfectly classified as non-exhausted; AUC = 1, the cluster can be perfectly classified as exhausted.
Extended Data Fig. 2 Comparison of Scriabin to agglomerative CCC analysis techniques and validation with spatial transcriptomic data.
a) Runtime of Scriabin and five published CCC methods on the 10X PBMC 5k dataset. For each dataset size, the dataset was randomly subsampled to six different indicated sizes and the same subsampled dataset was used for all methods. b) Runtime of Scriabin and Connectome comparative workflows. The 10X PBMC 5k and 10k datasets were merged into a single dataset which was subsampled as in (a), and the comparative workflows performed between cells from the 5k vs. 10k dataset. Six different dataset sizes were compared. c) Jaccard index heatmaps depicting the degree of overlap in the top 1,000 ligand-receptor CCC edges from each method-resource pair for four datasets: 10X PBMC 5k, Fluidigm C1 pancreas islets21,33, Smart-seq2 uterine decidua13, and Smart-seq2 HNSCC8. d, e) The procedure described in Fig. 3a was repeated for 11 datasets, and the median distance quantile of a percentile of the most highly interacting cell-cell pairs was calculated using real cell distances relative to randomly permuted cell distances. d) Each facet shows the median distance quantile of the top 0.1%, 0.5%, 2.5%, 5%, 10%, and 20% most highly interacting cell-cell pairs. e) Each facet shows the median distance quantile of cell-cell pairs within the range of interaction quantile shown. In each facet, an exact two-sided p-value from the Wilcoxon rank-sum test is shown.
Extended Data Fig. 3 Flow cytometry and transcriptional analysis of B and NK cell transfection and co-culture.
a) Flow cytometry gating scheme used to identify B and NK cells. b) Scatter plots of flow cytometry data showing expression of CD40 and CD40L by B cells and NK cells. Left: B cells and NK cells transfected with GFP-encoding mRNA at start of co-culture. Middle: B cells transfected with CD40-encoding mRNA and NK cells transfected with CD40L-encoding mRNA at start of co-culture. Right: B cells transfected with CD40-encoding mRNA and NK cells transfected with CD40L-encoding mRNA at end of co-culture. For (a-b), percentages of the parent gate are shown for each gate. c) UMAP projections of full dataset colored by cell condition of origin (left) or annotated cell type (right). d) Dot plot depicting average and percent expression of exogenous mRNAs in the four co-cultures.
Extended Data Fig. 4 Analysis of CCC between CD40LG-transfected NK cells and CD40-transfected B cells with Connectome and NicheNet.
a) Circos plot summarizing Connectome’s38 results of significantly differentially-expressed ligand-receptor pair edges between the CD40LG-CD40 transfected condition (shades of red) and GFP-GFP transfected condition (shades of blue). CCC is analyzed between ligands expressed by sender NK cells (bottom) and receptors expressed by receiver B cells (top). b) Dot plot depicting percentage and average expression of differentially-expressed receptors by B cells (top) and ligands by NK cells (bottom) returned by Connectome’s DifferentialConnectome workflow. c) NicheNet20 was applied to predict ligand activities in B cells between the CD40LG-CD40 transfected condition and the GFP-GFP transfected condition. The bar plot depicts pearson coefficient outputs of NicheNet for this analysis. d) Dot plot depicting percentage and average expression of potentially-active ligands shown in (c) by NK cells.
Extended Data Fig. 5 Additional analyses of the scRNA-seq dataset of leprosy granulomas.
a) Bar graph depicting cell proportions per granuloma in the dataset of Ma, et al.41. Author-provided cell type annotations are used for analysis. b) Subclustering resolutions of T cells (left) and myeloid cells (right) required for comparative CCC analysis by agglomerative methods. Pink bars indicate the percentage of subclusters containing at least one cell from an LL granuloma and one cell from an RR granuloma. Blue bars indicate the percentage of subclusters containing at least one cell from all nine analyzed granulomas. c) NicheNet20 was applied to predict ligand activities in myeloid cells between RR granulomas relative to LL granulomas. The bar plot depicts pearson coefficient outputs of NicheNet for this analysis. d) UMAP projections of T cells (top) and myeloid cells (bottom) colored by author-generated subcluster cell type annotation (left), granuloma type (middle), or if the cell falls into a cluster 2 perturbed bin (right; see Fig. 4f). e) We applied a binomial test to determine if cells from a cluster 2 perturbed bin were significantly enriched or depleted in any T cell or myeloid cell subcluster. The bar plot depicts the -log(p-value) of the exact binomial test. When p < 0.05, the bars are colored to indicate if perturbed cells are either enriched (red) or depleted (blue) from the cluster. The dotted line indicates the point at which p = 0.05. Calculated p-values are two-sided.
Extended Data Fig. 6 Co-expressed interaction programs in intestinal development.
a) Scatter plot depicting expression of LEC marker LYVE1 and RSPO3. Shown are Pearson’s r, and an exact two-sided P value. b) UMAP projections of ligand (shades of purple) or receptor (shades of green) expression in 3 gut endothelial cell-specific modules. c) UMAP projection of gut endothelial cells colored by expression of ligands in the interaction programs depicted in (b). d) Dot plot depicting the expression fold-change and Bonferroni-corrected Wilcoxon rank-sum test 2-sided p-values of interaction program expression in each anatomical location. e) Intramodular connectivity scores for each ligand-receptor pair in each anatomical location for the module indicated by the arrow in (d). The black arrow in (e) indicates the genes whose average and percent expression are plotted to the right. Shown is an exact two-sided Bonferroni-corrected p-value from the Wilcoxon rank-sum test as described in panel (d). f-g) Connectome38 was used to analyze CCC in the human intestinal development dataset49 using author-annotated cell types for aggregation. Results are plotted for communication between gut endothelial cells (senders) and intestinal epithelial cells (receivers; f) or between fibroblasts (senders) and intestinal epithelial cells (receivers; g).
Extended Data Fig. 7 scRNA-seq dataset of SARS-CoV-2 infected HBECs.
UMAP projections of 64,008 cells from the dataset published by Ravindra, et al.58 colored by time point (a), annotated cell type (b), or the percentage of UMIs per cell of SARS-CoV-2 origin (c).
Extended Data Fig. 8 Parameter tuning for ligand activity ranking and interaction program discovery workflows.
a) Heatmaps depicting Jaccard overlap index between DE testing results from CCIMs constructed with 217 different combinations of ligand activity ranking parameters. Three different datasets were used for testing: a pancreas islet dataset21,33, a uterine decidua dataset13, and a dataset of HNSCC8. b-d) 217 different parameter combinations were used to analyze CCC between NK cells transfected with CD40L-encoding mRNA and B cells transfected with CD40-encoding mRNA. Ligand activity-weighted CCIMs were calculated from each of these combinations and differential expression testing performed to identify which parameter combinations returned CD40L-CD40 as a differential edge with the highest specificity. b) Box plot depicting the difference between the log(fold-change) for CD40L-CD40 and the mean log(fold-change) for all other ligand-receptor pairs, with and without application of ligand activity ranking. n = 1 for analysis without ligand activity ranking; n = 216 for with ligand activity ranking. c) β coefficients and p-values from multiple regression analysis modeling the impact of each ligand ranking parameter on relative predictive power for the CD40L-CD40 edge. d) Scatter plots depicting relative predictive power for the CD40L-CD40 edge for all combinations of ligand ranking parameters. The mean for each parameter is shown within the plot. e) Example ligand activity distributions to aid in selection of the appropriate Pearson coefficient threshold. Generally, ligand activity coefficients form a right-skewed distribution, similar to the distributions shown here. The right tails of these distributions represent the putative biological activity and are the coefficients that should be used for CCIM weighting. We therefore encourage users to consider the number of ligands that are expected to display biological activity and the number of cells that are expected to have downstream signaling induced by those ligands. If there are very few ligands expected to be biologically active, and only a subset of cells responding to them, this threshold should be increased to include less of the right tail of the distribution. f) The interaction program discovery workflow was repeated on 35 random subsamples of the inDrop panc8 dataset21,34, using 19 different R2 thresholds to define the appropriate softPower parameter. Scatter plots depict association between R2 threshold and (clockwise from top left): recommended softPower, percentage of identified programs that failed significance testing, percentage of programs composed of only 1 ligand or receptor, and the average number of ligands and receptors composing a program. Shown are Pearson’s r, and an exact two-sided P value.
Extended Data Fig. 9 Highly perturbed samples require a higher degree of aggregation for dataset alignment.
A toy dataset of peripheral blood monocytes from a longitudinal dataset was analyzed. a) UMAP projection colored by time point. b-d) UMAP projections (left) colored by cluster identity, and bar plot depicting per timepoint cluster membership in the cluster principally occupied by sample Week 04 (right). Cluster resolutions: 1 (default, b), 0.3 (c), 0.05 (d).
Extended Data Fig. 10 Robustness analysis of Scriabin’s binning workflow.
a-d) Mouse and human PBMC scRNA-seq datasets from 10X Genomics were analyzed. a) UMAP projections of mouse and human PBMCs colored by the sample of origin (left) and by manually-annotated cell types (right). b) Heatmap depicting overlap between bin identity and cell type annotations. Each row sums to 100%, and the annotations at left show the number of cells within each bin and maximum degree of overlap of each bin with a given cell type identity (ie. the highest value in each row). c) UMAP projection highlighting cells in bin #191. d) Bar plot depicting differentially-expressed genes in bin #191 relative to other B cells shared between the human and mouse cells in bin #191. Differential expression tests were run individually for human and mouse cells. e-g) A toy dataset of ~14,000 peripheral blood mononuclear cells (PBMCs) with nine sub-datasets was analyzed. e) Density plot depicting the number of cells in each bin. The median bin size in this analysis is 25 cells. f) As in (b) An SNN graph was used to assess cell-cell connectivity for the binning workflow. Cell type annotations are transferred from a reference dataset and are thus orthogonal to the data used to generate the bins. g) Dot plot depicting the cell type annotations and scores for the anchor pairs used to generate the bins depicted in (f).
Supplementary information
Supplementary Information
Supplementary Text and Supplementary Tables 1 and 2
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wilk, A.J., Shalek, A.K., Holmes, S. et al. Comparative analysis of cell–cell communication at single-cell resolution. Nat Biotechnol 42, 470–483 (2024). https://doi.org/10.1038/s41587-023-01782-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-023-01782-z
