
Abstract
Antibiotic treatments have detrimental effects on the microbiome and lead to antibiotic resistance. To develop a phage therapy against a diverse range of clinically relevant Escherichia coli, we screened a library of 162 wild-type (WT) phages, identifying eight phages with broad coverage of E. coli, complementary binding to bacterial surface receptors, and the capability to stably carry inserted cargo. Selected phages were engineered with tail fibers and CRISPR–Cas machinery to specifically target E. coli. We show that engineered phages target bacteria in biofilms, reduce the emergence of phage-tolerant E. coli and out-compete their ancestral WT phages in coculture experiments. A combination of the four most complementary bacteriophages, called SNIPR001, is well tolerated in both mouse models and minipigs and reduces E. coli load in the mouse gut better than its constituent components separately. SNIPR001 is in clinical development to selectively kill E. coli, which may cause fatal infections in hematological cancer patients.
Similar content being viewed by others

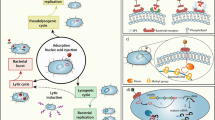

Main
Chemotherapeutic regimens used to treat hematological malignancies cause bone marrow suppression and gastrointestinal mucositis with associated increased intestinal permeability1,2,3,4. Translocation of gut bacteria, including Escherichia coli, from the gastrointestinal tract is a frequent cause of bloodstream infections5. The mortality related to bloodstream infections caused by enteric bacteria such as E. coli is 15–20%6; to decrease the chance of infection, antibiotics may be given before treatment in people at risk of low numbers of neutrophils in the blood7. Fluoroquinolones are used off-label in the United States, based on the results of two randomized trials demonstrating a decrease in bacterial infections in immunocompromised patients after use7,8,9. Fluoroquinolones have side effects, and their use in oncology patients has been accompanied by rising bacterial resistance10. In immunocompromised patients with hematological malignancies who develop chemotherapy-induced neutropenia, E. coli is responsible for 25.1–30% of all bacteremia cases11,12 and up to 65% of E. coli isolated as the causative pathogen from bloodstream infections in patients with hematological cancers undergoing hematopoietic stem cell transplantation were resistant to fluoroquinolones13. Other clinical options are needed that would prevent infections in these vulnerable patients, especially fluoroquinolone-resistant E. coli.
Bacteriophage therapy has been used before the broad availability of antibiotics14, but has now regained interest15 due to the rise in bacterial antimicrobial resistance combined with several successful individual case reports16,17,18. Still, few clinical trials with wild-type (WT) phages have been conducted19,20,21,22 and, although several have been directed toward E. coli, these have failed to produce convincing results in larger randomized controlled trials likely due to incomplete coverage of the target strains by the phage cocktail23. Recent efforts have used large-scale systematic screening of phages to broadly cover target strains, including characterization of phages (n = 41) targeting Klebsiella pneumoniae strains (n = 17)24 and phages (n = 248) targeting Vibrio strains (n = 294)25. T3 phage tail fibers have also been engineered to augmenting the spectrum of strains targeted by the engineered phage26. Finally, CRISPR–Cas systems can contribute to targeting efficacy as a complementary killing modality to the lytic activity of the phage. CRISPR–Cas complexes in some systems can bind to a homologous DNA target sequence and result in DNA degradation27,28. Because prokaryotes lack error-prone nonhomologous end-joining and rely on homologous recombination to repair DNA damage, they are prone to cell death following DNA degradation. This vulnerability has been exploited by using CRISPR–Cas as an antimicrobial modality for several bacteria, including Staphylococcus aureus, E. coli or Clostridioides difficile29,30,31,32,33,34,35.
To address the significant unmet medical need for new prophylactic agents for patients with hematological malignancies, we develop SNIPR001, which is a combination of four CRISPR–Cas-armed phages (CAPs) that specifically target a diverse spectrum of E. coli strains. Our research process for designing SNIPR001 includes several steps (Fig. 1). In short, a library (n = 162) of WT phages was tested in vitro on a panel of phylogenetically diverse E. coli strains representing the biology of the target bacterium E. coli. WT phages with the broadest and most complementary target strain coverage were selected for further engineering. Selected WT phages were subjected to both tail fiber engineering and CRISPR–Cas arming to create a library of CAPs. The CAP library was assessed for manufacturability, in vitro stability, spectrum of efficacy, in vivo pharmacokinetics and efficacy. A combination of four CAPs was selected to create the development candidate SNIPR001, which has now entered clinical development (ClinicalTrials.gov ID NCT05277350).

First, WT phages are screened against a panel of E. coli strains. Then, phages with broad activity against E. coli are tail fiber engineered and/or armed with CRISPR–Cas systems containing sequences specific to E. coli, creating CAPs. These CAPs are then tested for host range, in vivo efficacy and CMC specifications. SNIPR001 comprises four complementary CAPs and is a new precision antibiotic that selectively targets E. coli to prevent bacteremia in hematological cancer patients at risk of neutropenia.
Results
Phage screening
For the development of SNIPR001, we initially screened a library of 162 lytic phages derived from wastewater, phage banks and commercial phage cocktails (Supplementary Table 1). The host range and potency of the phages were assessed by a stringent in vitro growth kinetics assay against either an internal panel of 429 phylogenetically diverse E. coli strains, or an abbreviated panel of 82 E. coli strains (Fig. 2a), selected to adequately represent the full 429 strain panel. The E. coli strains originated from patients with bloodstream infections5 and urinary tract infections, from feces of humans with no known disease, and from the E. coli reference collection36. For a subset, we determined their receptors using efficiency of plating (EoP) assays on two broadly sensitive strains, their deep-core (∆rfaD) lipopolysaccharide (LPS) mutant derivatives and their surface protein knock-out mutants (Tsx, LamB, OmpC, OmpA, TolC and OmpF) from thereof (Supplementary Fig. 2). Based on the results, the eight phages α15, α17, α20, α31, α33, α46, α48 and α51 (all members of Tevenvirinae) were selected on their orthogonal and broad-spectrum effect, complementary binding to bacterial surface receptors, as well as engineerability to stably carry inserted cargo (Fig. 2b).

a, Heatmap showing the potency of 162 WT phages (horizontal axis) against 82 E. coli strains (vertical axis) based on growth kinetics. iAUC values are shown as a gradient of green. Phage genome size based on the sequencing data is shown in the top bar graph. For a selected group of phages, the cognate bacterial receptor protein was determined and shown in the bottom panel. The phage taxonomical classification based on the sequencing data is annotated in the bottom bar. The top tree shows the relationship between the phages based on their growth kinetics. The eight phages that were selected for engineering (α15, α17, α20, α31, α33, α46, α48 and α51) are highlighted by circles, four of those (α15, α20, α48 and α51) that form the basis of SNIPR001 are colored green. b, Overall development funnel of SNIPR001 starting with the 162 WT phages and, after engineering and selection assays, resulting in final cocktail of four CAPs in SNIPR001 with per CAP details described in Extended Data Table 1a.
Tail fiber engineering
We determined that α20 requires the presence of both LPS and maltoporin LamB, while the remaining selected phages were dependent on LPS or nucleoside transporter Tsx to infect their hosts (Fig. 2a). Given the dual receptor use of α20 and the conserved nature of the Tsx protein, we deemed the initial need for tail fiber engineering small for the majority of our phages. However, one of the broadest host-range phages on our test panels, α15, was solely dependent on LPS in propagation host E. coli b52 (Fig. 2a and Supplementary Fig. 2); and because LPS is extremely diverse and phage-resistant clones characterized by mutations in one of the many LPS biosynthesis genes can easily evolve during therapy37, we wanted to expand the receptor repertoire of α15. T-even phages bind to cell receptors using their long tail fibers or a monomeric adhesin that caps the distal tip of these trimeric fibers38. Thus, we chose a Tsx-binding adhesin from phage α17 and engineered it into α15.2 to consolidate both affinities in one phage. Virions of this phage are carrying stochastic combinations of two-receptor affinity, enabling them to infect bacterial cells via both receptors (Fig. 3a and Supplementary Fig. 2). We then hypothesized that α15.2 should select for a reduced number of resisters in comparison to the ancestor LPS-dependent WT α15. We selected clinical E. coli strains b1460, b1475 and b1813 where α15.2 outperformed WT α15 in the kinetic assays and subjected them to lawn kill assays. Indeed, α15.2 substantially led to a reduced number of survivors in comparison to WT α15 albeit with different levels per tested strain (Fig. 3b–d). Ten random purified colonies from WT α15 challenged group of each tested strain were as well tested for EoP with WT α15 and CAP α15.2. In accordance, results demonstrate a clear benefit of the tail fiber engineered α15.2 over LPS-dependent WT α15, as α15 survivors mostly retained sensitivity to CAP α15.2 despite being resistant WT α15 (Fig. 3b–d, insets).

a, EoP results of LPS-dependent WT α15, Tsx-dependent WT α17 and engineered CAP α15.2 that consolidates both WT phages’ receptors. Presented titers (PFU ml−1) were obtained from independent biological triplicates as dots, with averages illustrated as bars. b–d, Lawn kill assay results of E. coli are shown as boxplots, whiskers indicate maximum and minimum values, box bounds indicate 25th and 75th percentile, with center line indicating the median; b1460 (b), b1475 (c), b1813 (d) with phages WT α15 and CAP α15.2. Significances *P < 0.05 and ***P < 0.001, P values (two-sided Mann–Whitney U test) were calculated from two independent biological duplicates comprised of ten replicates. Holm’s method adjusted P values are 1.59 × 10−7, 3.36 × 10−2 and 1.6 × 10−7 for b1460, b1475 and b1813, respectively. Distribution of 10 EoP profiles of survivor colonies purified from WT α15 lawn kill assay (b–d insets). Resistant, no plaque formation with tested phage; sensitive, EoP similar to that of parental strain; reduced, EoP (>1–2 log10) lower than that of the parental strain.
CRISPR–Cas arming of phages to target E. coli
To CRISPR–Cas arm the selected lytic phages and generate a library of CAPs, the type I-E CRISPR–Cas system of E. coli39 was engineered (Supplementary Fig. 1) to target phylogenetically diverse E. coli strains. A CRISPR-guided vector (CGV-EcCas) was generated, containing the cas3 gene (ygcB) and a downstream cascade gene complex encoded by casA (ygcL, cas8e), casB (ygcK, cas11), casC (ygcJ, cas7), casD (ygcI, cas5) and casE (ygcH, cas6), and a CRISPR array targeting the E. coli genome (Fig. 1). To evaluate the killing efficiency of the CRISPR–Cas system, the CGV-EcCas was conjugated to E. coli strain b52, showing an average reduction of 3.5 log10 CFU ml−1, compared to the empty vector (Supplementary Fig. 3). As expected, no effect was observed after conjugating the CGV-EcCas to a nontarget E. coli strain (Supplementary Fig. 3). The killing efficiency of CGV-EcCas was further assessed on the abbreviated panel of 82 E. coli strains. Conjugative delivery of the empty vector was accomplished in 75% of the isolates (Fig. 4a). For all strains where the CGV-EcCas was delivered, bacterial counts were reduced below the limit of detection (LOD, 200 CFU ml−1) corresponding to a reduction of 1–6 log10, highlighting the potent CRISPR–Cas-mediated killing (Fig. 4a).

a, CRISPR–Cas-driven elimination of an abbreviated panel of 82 E. coli clinical isolates by conjugation of CGV-EcCAS (green) or empty vector (gray). The conjugation efficiency was determined by spotting a dilution series of the conjugation reaction on LB agar supplemented with antibiotics (n = 3 indicated by dots). The LOD was 200 CFU ml−1. b, Reduction of the metabolic activity of biofilms by CGVs that differed only in the promoter driving the expression of the CRISPR–Cas system, which are targeting the bacterial chromosome. Experiments were carried out in triplicate, measuring the relative metabolic activity between expression without a promoter and a given promoter. Average relative activities for PrelB and PbolA were significantly different and illustrated as dots, with averages illustrated as bars (two-sided Student’s t-test, P = 0.0052) 83.7 ± 6.7%, and 45.3 ± 2.5%. c–f, RT-qPCR showing increasing levels of cas3 transcripts relative to housekeeping gene gapA transcripts (replicates as dots, averages as bars) in negative correlation with decreasing number of unabsorbed phages over time in a synchronized infection (replicates as crosses, averages as lines). Cas3 activity is measured as ratio of cas3 transcripts relative to gapA transcripts, and number of unabsorbed phages in PFU ml−1. The results shown are the average of two independent biological replicates with technical triplicates. Bars or lines, respectively, indicate average values of these replicates, with error bars indicating standard deviation. g, Fraction of CAP and WT phage during coculture with a host strain susceptible to both phages. CAP α15.2 increases its relative abundance compared to the WT phage from 7% to 86% over two consecutive passages. h, CAP α20.4 outcompeted WT α20 by increasing its relative abundance during coculture with the common target E. coli strain b230 from 10% to 68% over four consecutive passages. g,h, The ratio of CAP and WT phage during coculture with a host strain (E. coli b230) susceptible to α15.2, α15, α20.4 and α20. CAP α15.2 increase its relative abundance compared to the WT phage from 7% to 86% (g) while CAP α 20.4 outcompeted WT α20WT from 10% to 68% (h).
We aimed to engineer our CRISPR–Cas systems to be functional under restricted bacterial growth conditions, which have been observed in the gut or in biofilms40. We tested two relevant promoters (PrelB41 and PbolA42) for their performance, both in planktonic cells grown in standard growth conditions (lysogeny broth (LB), 37 °C) and in biofilms, grown on peg lids in 96-well plates. Significant killing, measured as reduction of metabolic activity, was observed in E. coli biofilms when the CRISPR–Cas system was expressed from PbolA compared to PrelB (Fig. 4b). As promoter PbolA showed the best overall performance in the different conditions, it was chosen for transcription of the CRISPR–Cas system in the CAPs.
The eight selected WT phages were CRISPR–Cas-armed to generate 15 CAP variations (Extended Data Table 1a). In addition to promoter PbolA, the CRISPR–Cas systems were engineered to express from a synthetic constitutively expressed E. coli promoter (PJ23100) to further strengthen the CRISPR–Cas expression (Supplementary Fig. 1). CRISPR arrays were designed to target multiple virulence (spacers 1, 2 and 3) or essential genes (spacers 4 and 5; Extended Data Table 1b), as targeting multiple regions has been shown to prevent resistance evolution43. To confirm the CRISPR–Cas activity in the CAPs, we measured the cas3 transcripts in samples obtained at 5, 15 and 30 min following a synchronized infection with the equal multiplicity of infection (MOI) of CAP α15.2 in comparison to WT α15 using RT-qPCR and observed increasing levels of cas3 RNA only upon CAP α15.2 infection (Supplementary Fig. 4). Next, we extended this assay to all four CAPs (α15.2, α20.4, α48.4 and α51.5) and demonstrated increasing levels of cas3 transcripts highlighting that the CAPs expressed the CRISPR–Cas system during infection of a target strain (Fig. 4c–f).
To demonstrate the competitive superiority of the CAPs, we performed competition experiments in which CAPs (α20.4 and α15.2) and their WT ancestral phages were cocultured with E. coli strain b230, serving as a target for both competing phages. Approximate initial ratios of 1 CAP to 9 WT phages were cocultured and passaged four times on fresh target cells in liquid cultures. After each passage, the relative abundance of CAP and WT phage particles was evaluated. Both CAPs outcompeted their WTs within four rounds; CAP α20.4 reached 68% after four rounds and CAP α15.2 reached 86% after two rounds (Fig. 4g–h), demonstrating an improved fitness compared to the WT phages.
Selection and characterization of the optimal CAP cocktail
The activity of the 15 CAPs was tested against the E. coli panel (n = 429) using the growth kinetics assay (Supplementary Fig. 5). The individual CAPs showed activity toward 4.1–29.4% of the strains tested. To maximize our coverage, we sought to rationally combine CAPs with a broad and complementary spectrum of activity. Thus, we made subsets of CAP cocktails based on our in silico predictions using individual performances and tested their combinatorial in vitro performance. These results showed good compliance with our predictions (Supplementary Fig. 6). The initial 15 CAPs could be classified into four clusters based on their host-range profiles (Supplementary Fig. 5). We then excluded the seven lowest-ranking CAPs based on their redundant host-range in our cocktail predictions (Supplementary Fig. 7). Thus, eight CAPs (α15.2, α15.4, α17.2, α20.4, α46.4, α48.4, α51.5 and α51.6) were chosen for further assessments. First, all eight CAPs were individually orally dosed to mice (n = 3) and their normalized recovery (Supplementary Fig. 8) showed that all CAPs could be retrieved from fecal matter. Next, in vitro stability was assessed at accelerated conditions (40 °C, n = 3). Based on these results, two CAPs (α15.4, α17.2) were deselected as their titer dropped to below 1% of the starting material (Supplementary Fig. 9). The resulting six CAPs were individually tested (n = 6) in a mouse efficacy model (Supplementary Fig. 10), these results were combined with the predicted host range of the simulated cocktails (Supplementary Fig. 11; n = 15) and verification of complementing use of surface receptors for infection, resulting in the selection of CAPs α15.2, α20.4, α48.4 and α51.5 as the optimal CAPs for SNIPR001.
The ancestors of CAPs α15.2, α20.4, α48.4 and α51.5 are classified under the Tevenvirinae subfamily. Specifically, α15, α48 and α51 share 96.4%, 96.6% and 96.1% sequence similarity to E. coli phage T2, respectively, whereas α20’s closest relative is E. coli phage RB69 (96.8%; Supplementary Fig. 12). In silico analyses of the genomes of SNIPR001 showed that the CAPs encode no known transposase or integrase genes, indicating that the phages are not temperate, and thus not predicted to be capable of inserting their DNA in bacterial cells. In addition, we observed no antimicrobial resistance markers or virulence genes in the phage genomes (Supplementary Table 2). We investigated whether SNIPR001 CAPs cause generalized transduction and found no evidence of transduction with the LOD of 2 × 10−7 for frequency of transduction (Supplementary Table 3).
Developing a drug product from individual CAPs
Manufacturing a stable drug product comprised of four engineered phage particles requires establishing a phage and bacterial host collection, creating a Bacterial Master Cell Bank and a Master Phage Seed and turning the four resulting individual drug substances into a final SNIPR001 drug product (Supplementary Fig. 13). An important aspect of the chemistry, manufacturing and control (CMC) process is maintaining the stability of the individual components over time. We measured the titer of the individual CAPs at the stage of drug substances and found no indication of stability issues over 5 months of storage (Supplementary Fig. 14). To confirm the presence of the engineered phage parts during the CMC process, we established test criteria (Supplementary Table 2) based on whole genome sequencing of the samples. All four CAPs passed the acceptance tests, validating the presence of the CRISPR–Cas system and overall sequence identity to the CAP references (Supplementary Table 2). The final release testing criteria for the drug substances are listed in Supplementary Table 4.
SNIPR001 does not affect other gut-associated bacteria
Ideally, a phage-based therapy should not disturb the nontargeted genera of the microbiome, thus the specificity of SNIPR001 toward E. coli was assessed by investigating its effects on a panel of strains, which includes non-E. coli species that are E. coli relatives, as well as a range of families associated with the commensal bacterial community in the gut bacteria (and E. coli as a positive control). The bacteria were cultured without CAPs, with the SNIPR001 cocktail or with individual SNIPR001 CAPs (n = 4). The growth in CFU ml−1 was evaluated over a 4-h period (ΔCFU ml−14 h–0 h). In parallel, E. coli b2480 was grown under the same conditions as a positive control (Supplementary Fig. 15). We observed no significant effect (P > 0.05, two-sided Student’s t-test, FDR corrected with Holm’s method) of the SNIPR001 cocktail or any of the SNIPR001 CAPs on non-E. coli strains, while the growth of E. coli was significantly inhibited (P < 0.05, two-sided Student’s t-test, FDR corrected with Holm’s method). Thus, SNIPR001 is not expected to impact the gut microbiome beyond the target E. coli.
SNIPR001 in vitro host-range in clinical target population
To understand the potential effect in strains relevant to hematological cancer patients, the coverage of SNIPR001 was tested against our internal E. coli panel (429 strains) and a set of 382 clinical E. coli strains (JMI Laboratories). These JMI strains originated from patients with bloodstream infections hospitalized in hemato-oncology units across four different regions from 2018–2020 (Asia-Pacific 54 isolates, Europe 161 isolates, Latin America 26 isolates and North America 141 isolates; Supplementary Fig. 16). The genotypic distribution of E. coli strains in the patient population was determined using whole genome sequencing and was found to be diverse, representing nine phylogroups and 118 multilocus sequence types (MLSTs; Fig. 5a and Supplementary Fig. 17). We recorded phage infectivity against the E. coli panel using a spotting assays. Visible single plaques were differentiated from lysis zones in cases where single plaques could not be verified. All spotting assays were run in duplicates. We observed overall coverages of 90.4 ± 1.6% of SNIPR001 in the 382 JMI E. coli panel, and of 95.6 ± 0.3% of SNIPR001 on the internal E. coli panel (429 strains). Furthermore, we observed plaques in 53.1 ± 7.7% and lysis zones in 37.3 ± 6.1% of the JMI panel strains, and similarly, plaques in 60.1 ± 6.6% and lysis zones in 35.4 ± 6.3% of the internal panel strains (Fig. 5b). SNIPR001 showed 100% coverage in the B2 phylogroup, representing 53% of the JMI panel. This phylogroup is correlated with multidrug resistance and virulence. Additionally, we observed that SNIPR001 covered 91.7% (n = 55) of strains classified as multidrug resistant (MDR), 100% (n = 5) of carbapenem-resistant strains, 92.2% (n = 95) of extended-spectrum β-lactamases producing strains and 88.9% (n = 176) of strains that are resistant to fluoroquinolones, such as ciprofloxacin and levofloxacin (Fig. 5c).

a, An unrooted phylogenetic tree of the JMI strains displaying a clinical panel of 382 E. coli strains encompassing nine phylogroups and 118 MLSTs. Plaquing data reflects a single plaquing replicate. One strain, E. coli b4038, with a long branch (indicated by a break) has been truncated to 37% of the original length. Phylogenetic distance scale indicated below the phylogenetic tree, as computed by MASH. b, A spot assay was used to analyze the efficacy of SNIPR001 against the clinical panel of 382 E. coli strains (from JMI Laboratories) isolated from bloodstream infections and the internal 429 E. coli strain panel. The spot assay was conducted as two independent experiments, with bars indicating average cumulative panel fraction and dots indicating the results of each duplicate relative to prior means. c, Coverage of SNIPR001 does not depend on antibiotic-resistant phenotypes; consequently, SNIPR001 targets > 90% of E. coli strains that are carbapenem resistant, ESBL-producing or MDR, and 89% of fluoroquinolone-resistant E. coli strains. Numbers indicated in each green or gray bar indicate the number of bacteria susceptible or resistant to SNIPR001, respectively, for each resistance category generated from a screening of 382 strains and subset to the number of strains with a given resistance. d, A midpoint-rooted phylogenetic tree of the 72 fluoroquinolone-resistant E. coli strains isolated from fecal samples of hematological cancer patients. A total of 67 of the 72 strains are susceptible to at least one of the four CAPS in SNIPR001. Plaquing results are generated by the conservative consensus between two runs of plaquing, that is displaying the outcome with lower plaquing efficiency. e, Redundancy distribution showing 82% of the fluoroquinolone-resistant E. coli strains (n = 72) from d are targeted by at least two different CAPs.
Finally, we validated SNIPR001 on a clinical panel (n = 72) of fluoroquinolone-resistant E. coli strains that were isolated from either a fecal sample or a perianal swab from hematological cancer patients. This population represents the expected clinical target patient population being pursued (SNIPR001 has been designated fast-track status by the FDA). A subset of these strains gave rise to bloodstream infection (Fig. 5d). 82% of the E. coli strains (n = 72) were susceptible to at least two or more of the CAPs in SNIPR001, and 93% of the strains were susceptible to the whole SNIPR001 cocktail (Fig. 5e). These data demonstrate the benefit of SNIPR001 compared to the individual CAPs with regards to improving the spectrum of efficacy.
Tolerability and recovery of SNIPR001 in minipigs
The tolerability and gastrointestinal recovery of SNIPR001 were evaluated in female Göttingen minipigs. Blood and feces were sampled over 7 d following oral administration of 2 × 1012 PFU of SNIPR001 or vehicle. No CAPs were recovered from plasma, indicating no systemic exposure, while CAPs were recovered in the feces up to 7 d after SNIPR001 administration with a peak of 2 × 107 PFU 24 h postdosing (Fig. 6a). The minipigs exhibited no clinical signs and no significant changes were observed in hematology or biochemistry parameters, in particular, no changes were seen in any immune cells (Supplementary Fig. 18), compared to vehicle treatment, supporting that SNIPR001 was well tolerated (Supplementary Figs. 19, 22–25, and Supplementary Table 5). Similar recoveries were obtained with the individual CAPs (Fig. 6b). In conclusion, SNIPR001 appears to be well tolerated in Göttingen minipigs with gastrointestinal recovery.

a, CAP recovery in minipigs feces after a single p.o. dose of 2 × 1012 PFU of SNIPR001 (n = 8, green) or vehicle (n = 6, gray) over 1 week with daily sampling. Trend lines indicate average recovered phage in PFU per gram feces, dots indicate individual measurement points. LOD of 33 PFU g−1 feces indicated by the dotted line. b, CAP recovery in minipig feces after a single p.o. dose of 2 × 1012 PFU of a single CAP (n = 8 minipigs received either α15.2, α20.4 or α51.5; n = 7 minipigs received α48.4) over 1 week with daily sampling. Trend lines indicate average recovery, while points indicate individual measurements. Recovery was measured in PFU per gram feces. LOD of 33 PFU g−1 feces (dotted line). c, CAP recovery in mouse feces 8 h, 24 h and 48 h after the start of treatment with three times daily administration of varying doses of SNIPR001 (n = 10 for low, medium and high, green), vehicle (n = 10, gray), or gentamicin (n = 4, gray). Recovery is measured in PFU g−1 feces, LOD of 371 PFU g−1 feces (dotted line). d, E. coli b17 recovery in mouse feces indicates increased SNIPR001 effect with increased dose; color legend and group sizes are the same as in c. *P < 0.05, **P < 0.01, ***P < 0.001; statistical analyses were performed using two-sided Kruskal–Wallis tests for comparison of all SNIPR001-treated groups, two-sided Mann–Whitney U test was used for comparison of treated groups with vehicle corrected using Holm’s method separately for each day. The exact P values are shown in Extended Data Table 5. Recovery is measured in CFU per gram feces, with a LOD of 371 CFU g−1 feces. Animals that have begun SNIPR001 treatment are indicated in green, others in gray. e, E. coli b17 recovery in mouse feces 8 h and 24 h after the start of treatment with three times daily administration of CAPs α15.2, α20.4, α48.4 or α51.5 (n = 6 for each CAP) and in combination as SNIPR001 (n = 6) confirming synergy of the CAPs, as well as vehicle (n = 6), and gentamicin (n = 3). Differences in CFU per gram tested by a two-sided Mann–Whitney U test, P values corrected with Holm’s method. Adjusted P values for comparisons of vehicle and SNIPR001 are both 0.022 for days 2 and 3.
Efficacy in a mouse colonization model
To assess the in vivo efficacy of the four selected CAPs in reducing E. coli, we adapted a mouse gut colonization model from ref. 44 for E. coli strain b17 (Supplementary Fig. 20). Streptomycin was administered for 3 d to reduce Gram-negative bacteria from the mouse gastrointestinal tract, after which streptomycin administration was stopped and animals were inoculated once perorally with E. coli b17 (1 × 107 CFU). This allowed stable colonization for 3–4 d. Aiming at assessing the efficacy of CAPs on established colonization, treatment was started 2 d after inoculation and the study was terminated on day 4 after inoculation, as the colonization starts to drop. To ensure maximum exposure to CAPs, mice were treated with three daily doses, administered 8 h apart, for a total of six doses over 2 d.
Mice were treated by oral gavage with a high, medium or low dose (2 × 1011 PFU, 2 × 109 PFU and 1 × 107 PFU, respectively) of SNIPR001, vehicle (negative control) or gentamicin (positive control). CAP recovery in the feces ranged from 3 × 107 PFU g−1 in the low dose to 1 × 1010 PFU g−1 in the high dose, confirming successful GI passage (Fig. 6c). These levels of CAPs were associated with a significant (P < 0.05, two-sided Mann–Whitney U test, FDR corrected) dose-dependent reduction in the target E. coli population compared to vehicle treated mice, after 24 h of treatment (day 3). At the high dose, SNIPR001 led to a 4 log10 CFU g−1 reduction (Fig. 6d). Despite an increased variability in bacterial recovery on day 4, possibly due to clearance of the colonizing strain as illustrated in the vehicle group, similar reductions were observed after 2 d of treatment (day 4). While the medium dose did not reach statistical significance (P < 0.05, two-sided Mann–Whitney U test), there was nevertheless a numerical reduction in comparison to the vehicle group. Subsequently, the efficacies of the individual CAPs were compared to the SNIPR001 cocktail in this model. In this experiment, a greater reduction in the colonization of the target strain was observed with SNIPR001 compared to any single CAP (which showed a numerical, but not statistically significant reduction) highlighting a benefit in efficacy from the combination (Fig. 6e). We also assayed the resistance profile of randomly sampled surviving bacteria and found no isolates that were resistant to the SNIPR001 cocktail. We did identify one isolate from one animal which was resistant to three of the four phages of the cocktail (Supplementary Fig. 21). Overall, these data demonstrate the ability of SNIPR001 to decrease the target E. coli in the GI tract of colonized mice.
Discussion
Here we describe the development of SNIPR001 designed to target gut E. coli that frequently translocate in the bloodstream to cause bloodstream infections in patients with hematological cancers who are neutropenic. While fluoroquinolones are being used off-label, these patients continue to have high morbidity and mortality. The use of traditional antibiotics has led to significant bacterial resistance development, and the number of deaths attributable to bacterial antimicrobial resistance in 2019 has been estimated to be 1.27 million, with E. coli being the leading pathogen45. In this study, we describe the development of SNIPR001, a combination of engineered phages with the potential to address challenges related to antibiotic resistance in immunocompromised patients.
SNIPR001 combines state-of-the-art phage screening, with phage tail fiber engineering and CRISPR–Cas arming. Traditionally, phage therapy has been used experimentally with limited characterization and often applied in a highly individualized way because of the often narrow host range of individual phages46. Building on recent advances in phage engineering that have enabled the manipulation of virulent phages47 and the ability to engineer tail fibers26 and CRISPR–Cas arm the phages, we enhanced the potency of the phages comprising SNIPR001 to enable it to target a broader range of clinically relevant E. coli, including strains that are resistant to current therapies.
To deliver a development candidate ready for clinical testing, we established a traceable manufacturing process resulting in stable CAP substances, and final confirmation of the efficacy of SNIPR001 on large and clinically relevant strain panels supports the clinical potential of the SNIPR001 cocktail. The observed 4 log10 reduction of E. coli in our in vivo model is a clear improvement over the previous studies35,48.
SNIPR001 is an orthogonal antimicrobial approach as it has shown activity in MDR strains. In addition, there is emerging evidence that maintaining a normal microbiome is important for upholding immunological tonus and potentially benefiting the outcome of oncology treatments49, and this has also been recognized in the most recent guidance on prophylactic management of patients at risk of febrile neutropenia7. In this context, in vitro studies with SNIPR001 have shown specificity toward E. coli with no off-target effects toward any of the tested non-E. coli strains, thereby having a less detrimental effect on the microbiome. In the future, individualized combinations of narrow-spectrum antibiotics such as SNIPR001 may be used first-line rather than use in addition to broad-spectrum antibiotics such as fluoroquinolones.
As with any nonclinical study, the translatability of the in vitro and preclinical findings into humans requires investigation, in particular for MDR strains. Although we did not observe structural resistance toward SNIPR001 in mice, resistance development, and the synergy that a combination of CAPs provides, are challenging to study in vivo with a complex drug product like SNIPR001. Furthermore, part of the activity spectrum of SNIPR001 is driven by lysis zone formation and not plaquing, and it is to be investigated how this phenotype translates into clinical efficacy. Therefore, a clinical study to evaluate the ability of SNIPR001 to ascertain safety and its ability to reduce E. coli in the gut without perturbing the overall gut microbiome is currently ongoing in the United States (NCT05277350). SNIPR001 exemplifies a potentially significant therapeutic advance in the field of antimicrobials for high-risk patient populations and can serve as a blueprint for narrow-spectrum therapies for other life-threatening antimicrobial-resistant pathogens in high-risk patient populations.
Methods
Phage collection and isolation procedures
The starting point for the phage screening was a collection of 162 lytic WT phages, 82 were isolated in-house from commercial cocktails and environmental sources, 71 phages were obtained from a phage bank (LyseNTech, Korea) and two phages from ATCC, one phage was donated by the University of Copenhagen and six were obtained from Kirikkale University, Turkey50 (Supplementary Table 1). Phage isolation was carried out by using E. coli strain panels (see E. coli panels and isolation procedures). In brief, 100 μl of overnight cultures of each E. coli strain were mixed with 100 μl of each phage cocktail or wastewater sample. Following 6 min incubation at room temperature (in this period infection should occur), 3 ml of prewarmed top agar containing Ca2+ were added to the E. coli/phage or wastewater mixtures and poured immediately on an LB plate. Alternatively, tenfold dilutions of each cocktail were spotted on lawns prepared with isolation strains. After drying, plates were incubated at 37 °C overnight. Plaques were picked from each plate and resuspended in 500 μl of SM buffer, vortexed and stored at 4 °C. Tenfold dilutions were spotted on the isolation strain which the plaque was originally picked from. To increase the likelihood of obtaining plaques corresponding to single phages, the procedure was repeated at least three times. Lysates were prepared from single plaques picked at the previous round of propagation, DNA was extracted and their genomes were sequenced.
E. coli panels and isolation procedures
Three E. coli panels, one internal SNIPR Biome panel and two clinically relevant panels were included in this study. The internal panel consists of 429 phylogenetically diverse E. coli strains, isolated from the blood of patients with bloodstream infections and urinary tract infections, from feces of humans with no known disease, animals and the environment. The strains cover seven different phylogroups (A, B1, B2, C, D, E and F), 114 MLST groups, serotypes (K- and O-type), antibiotic resistance profiles and different geographical locations of isolation.
The JMI panel comprises 382 strains E. coli clinical collection obtained from JMI Laboratories. These strains were isolated from patients with bloodstream infections hospitalized in hematology and oncology units across four different regions (Asia-Pacific 54 isolates, Europe 161 isolates, Latin America 26 isolates and North America 141 isolates), sourced through the SENTRY Antimicrobial Surveillance Program (2018–2020), which is composed of a network of more than 150 medical centers in more than 28 countries worldwide (https://www.jmilabs.com/sentry-surveillance-program).
Finally, the panel comprising 72 fluoroquinolone-resistant E. coli strains is isolated from either fecal samples or perianal swabs of hematological cancer patients hospitalized for hematopoietic cell transplantation51,52.
E. coli strains were cultivated at 37 °C in LB at 250 rpm in liquid media or on agar plates containing 1.5% (wt/vol) agar. When necessary, cultures were supplemented with ampicillin (100 μg ml−1), kanamycin (50 μg ml−1), gentamicin (15 μg ml−1) or amikacin (50 μg ml−1). All media for the growth of conjugation donor E. coli JKE201 (ref. 53) and its derivatives were supplemented with 1,6-diaminopimelic acid (80 μg ml−1) to complement their auxotrophy.
Both E. coli strain b52, which was used to produce α15.2, α48.4 and α51.5, and E. coli strain b2479, which was selected to produce α20.4, belong to phylogroup A. Strain E. coli b17 was used as colonizing strain in the in vivo efficacy models as the strain is susceptible to all SNIPR001 CAPs and is part of the SNIPR Biome strain bank.
Phage screening by growth kinetics
In vitro susceptibility of the internal E. coli panel (n = 429) to the 162 WT phages was evaluated using a growth kinetics assay. The assay measures the metabolic activity of a bacteria by tracking the reduction of a tetrazolium dye to a purple compound that aggregates during bacterial growth. The colorimetric reading was recorded every 15 min over a 24-h period by using the OmniLog (Biolog)—adapted from ref. 54. The inhibitory area under the curve (iAUC) was calculated from the kinetic curves over the course of the experiment and was defined as the ratio between the normalized AUC of the phage-treated bacterial growth curve and the bacteria-only control. Susceptibility was defined at iAUC values ≥0.2. Prescreening, including 48 phages, was carried out at MOI 10, after which 114 phages were screened at MOI 1.
Calculation of bacterial growth inhibition using iAUC
The growth inhibitory effect of SNIPR001 was determined using growth kinetic curves constructed using the OmniLog apparatus. To limit technical variability in measurement between timepoints, a cubic smoothing spline function was applied to the data in Scala using the ‘umontreal.ssj.functionfit’ package. To identify appropriate ρ and weight variables, every combination of ρ and weight 0.1 and 0.5 was applied in 0.1 increments (that is, 0.1, 0.2, … 0.5). The spline with the lowest mean absolute error was chosen for area under the curve (AUC) calculation. The initial cumulative amount of fluorescent dye at the initial timepoint varies slightly from well to well, leading to artificial inflation of the AUC of certain wells. Using the best smoothed square spline, the mean signal for the first 1.5 h, before any measurable growth, was removed from all growth curves to approximate a zero-growth signal intercept. The total iAUC was calculated as the sum of the Riemann midpoint sums for each timepoint along the smoothed square spline. Lastly, we calculated the iAUC as iAUC = 1 − AUCSample/AUCControl, where AUCSample is the AUC of the spline created by a given bacteria and SNIPR001, while AUCControl refers to the AUC of the spline created with a given bacteria without a given phage or CAP, or a combination of those. Thus, iAUC values usually lie between 0 and 1, where 0 indicates no growth inhibition and 1 indicates complete growth inhibition. Some biological and technical noise does result in iAUC values outside these bounds on occasion but is considered negligible.
Host range was calculated as the fraction of a panel that had an iAUC < 0.2 for each repeat. Reported standard deviations were calculated as the deviance in the number of strains with an iAUC < 0.2, and then normalized to the size of the panel, by dividing the s.d. with the size of the panel.
Combination complementarity prediction
Phage and CAP complementarity were evaluated in silico under the assumption of complementarity—if at least one CAP in a combination of phages can strongly inhibit a given bacterial strain, the combination of CAPs is assumed to strongly inhibit that bacterial strain. In in vitro studies, the total host range was estimated by calculating the fraction of a panel that was inhibited by one or more of the members of a given CAP or phage combination. In OmniLog screenings, a strain was considered inhibited if the iAUC of phage was above 0.2 compared to control. When using plaquing results, a strain was considered inhibited if a plaque or lysis zone was observed.
In in vivo studies, the effect of CAP combinations was considered complementary, and the efficacy of individual CAPs was assessed as the log10-transformed difference in CFU per gram between a vehicle and a given CAP. The predicted effect of a combination was thus evaluated as the sum of these log10 reductions for each member of a combination.
In silico marginal host-range calculation
To get an overview of the ability of a CAP to participate in an efficacious CAP combination, we evaluate the marginal host ranges for each CAP. The marginal host range is a measure of the gained host range when a given CAP is incorporated in a combination. This is calculated as the difference in host range between a combination with and without a given CAP of interest. By calculating the marginal host ranges of each combination for each CAP, we can compare the different CAPs with regard to their utility in adding host ranges. However, the composition of the CAP panel can lead to unfair scoring—the addition of a CAP to a combination, where one of the composing CAPs has a very similar inhibitory profile, would have an unfairly low marginal host range. Similarly, if a CAP is added to a combination of CAPs that all have very similar inhibitory profiles, the marginal utility gain would be unfairly high. If the set of CAPs being screened does not equally represent different types of inhibitory profiles, some CAPs will have misleading marginal host-range distributions. To avoid this issue, we do not generate combinations of CAPs that contain multiple CAPs that originate from the same WT phage.
To identify CAPs whose marginal host range tended to be good, we used the mode to differentiate the CAPs. The mode of the distribution for each phage was used to calculate the overall utility of phage using the density() function in R v. 4.1.0.
Engineering phages with a CRISPR-Cas system
Phages were CRISPR–Cas armed by using homologous recombination. We inserted the payload in the region between the pin (encoding the inhibitor of host Lon protease) and vs.7 (encoding a conserved hypothetical protein) gene. Recombination was carried out in bacterial cells during phage propagation. Cells carried a plasmid that served as a recombination template. Recombination template plasmids carried the sequences that were aimed to be inserted into the phage genome between ~200 bp and 700 bp flanking sequences that were homologous to the phage sequences at the insertion site. For each phage, we inserted the type I-E CRISPR–Cas system endogenous to E. coli (Genbank CP032679.1), that is, the cas3 gene (ygcB) and the downstream genes encoding the cascade complex, casA (ygcL), casB (ygcK), casC (ygcJ), casD (ygcI) and casE (ygcH), as well as a CRISPR array targeting selected E. coli sequences. For all CAPs selected, the cas genes originating from E. coli are identical. Insertion of the CRISPR–Cas system resulted in the deletion of ~7 kbp deletion of phage DNA in the pin - vs.7. The sequences of the resulting CAPs were verified by NGS (BaseClear).
Transduction of CGVs in biofilms
E. coli b52 cells were grown in 96-well plates, and biofilms were allowed to develop on peg lids. Each well contained 180 µl M9 medium (Sigma-Aldrich, M6030) supplemented with 20 mM glucose, 2 mM MgSO4, 0.1 mM CaCl2, 0.1% Amicase (Sigma-Aldrich) and 0.1% mannitol. Wells were inoculated with 1 µl of overnight b52 culture. The peg lid was inserted, and the microtiter plate was incubated statically for 24 h at 37 °C. Next, the peg lid was transferred to a new plate with fresh media without washing, and the plate was incubated for an additional 24 h. After incubation, a new plate was prepared with 100 µl media and 100 µl of CGV transducing particles (~108 particles) in each well (three replicates). Biofilms grown on the pegs were rinsed three times in sterile H2O (200 µl) before transferring them on the new plate. The plate was incubated statically for 5 h at 37 °C.
To assay the metabolic activity of cells in the biofilms, lids were rinsed three times in sterile H2O (200 µl) before placing them in a plate with 20 µl Alamarblue stain (Thermo Fisher Scientific) and 180 µl media in each well. Plates were incubated for 1.5 h at 37 °C and moved to a microplate reader (Synergy H1, Biotek). Fluorescence (excitation, 560 nm; emission, 590 nm) and absorbance (600 nm) were recorded for each well.
The metabolic activities of the biofilms treated with CGVs carrying one of the promoters (PrelB or PbolA) were reported relative to the metabolic activities of biofilms treated with a CGV not carrying a promoter transcribing the cas genes.
Plasmid and strain construction
To construct CGV-EcCas, cas3 and cascade genes from E. coli were amplified and cloned into a ColE1-type plasmid, pZE21 (ref. 55), containing kanamycin, gentamycin and amikacin resistance markers, and oriT RP4.
DNA fragments encoding a 3-spacer array targeting genes in E. coli were synthesized as gBlock fragments (IDT) flanked by AarI restriction enzymes (gB149, gB150, gB152 and gB153; Supplementary Table 6). Similarly, constitutive promoter J23100 (ttgacggctagctcagtcctaggtacagtgctagc) was synthesized as a gBlock fragment (IDT) (gB-d2; Supplementary Table 6) to drive the expression of the CRISPR array. The array contains nucleotides from the genome of E. coli per target locus separated by direct repeats. The protospacer adjacent motif is located adjacent to the selected target sequences in the genome of E. coli.
cas3 and cascade genes from E. coli were amplified with primers containing AarI restriction sites (TH556 and TH558; Supplementary Table 6). Similarly, pM0 constitutive promoter to drive the expression of the cas genes (ggattaacaatataagctgaccttcaagtattgaat) was amplified with primers TH402 and TH403 (Supplementary Table 6). To combine cas3 and cascade genes with the CRISPR array, all plasmids were digested with BsaI and ligated with T4 DNA ligase. Finally, to generate CGV-EcCas, the CRISPR–Cas system was moved into conjugative plasmid pZE21 by InFusion HD cloning using primers TH712 to TH715 (Supplementary Table 6).
Transformation assays
Overnight cultures were diluted (1:100) in fresh LB medium and grown to mid-exponential phase (OD600 ≈ 0.6). Subsequently, cells were prepared for electroporation and concentrated 50-fold in ice-cold MilliQ water. Cells were then electroporated with appropriate plasmids, allowed to recover for 1 h at 37 °C in super optimal broth, and plated on LB plates supplemented with antibiotics.
Conjugation assays
Conjugation experiments assessing the transfer and killing efficiency of CGV-EcCas were established using E. coli JKE201 as the donor and E. coli clinical isolates as recipients (including target and nontarget and E. coli strains as controls). Plasmids were conjugated into E. coli recipients by liquid mating. Briefly, overnight cultures were diluted (1:100) in fresh LB medium, grown to OD600 ≈ 0.4, washed, and suspended in fresh LB to OD600 ≈ 0.25. 125 μl of donor and 25 μl of recipient cell suspensions were mixed for 5:1 mating in a 96-well microplate and incubated for 16 h at 37 °C. The conjugation efficiency was determined by plating a dilution series of conjugation reactions onto LB agar supplemented with antibiotics (to select for the transconjugants). The specific killing efficiency was quantified by plating 90 μl of the conjugation reactions on selective plates. The CGV-EcCas plasmid encodes kanamycin, gentamycin and amikacin resistance to enable selection for transconjugants. Viability was calculated by counting CFUs on the plates, and data were recorded as viable cell concentration (CFU ml−1).
Synchronized CAP infection and cas3 expression assay
An overnight culture of the test strain in LB was 100-fold diluted and incubated to stationary phase in LB at 37 °C with shaking, and 10-ml aliquots were subsequently separated into 50-ml falcon tubes. Each aliquot was then seeded with 50 µl of high-titer lysate of the individual CAPs, and incubation was continued under the same conditions. Additionally, a mock 10 ml LB volume for each CAPs was also seeded with 50 µl of CAP lysates and used for 0 min phage enumeration. At 5 min, 15 min and 30 min postseeding, aliquots were collected for total RNA extraction and phage enumeration. Phage enumeration aliquots were syringe filtered (0.2 µm, Sartorius AG) and subjected to an EoP assay. For total RNA extraction, 1 ml aliquots of individual cultures were centrifuged at 13.3kg using a table-top centrifuge for 15 s, and supernatants were discarded. Then, pellets were immediately resuspended in cold RNA Later (Thermo Fisher Scientific, AM7020) and stored at −20 °C until extraction. Total RNA was extracted using a GeneElute Total RNA kit (Sigma-Aldrich) following the manufacturer’s protocol for extraction of RNA from bacteria. After the first elution, 1 µl of Dnase I (1 U µl−1) was added and incubated overnight at 37 °C. The reaction was terminated by incubation at 70 °C for 15 min. The RNA was re-purified on a GeneElute column and eluted in 35 µl of kit elution buffer. Total RNA concentration was estimated on a NanoDrop instrument (Thermo Fisher Scientific, One/OneC), and 0.5–2 µg of RNA was added to a cDNA synthesis reaction containing SuperScriptIII RT enzyme (Thermo Fisher Scientific) and random decamers to prime synthesis in a 20-µl reaction volume. The cDNA reaction was diluted to 100 µl in water. RT-PCR was conducted in triplicate using 5 µl of cDNA as template, 10 µl of Power SYBR Green PCR Master mix (Thermo Fisher Scientific) and 0.2 µM of each PCR primer. PCRs were performed on an AB QuantStudio5 system (Applied Biosystems) using the standard two-step thermocycling protocol for Power SYBR Green PCR Master Mix with 60 °C annealing/extension. The forward and reverse primers for gapA (reference gene) were 5′-cgctaacttcgacaaatatgctggc-3′, and 5′-aggacgggatgatgttctgggaa-3′, and for cas3 were 5′-caagtatgctaccaacggctaaag-3′ and 5′- ccaatcaaaatcaacgtcgagtga-3′. Single PCR products were confirmed for these primer pairs by melting curve analysis. Relative levels of transcripts were estimated using tenfold dilutions of purified PCR products as standards, and values were expressed as the ratio of cas3 to gapA transcripts.
Phage competition assay
Lysates of the two phages were mixed at 9:1 (WT:CAP) ratio and the phage mixtures were added to 10 ml 2xYT medium containing 10 mM CaCl2 and 20 mM MgCl2, and 100 µl overnight of E. coli strain b230, serving as a target for both competing phages. After 2 h incubation in a 37 °C shaking incubator, the cultures were centrifuged and 1 µl of the supernatant was added to a new b230 culture. The same steps were repeated twice.
The ratio of phages was assessed by PCR with three primers, resulting in two specific products, one for the WT phage and one for the CAP (α15/15.2—5′-ttcattgcgtatttgtagatgaagctc-3′, 5′-cttttcagacttatcttgcgtttcttaagaagttctacaagttct-3′, 5′-gtacgactgattgatcccaccagc-3′; α20/20.4—5′-atggcttttattgctaccgggt-3′, 5′-aaatctagagcggttcagtactcaaggaaatcatcccagaaactc-3′, 5′-tgctatctttggctccactgtgat-3′). PCR products were separated on a 1% agarose gel and DNA bands were stained by SYBRsafe and visualized and quantified by the ChemiDoc XRS + System (model 1708265, Bio-Rad). The background-corrected intensity of the band corresponding to the WT phage was divided by the intensity of the band corresponding to the CAP in the same lane, to obtain the ratio of the two band intensities (WT/CAP). The fraction of CAP compared to the total phage content (WT + CAP) was determined based on the calibration curve, which was made by using a set of different mixtures of the two phages and fitting a curve to the measured band intensity ratios (WT/CAP). The estimated error of the reported values is less than 20%.
Lawn killing assay
An overnight culture of the test strain in LB was adjusted to 109 CFU ml−1. Hundred µl aliquots of CFU ml−1 adjusted strain was mixed with 100 µl of 109 PFU ml−1 to achieve a multiplicity of infection of 1 of either CAP α15.2 or WT α15 in 15 ml falcon tubes, mixed with 3 ml of molten and pretempered top agar and spread on LB plates. After lawns were solidified, plates were incubated at 37 °C overnight, and the total number of surviving colonies was counted for CAP α15.2 or WT α15 groups the next day. Assays were performed as independent biological duplicates where each experiment comprised ten technical replicates. Statistical significance was established using both replicates using a two-sided Mann–Whitney U test.
Generalized transduction assay
The transduction ability of each CAP was evaluated via the generalized transduction assay. Briefly, transducing lysates were prepared by propagating each CAP on E. coli MG1655 lamB::Cm. This strain was modified from the WT MG1655 (700926, American Type Culture Collection) to carry a chloramphenicol selection marker. Experiments were conducted in parallel with the well-characterized lytic T4 phage (negative control), and its transducing mutant T4GT7 (ref. 56; positive control). Following this step, the WT E. coli MG1655 strain was infected at an OD600 of 0.3 with each transducing lysate at MOI of 0.5, 0.1 and 0.01, and spread on LB plates containing chloramphenicol. Next day, the number of transductant colonies was recorded for each CAP and control and different MOIs. The frequency of transduction was calculated as the number of transductants divided by the titer of the transducing lysate.
Sequence analysis of CAPs
Sequences of the individual SNIPR001 CAPs were analyzed for the presence of antibiotic resistance, virulence genes and lysogeny associates genes (transposases and integrases) using databases (Extended Data Table 2). Furthermore, for release criteria during the CMC process (Supplementary Table 2), phage samples were analyzed using whole genome sequencing. This typically results in >1000× coverage of the whole phage genome. Assemblies are constructed by down-sampling the data to 1000× average coverage for the phage and assembling using SKESA. To detect differences between samples and to detect nonmajority mutations the raw reads were mapped back to the assembly using BWA (version 0.7.17).
Phage specificity assay using liquid killing assay
SNIPR001 CAPs (α15.2, α20.4, α48.4 and α51.5) and SNIPR001 killing specificity were evaluated via a biopotency assay against a panel of human-relevant, aerobic (n = 6) and anaerobic (n = 3) bacterial strains. An E. coli strain b2480 was included as a positive control for phage-mediated killing (Extended Data Table 3).
In brief, overnight cultures were adjusted to 106 CFU ml−1 in LB broth. SNIPR001 CAPs or SNIPR001 (in which each CAP was combined in equal ratio) were added at an MOI of 1 before incubation for 4 h. Untreated bacteria were cultured in parallel as controls for bacterial growth. CFU counts were recorded at 0 h and 4 h post phage treatment, and data are represented as Δlog10 CFU ml−1 by subtracting the initial inoculum (0 h) from the assay endpoint CFU per milliliter (4 h).
CMC
The in vitro stability of phages was assessed by following the potency of CAPs in the formulation buffer overtime at 2–8 °C and at accelerated temperature (40 °C). Polypropylene cryovials were filled with one milliliter of each phage for storage at the appropriate temperature. At each timepoint, the potency of each phage was assessed by EoP method in triplicates. T0 was measured before the initiation of storage.
Spotting assay and EoP
For counting of phage titers, phage lysates or the equal volume mix of SNIPR001 CAPs were serially diluted tenfold in SM buffer or PBS, respectively. Bacterial lawns were prepared by adding 100 µl or 300 µl of bacterial overnight culture to 3 ml or 10 ml of 0.5% top agar (containing Ca2+ and Mg2+), which was vortexed briefly and poured onto a round or square LB plate. Five microliters of the dilution series of test phages were then spotted on lawns and left to dry at room temperature with an open lid before incubation at 37 °C overnight. The strains b52, b2479 and b17 were used as controls of the assay and included in each round of assays.
The next day, results were assessed (Extended Data Table 4). In this assay, a susceptible strain is defined as one producing plaques that are countable in PFU per milliliter as well as one without visible plaques but demonstrating impairment of bacterial growth (that is, lysis zones). Coverage defines the percentage of the total number of susceptible strains. Images of all plates were recorded. Figures illustrating EoP results first had titers log10 transformed and then standard deviances and averages were calculated subsequently. The clinical panels and control strains were tested in two independent experiments.
Animals and housing
Mouse studies were performed with female CD-1 IGS mice (approximately 6–7 weeks of age upon arrival) from Charles River. The animals were housed in groups of three to five mice per cage within a climate-controlled room (temperature, 20–23 °C; relative humidity, 30–70%) under a 12 h light/12 h dark cycle (illuminated, 07:00–19:00). Standard pelleted chow and tap water were available ad libitum. Animals were allowed an acclimatization period of at least 7 d before the start of the experimental procedures. Thirty female Göttingen minipigs (approximately 4–7 months of age upon arrival) from Ellegaard Göttingen minipigs A/S were used for tolerability and kinetic studies. Animals were allowed an acclimatization period of at least 14 d before the start of experiments. Pigs were housed in groups of two to three animals and given standard pig diet twice daily and tap water was available ad libitum. All procedures were conducted in accordance with guidelines from the Danish Animal Experiments Inspectorate, Ministry of Environment and Food of Denmark and in accordance with the institutional license (BioAdvice, animal license 2015-15-0201-00540).
Mouse gut colonization model
The mouse gut colonization model was adapted from ref. 44. Briefly, pretreatment with streptomycin (5 g l−1) in the drinking water was given 3 d before inoculation with E. coli b17 to decrease the level of native bacteria. On day 0, an inoculum of 3 × 107 CFU of E. coli b17 was prepared from a frozen glycerol stock and administered to all mice in 0.25 ml by oral gavage.
Treatment was administered three times daily for 2 d starting 2 d after inoculation. Right before each administration, the four CAPs were mixed in a 1:1:1:1 ratio to form SNIPR001 at a high, medium or low concentration resulting in dose levels of 2 × 1011, 2 × 109 and 1 × 107 PFU. At the time of treatment, mice were administered 0.1 ml of 10% sodium bicarbonate by oral gavage followed by the oral administration of 0.3 ml of SNIPR001, saline (vehicle) or 43.5 mg kg−1 gentamicin.
CAP recovery and tolerability studies
Göttingen minipigs were first given a cocktail of antibiotic comprising neomycin (60 mg kg−1, orally, once daily for 4 d) and cefquinome (2 mg kg−1, intramuscular once daily for 3 d) before SNIPR001 or single CAP administration to decrease the level of Gram-negative bacteria in the GI tract and therefore limiting phage replication. Animals were then fasted overnight and lightly sedated before administration of a single CAP, or SNIPR001 cocktail, once orally at 2 × 1012 PFU in 100 ml, following an oral administration of 50 ml of 10% sodium bicarbonate. Fecal samples were collected daily for CAPs quantification by plaque assay. In addition, for the tolerability study, blood samples were collected for hematology and blood chemistry analysis, including C-reactive protein, and plaque assay. Animals were closely monitored following SNIPR001 administration, and their body temperature was recorded regularly.
Quantification of E. coli b17 and CAPs in feces
Fecal samples were homogenized and serially diluted in SM buffer. Triplicates of 10 μl of each dilution were then spotted on McConkey agar plates (Sigma-Aldrich, M7408) supplemented with streptomycin (1 mg ml−1) and incubated for 12–16 h at 37 °C for E. coli enumeration.
Plaque assays were performed for enumeration of CAPs in feces samples. Briefly, homogenized samples were centrifuged at 10,000g for 10 min, and the supernatant was serially diluted. Triplicates of 10 μl of each dilution were spotted on an E. coli b17 overlay and incubated for 12–16 h at 37 °C.
To quantify the presence of in vivo resistors, three colonies from each mouse fecal sample in the medium dose group at three different time points were picked from the McConkey agar plates. Colonies were incubated for 12–16 h at 37 °C in LB broth and used to make top agar overlays on LB agar plates. Then, plates were dried for 15 min in the LAF bench. The SNIPR001 cocktail, as well as the four individual CAPs, were spotted as a dilution series from 1 × 105 PUF ml−1 stocks. As a control, a top agar overlay of colonization strain E. coli b17 was spotted in the same way. Plates were left to dry in the LAF bench with the lid on and subsequently incubated upside down for 12–16 h at 37 °C.
Whole genome sequencing of E. coli strains from JMI
Total genomic DNA was extracted and purified using the KingFisher Cell and Tissue DNA kit (Thermo Fisher Scientific) in a robotic KingFisher Flex Magnetic Particle Processor (Thermo Fisher Scientific) workstation.
Total genomic DNA was used as input material for library construction. DNA libraries were prepared using the Nextera XT library construction protocol and index kit (Illumina) and sequenced on a MiSeq Sequencer (Illumina) using MiSeq Reagent Kits v3 (600 cycles).
Resistance phenotype definitions
The extended-spectrum β-lactamase-phenotype was defined for E. coli as a minimum inhibitory concentration (MIC) value ≥2 mg l−1 for ceftriaxone, ceftazidime and/or aztreonam (https://clsi.org/).
Carbapenem-resistant Enterobacterales was defined as any isolate displaying imipenem, doripenem and/or meropenem resistance with MIC > 2 mg l−1 (https://clsi.org/).
Assembly of whole-genome sequencing data
Raw sequencing reads were trimmed using Trimmomatic57 (version 0.39) with the settings ‘LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36’. Trimmed reads were assembled using SPAdes58 (version 3.14.1) with default settings. Contigs shorter than 500 bp or with a sequencing depth below two times were removed from the final assemblies.
Comparative genomic methods for clinical E. coli strains
MLST was performed using MLST2 (ref. 59) on the assembled genomes of the E. coli bacteria using default settings, with the MLST database downloaded on 1 July 2021, from the MLST2 repository (https://bitbucket.org/genomicepidemiology/mlst_db/src/master/). Phylogroup classification was conducted using ClermonTyping60 on the assembled E. coli genomes using default settings. Distance matrices for phylogenetic tree construction were generated using MASH61 with a k-mer size of 21 and 10,000 sketches per genome. Sketches were then compared to create the MASH distance in a pairwise manner to create a distance matrix of E. coli genomes.
Phage synteny analysis
To generate the synteny plot, WT sequences of the four phages included in the final cocktail, plus the two closely related and well-known reference phages (RB69 AY303349.1 and T2 NC_054931.1) were annotated with RAST to extract predicted protein sequences. All protein sequences for each phage were queried again all other phage genomes using tblastn (v 2.12.0), with an E-value cutoff of 1 × 10−10. The synteny plot was then generated using a custom Python (v 3.7.10) script (see Data availability), using the drawSvg library (v 1.9.0). The plot shows the phage genomes in order of similarity and displays all tblastn hits as synteny blocks shaded by their protein identity. The proteins of the two reference phages were manually classified as belonging to each of the functional groups ‘DNA metabolism’, ‘structure’ or ‘other’ and colored accordingly.
Data processing and visualization
Figures and key statistics were generated using R version 4.1.0. For figure generation, the following packages were used: RcolorBrewer v. 1.1-2, ape v. 5.5, ggsignif v. 0.6.2, ggpubr v. 0.4.0, matrixStats 0.59, reshape2 v. 1.4.4, ggimage v. 0.3.0, here v. 1.0.1, purr v. 0.3.4, ggtree62 v. 3.0.2, systemfonts v. 1.0.2, Cairo v. 1.5-12.2, cowplot v. 1.1.1, reaxxl v. 1.3.1, ggplot2 v.3.3.3, openxlsx, v. 4.2.3, patchwork v. 1.1.1, dplyr v. 1.0.7 and ggh4x v. 0.2.3. Averages and standard deviations are calculated after transforming the values to the scale shown on a given figure, for example when a log10 scale is used, the averages and standard deviations are calculated after log10 transformation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data and results that were generated during this study are deposited at https://github.com/sniprbiome/SNIPR001_paper. Additional data are available in the Article, Online methods and Supplementary tables. To reproduce the results, no further data is needed.
Phage genome sequences are deposited at Genbank under access numbers OQ067373 – 76.
The MLST database was downloaded on July 1, 2021, from the MLST2 repository (https://bitbucket.org/genomicepidemiology/mlst_db/src/master/). For annotation of the CAP sequences, the following tools and datasets were used ResFinder 4.1 (https://cge.cbs.dtu.dk/services/ResFinder), VirulenceFinder-2.0 (https://cge.cbs.dtu.dk/services/VirulenceFinder/), PHASTER Prophage/Virus DB (https://phaster.ca/).
Code availability
All code needed to produce this study is available at https://github.com/sniprbiome/SNIPR001_paper.
References
Pulte, D., Jansen, L. & Brenner, H. Changes in long term survival after diagnosis with common hematologic malignancies in the early 21st century. Blood Cancer J. 10, 56 (2020).
Riley, M. et al. Epidemiology and burden of mucosal barrier injury laboratory-confirmed bloodstream infections in bone marrow transplant and hematology-oncology units. Am. J. Infect. Control 45, S38 (2017).
Dandoy, C. E. et al. Incidence, risk factors, and outcomes of patients who develop mucosal barrier injury–laboratory confirmed bloodstream infections in the first 100 days after allogeneic hematopoietic stem cell transplant. JAMA Netw. Open 3, e1918668 (2020).
Velden, W. J. F. M., Herbers, A. H. E., Netea, M. G. & Blijlevens, N. M. A. Mucosal barrier injury, fever and infection in neutropenic patients with cancer: introducing the paradigm febrile mucositis. Br. J. Haematol. 167, 441–452 (2014).
Girmenia, C. et al. Incidence, risk factors and outcome of pre-engraftment gram-negative bacteremia after allogeneic and autologous hematopoietic stem cell transplantation: an italian prospective multicenter survey. Clin. Infect. Dis. 65, 1884–1896 (2017).
Klastersky, J. et al. Bacteraemia in febrile neutropenic cancer patients. Int. J. Antimicrob. Agents 30, 51–59 (2007).
Taplitz, R. A. et al. Antimicrobial prophylaxis for adult patients with cancer-related immunosuppression: ASCO and IDSA clinical practice guideline update. J. Clin. Oncol. 36, JCO.18.00374 (2018).
Cullen, M. et al. Antibacterial prophylaxis after chemotherapy for solid tumors and lymphomas. N. Engl. J. Med. 353, 988–998 (2005).
Bucaneve, G. et al. Levofloxacin to prevent bacterial infection in patients with cancer and neutropenia. N. Engl. J. Med. 353, 977–987 (2005).
See, I., Freifeld, A. G. & Magill, S. S. Causative organisms and associated antimicrobial resistance in healthcare-associated, central line-associated bloodstream infections from oncology settings, 2009–2012. Clin. Infect. Dis. 62, 1203–1209 (2016).
Trecarichi, E. M. et al. Current epidemiology and antimicrobial resistance data for bacterial bloodstream infections in patients with hematologic malignancies: an Italian multicentre prospective survey. Clin. Microbiol. Infect. 21, 337–343 (2015).
Marín, M. et al. Factors influencing mortality in neutropenic patients with haematologic malignancies or solid tumours with bloodstream infection. Clin. Microbiol. Infect. 21, 583–590 (2015).
Averbuch, D. et al. Antimicrobial resistance in gram-negative rods causing bacteremia in hematopoietic stem cell transplant recipients: intercontinental prospective study of the infectious diseases working party of the European bone marrow transplantation group. Clin. Infect. Dis. 65, 1819–1828 (2017).
Międzybrodzki, R. et al. Bacteriophages (Springer, 2018) https://doi.org/10.1007/978-3-319-40598-8_31-1
Międzybrodzki, R. et al. Bacteriophages (Springer, 2021) https://doi.org/10.1007/978-3-319-41986-2_31
Schooley, R. T. et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant acinetobacter baumannii infection. Antimicrob. Agents Chemother. 61, e00954-17 (2017).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019).
Jennes, S. et al. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—a case report. Crit. Care 21, 129 (2017).
Jault, P. et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 19, 35–45 (2018).
Sarker, S. A. et al. Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: a randomized trial in children from Bangladesh. eBiomedicine 4, 124–137 (2016).
Leitner, L. et al. Intravesical bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: a randomised, placebo-controlled, double-blind clinical trial. Lancet Infect. Dis. 21, 427–436 (2020).
Sabino, J, Hirten, R. P & Colombel, J. Alimentary Pharmacology & Therapeutics, Vol. 51 (Wiley Online Library, 2020).
Górski, A., Borysowski, J. & Międzybrodzki, R. Phage therapy: towards a successful clinical trial. Antibiotics 9, 827 (2020).
Federici, S. et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell 185, 2879–2898 (2022).
Kauffman, K. M. et al. Resolving the structure of phage–bacteria interactions in the context of natural diversity. Nat. Commun. 13, 372 (2022).
Yehl, K. et al. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell 179, 459–469 (2019).
Westra, E. R. et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and Cas3. Mol. Cell. 46, 595–605 (2012).
Sinkunas, T. et al. Cas3 is a single-stranded DNA nuclease and ATP‐dependent helicase in the CRISPR/Cas immune system. EMBO J. 30, 1335–1342 (2011).
Bikard, D. et al. Development of sequence-specific antimicrobials based on programmable CRISPR–Cas nucleases. Nat. Biotechnol. 32, 1146–1150 (2014).
Citorik, R. J., Mimee, M. & Lu, T. K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141–1145 (2014).
Neil, K. et al. High‐efficiency delivery of CRISPR–Cas9 by engineered probiotics enables precise microbiome editing. Mol. Syst. Biol. 17, e10335 (2021).
Edgar, R. & Qimron, U. The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J. Bacteriol. 192, 6291–6294 (2010).
Hsu, B. B. et al. In situ reprogramming of gut bacteria by oral delivery. Nat. Commun. 11, 5030 (2020).
Yosef, I., Manor, M., Kiro, R. & Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl Acad. Sci. USA 112, 7267–7272 (2015).
Selle, K. et al. In vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 antimicrobials. mBio 11, e00019-20 (2020).
Ochman, H. & Selander, R. K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 157, 690–693 (1984).
Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 10, 351 (2018).
Dunne, M. et al. Salmonella phage S16 tail fiber adhesin features a rare polyglycine rich domain for host recognition. Structure 26, 1573–1582 (2018).
Brouns, S. J. J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008).
Knight, D. & Girling, K. Gut flora in health and disease. Lancet 361, 1831 (2003).
Christensen, S. K., Mikkelsen, M., Pedersen, K. & Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl Acad. Sci. USA 98, 14328–14333 (2001).
Vieira, H. L. A., Freire, P. & Arraiano, C. M. Effect of Escherichia coli morphogene bolA on biofilms. Appl. Environ. Microbiol. 70, 5682–5684 (2004).
Uribe, R. V. et al. Bacterial resistance to CRISPR–Cas antimicrobials. Sci. Rep. 11, 17267 (2021).
Galtier, M. et al. Bacteriophages to reduce gut carriage of antibiotic resistant uropathogens with low impact on microbiota composition. Environ. Microbiol. 18, 2237–2245 (2016).
Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Hyman, P. & Abedon, S. T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 70, 217–248 (2010).
Lemay, M.-L., Renaud, A., Rousseau, G. & Moineau, S. Targeted genome editing of virulent phages using CRISPR–Cas9. Bio. Protoc. 8, e2674 (2018).
Lam, K. N. et al. Phage-delivered CRISPR–Cas9 for strain-specific depletion and genomic deletions in the gut microbiome. Cell Rep. 37, 109930–109930 (2021).
Lee, K. A. et al. The gut microbiome: what the oncologist ought to know. Br. J. Cancer 125, 1197–1209 (2021).
Gencay, Y. E., Ayaz, N. D., Copuroglu, G. & Erol, I. Biocontrol of shiga toxigenic Escherichia coli O157:H7 in Turkish raw meatball by bacteriophage. J. Food Saf. 36, 120–131 (2016).
Satlin, M. J. et al. Colonization with fluoroquinolone-resistant enterobacterales decreases the effectiveness of fluoroquinolone prophylaxis in hematopoietic cell transplant recipients. Clin. Infect. Dis. 73, 1257–1265 (2021).
Satlin, M. J. et al. Colonization with levofloxacin-resistant extended-spectrum β-lactamase-producing enterobacteriaceae and risk of bacteremia in hematopoietic stem cell transplant recipients. Clin. Infect. Dis.67, 1720–1728 (2018).
Harms, A. et al. A bacterial toxin-antitoxin module is the origin of inter-bacterial and inter-kingdom effectors of Bartonella. PLoS Genet. 13, e1007077 (2017).
Henry, M. et al. Development of a high throughput assay for indirectly measuring phage growth using the OmniLogTM system. Bacteriophage 2, 159–167 (2014).
Lutz, R. & Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210 (1997).
Young, K. K., Edlin, G. J. & Wilson, G. G. Genetic analysis of bacteriophage T4 transducing bacteriophages. J. Virol. 41, 345–347 (1982).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Larsen, M. V. et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361 (2012).
Beghain, J., Bridier-Nahmias, A., Nagard, H. L., Denamur, E. & Clermont, O. ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 4, e000192 (2018).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Yu, G. Using ggtree to visualize data on tree‐like structures. Curr. Protoc. Bioinform. 69, e96 (2020).
Gibson, S. B. et al. Constructing and characterizing bacteriophage libraries for phage therapy of human infections. Front. Microbiol. 10, 2537 (2019).
Acknowledgements
SNIPR Biome acknowledges funding from Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator (CARB-X). Research reported in this publication is supported by CARB-X. CARB-X’s funding for this project is sponsored by the Cooperative Agreement Number IDSEP160030 from ASPR/BARDA and by an award from Wellcome Trust. The content is solely the responsibility of the authors and does not necessarily represent the official views of CARB-X or any of its funders. We would also like to thank our collaborators at SSI (Denmark) and Minerva Imaging (Denmark) for their contribution to the manuscript. We would like to acknowledge E. Søndberg (SNIPR BIOME) who illustrated Fig. 1.
Author information
Authors and Affiliations
Contributions
E.v.d.H., J.K.H., K.B., Y.E.G., D.J., V.M., C.R., A.S., A.S.T., S.S., A.T., J.C., L.J., B.D., M.G., J.H., A.T., J.C., C.G., M.Z. and M.O.A.S. conceptualized and designed the work. K.B., Y.E.G., D.J., V.M., C.R., A.S., A.S.T., S.S., A.T., L.B., T.B., M.K.E., K.C.J., L.K., R.P., T.S.S., I.T., A.Ø.P., E.G.B., B.H., A.G., M.J.S., J.K.H. and E.v.d.H. were responsible for data acquisition and analysis. All authors contributed to data interpretation and finalization.
Corresponding author
Ethics declarations
Competing interests
All authors affiliated with SNIPR Biome are present or past employees of SNIPR Biome and maybe share- or warrant holders. F.S. and T.B.D. are subcontractors of SNIPR Biome. M.J.S. received research funding from Merck, Biomérieux and SNIPR Biome. M.J.S. is an unpaid consultant for SNIPR Biome and has been consulting for Shionogi and participated on a Data Safety Monitoring Board for AbbVie. Patent applications have been filed based on material described in this article. SNIPR, CRISPR-Guided Vectors and CGV are trademarks of SNIPR Biome ApS. Data not included in the publication is commercially sensitive as SNIPR Biome is in the process of securing patent protection for these aspects, which precludes their inclusion in the paper at this stage. Upon request, SNIPR Biome is willing to share all data with other parties with no competing interest.
Peer review
Peer review information
Nature Biotechnology thanks Antonia Sagona and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Supplementary Information
Supplementary Figs. 1–25 and Supplementary Tables 1–6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gencay, Y.E., Jasinskytė, D., Robert, C. et al. Engineered phage with antibacterial CRISPR–Cas selectively reduce E. coli burden in mice. Nat Biotechnol 42, 265–274 (2024). https://doi.org/10.1038/s41587-023-01759-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-023-01759-y
This article is cited by
-
A master phage cocktail
Nature Reviews Microbiology (2023)
-
Enhancing bacteriophage therapeutics through in situ production and release of heterologous antimicrobial effectors
Nature Communications (2023)
-
Role of the microbiota in response to and recovery from cancer therapy
Nature Reviews Immunology (2023)
-
Armed phages are heading for clinical trials
Nature Microbiology (2023)
-
The Y-ome Conundrum: Insights into Uncharacterized Genes and Approaches for Functional Annotation
Molecular and Cellular Biochemistry (2023)
