
Abstract
Circulating tumor-derived DNA (ctDNA) is an emerging biomarker for many cancers, but the limited sensitivity of current detection methods reduces its utility for diagnosing minimal residual disease. Here we describe phased variant enrichment and detection sequencing (PhasED-seq), a method that uses multiple somatic mutations in individual DNA fragments to improve the sensitivity of ctDNA detection. Leveraging whole-genome sequences from 2,538 tumors, we identify phased variants and their associations with mutational signatures. We show that even without molecular barcodes, the limits of detection of PhasED-seq outperform prior methods, including duplex barcoding, allowing ctDNA detection in the ppm range in participant samples. We profiled 678 specimens from 213 participants with B cell lymphomas, including serial cell-free DNA samples before and during therapy for diffuse large B cell lymphoma. In participants with undetectable ctDNA after two cycles of therapy using a next-generation sequencing-based approach termed cancer personalized profiling by deep sequencing, an additional 25% have ctDNA detectable by PhasED-seq and have worse outcomes. Finally, we demonstrate the application of PhasED-seq to solid tumors.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
WGS data were obtained from the PCAWG dataset available at https://dcc.icgc.org/pcawg/. Additional lymphoma WGS data were obtained from the ICGC website (malignant lymphoma project, MALY-DE), as well as from work by Morin et al.33 and Qian et al.34. Raw data for samples used in the dilution series are available at http://phasedseq.stanford.edu/. This website provides users with software for phased variant monitoring (PhasED-seq v.1.0). Included here are the list of SNVs and PVs used to detected tumor DNA from each of the three samples used in this dilution series, as well as the sequencing files for each concentration condition. Owing to restrictions related to dissemination of germline sequence information included in the informed consent forms used to enroll study participants, we are unable to provide access to other raw sequencing data. Reasonable requests for additional data will be reviewed by the senior authors to determine whether they can be fulfilled in accordance with these privacy restrictions. Requests for additional materials related to this work should be directed to A.A.A.
Code availability
PhasED-seq (v.1.0) was used to generate the results in this work and is freely available for academic research use at http://phasedseq.stanford.edu/, including a downloadable compiled version of the software. For academic users, the source code is available upon request.
References
Diehl, F. et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008).
Newman, A. M. et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 34, 547–555 (2016).
Scherer, F. et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 8, 364ra155 (2016).
Chabon, J. J. et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 7, 11815 (2016).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 (2014).
Sozzi, G. et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res. 61, 4675–4678 (2001).
Thierry, A. R. et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 20, 430–435 (2014).
Tie, J. et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 8, 346ra392 (2016).
Kalinich, M. & Haber, D. A. Cancer detection: seeking signals in blood. Science 359, 866–867 (2018).
Phallen, J. et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan2415 (2017).
Abbosh, C., Birkbak, N. J. & Swanton, C. Early stage NSCLC—challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 15, 577–586 (2018).
Abbosh, C. et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017).
Newman, A. M. et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554 (2014).
Kurtz, D. M. et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B cell lymphoma. J. Clin. Oncol. 36, 2845–2853 (2018).
Roschewski, M. et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B cell lymphoma: a correlative biomarker study. Lancet Oncol. 16, 541–549 (2015).
Garcia-Murillas, I. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 7, 302ra133 (2015).
Schmitt, M. W. et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA 109, 14508–14513 (2012).
Kennedy, S. R. et al. Detecting ultralow-frequency mutations by duplex sequencing. Nat. Protoc. 9, 2586–2606 (2014).
Medina Diaz, I. et al. Performance of Streck cfDNA blood collection tubes for liquid biopsy testing. PLoS ONE 11, e0166354 (2016).
de Yebenes, V. G. & Ramiro, A. R. Activation-induced deaminase: light and dark sides. Trends Mol. Med. 12, 432–439 (2006).
Pasqualucci, L. et al. Hypermutation of multiple proto-oncogenes in B cell diffuse large-cell lymphomas. Nature 412, 341–346 (2001).
Jiang, P. et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl Acad. Sci. USA 112, E1317–E1325 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Burns, M. B., Temiz, N. A. & Harris, R. S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 (2013).
Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015).
Khodabakhshi, A. H. et al. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget 3, 1308–1319 (2012).
Schmitz, R. et al. Genetics and pathogenesis of diffuse large B cell lymphoma. N. Engl. J. Med. 378, 1396–1407 (2018).
Rowley, J. D. Chromosome studies in the non-Hodgkin’s lymphomas: the role of the 14;18 translocation. J. Clin. Oncol. 6, 919–925 (1988).
Schmitz, R. et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120 (2012).
Alizadeh, A. A. et al. Distinct types of diffuse large B cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Lenz, G. et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J. Exp. Med. 204, 633–643 (2007).
Morin, R. D. et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 122, 1256–1265 (2013).
Qian, J. et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537 (2014).
Richter, J. et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 44, 1316–1320 (2012).
Puente, X. S. et al. Noncoding recurrent mutations in chronic lymphocytic leukaemia. Nature 526, 519–524 (2015).
Pasqualucci, L. et al. Analysis of the coding genome of diffuse large B cell lymphoma. Nat. Genet. 43, 830–837 (2011).
Steidl, C. et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471, 377–381 (2011).
Nakamura, N. et al. Analysis of the immunoglobulin heavy chain gene variable region of CD5-positive and -negative diffuse large B cell lymphoma. Leukemia 15, 452–457 (2001).
Wan, J. C. M. et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aaz8084 (2020).
Chabon, J. J. et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251 (2020).
Razavi, P. et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 25, 1928–1937 (2019).
Bratman, S. V. et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat. Cancer 1, 873–881 (2020).
Reinert, T. et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol https://doi.org/10.1001/jamaoncol.2019.0528 (2019).
Leary, R. J. et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci. Transl. Med. 2, 20ra14 (2010).
Leary, R. J. et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 4, 162ra154 (2012).
Li, Y. et al. Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Kurtz, D. M. et al. Reply to J. Wang et al. J. Clin. Oncol. 37, 755–757 (2019).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17, 31 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Bozdech, Z. et al. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 4, R9 (2003).
Jaeger, J. A., Turner, D. H. & Zuker, M. Improved predictions of secondary structures for RNA. Proc. Natl Acad. Sci. USA 86, 7706–7710 (1989).
Sugimoto, N., Nakano, S., Yoneyama, M. & Honda, K. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 24, 4501–4505 (1996).
Lu, Z. et al. BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood 121, 4551–4554 (2013).
Robbiani, D. F. et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 135, 1028–1038 (2008).
Lieber, M. R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 16, 387–398 (2016).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Moding, E. J. et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nat. Cancer 1, 176–183 (2020).
Acknowledgements
This work was supported by the National Cancer Institute (R01CA233975 and R01CA188298 to A.A.A. and M.D.; K08CA241076 to D.M.K.), the Virginia and D. K. Ludwig Fund for Cancer Research (A.A.A. and M.D.), the Bakewell Foundation (A.A.A. and M.D.), the Damon Runyon Cancer Research Foundation (PST no. 09-16 to D.M.K. and DR-CI no. 71-14 to A.A.A.), the American Society of Hematology Scholar Award (A.A.A.), the V Foundation for Cancer Research Abeloff Scholar Award (A.A.A.), the Conquer Cancer Foundation of the American Society of Clinical Oncology (D.M.K.), the Emerson Collective Cancer Research Fund (A.A.A.), the Stinehart/Reed Award (A.A.A.), the CRK Faculty Scholar Fund (M.D.) and the SDW/DT and Shanahan Family Foundations (A.A.A.). A.A.A. is a Scholar of The Leukemia & Lymphoma Society.
Author information
Authors and Affiliations
Contributions
D.M.K., J.S., M.D. and A.A.A. developed the concept, designed the experiments and analyzed the data. D.M.K., J.S., M.D. and A.A.A. wrote the manuscript. D.M.K., J.S. and L.C.T.K. developed the phased variant identification and monitoring workflows with input from M.D. and A.A.A. D.M.K., J.S., S.A., J.J.C., B.J.S, M.C.J., F.S., C.W.M., M.O., A.F.M.C. and E.J.M. performed molecular biology experiments related to the technical performance of PhasED-seq and detection of ctDNA in clinical samples. D.M.K., J.S., L.C.T.K., A.S., A.G., E.G.H., B.C., J.G.S.-M., A.F.M.C., M.S.E. and C.L.L. performed bioinformatic analyses. Participant specimens were provided by D.M.K., E.J.M., U.D., A.H., R.-O.C., J.R.W., M.R., W.H.W., G.G., D.R., M.D. and A.A.A. Participant enrollment, sample collection and data curation were organized and performed by D.M.K., U.D., A.H., R.-O.C., J.R.W., M.R., W.H.W., G.G., D.R., M.D. and A.A.A. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.M.K. reports paid consultancy from Roche and Genentech. A.A.A. reports research funding from Celgene and Pfizer, ownership interests in FortySeven and CiberMed, and paid consultancy from Roche, Genentech, Janssen, Pharmacyclics, Gilead, Celgene and Chugai. M.D. reports research funding from Varian Medical Systems, AstraZeneca and Illumina, ownership interest in CiberMed, and paid consultancy from Roche, AstraZeneca, Novartis, Genentech, Illumina, RefleXion, Gritstone Oncology, Boehringer Ingelheim and BioNTech. M.D., A.A.A., D.M.K., J.C. and M.S.E. report patent filings related to cancer biomarkers. M.D., A.A.A., D.M.K. and J.C. report ownership interest in Foresight Diagnostics. R.O.C. is a member of the Roche advisory board and received research support from Roche. D.R. received research support from Gilead, Janssen, Roche and AbbVie, outside the submitted work. E.J.M. has served as a paid consultant for DeciBio. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Biotechnology thanks Paul Spellman, Christian Steidl and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison of duplex sequencing to phased variant sequencing.
a, A schema comparing error-suppressed sequencing by duplex sequencing vs. recovery of phased variants. In duplex sequencing, recovery of a single SNV observed on both strands of an original DNA double-helix (that is, in trans) is required. This requires independent recovery of two molecules by sequencing as the plus and minus strands of the original DNA molecule go through library preparation and PCR independently. In contrast, recovery of PVs requires multiple SNVs observed on the same single strand of DNA (that is, in cis). Thus, recovery of only the plus or the minus strand (rather than both) is sufficient for identification of PVs. b, A model showing the two possible reasons for limited sensitivity for ctDNA MRD assays. An assay can be limited by either having i) an insufficient number of cfDNA fragments evaluable for tumor content, or ii) an inadequate error-profile. This plot demonstrates the analytical sensitivity as the number of evaluable cfDNA fragments increase with either the amount of plasma input or the number of mutations tracked, until eventually becoming limited by the background signal (grey). Separate plots shown for single-stranded and double-stranded SNV based methods, assuming 8.92 ng cfDNA/mL plasma; 50% efficiency of library preparation, and 20% efficiency of duplex sequencing.
Extended Data Fig. 2 Enumeration of SNVs and PVs in diverse cancers from WGS.
a–d, Univariate scatter plots showing the number of a) SNVs, b) 2x-PVs (2 SNVs in phase), c) 3x-PVs, and d) total 2x-PVs, controlling for total number of SNVs, from WGS data for 24 different histologies of cancer. Data are presented as median and interquartile range. (FL-NHL, follicular lymphoma; DLBCL-NHL, diffuse large B cell lymphoma; Burkitt-NHL, Burkitt lymphoma; Lung-SCC, squamous cell lung cancer; Lung-Adeno, lung adenocarcinoma; Kidney-RCC, renal cell carcinoma; Bone-Osteosarc, osteosarcoma; Liver-HCC, hepatocellular carcinoma; Breast-Adeno, breast adenocarcinoma; Panc-Adeno, pancreatic adenocarcinoma; Head-SCC, head and neck squamous cell carcinoma; Ovary-Adeno, ovarian adenocarcinoma; Eso-Adeno, esophageal adenocarcinoma; Uterus-Adeno, uterine adenocarcinoma; Stomach-Adeno, stomach adenocarcinoma; CLL, chronic lymphocytic leukemia; ColoRect-Adeno, colorectal adenocarcinoma; Prost-Adeno, prostate adenocarcinoma; CNS-GBM, glioblastoma multiforme; Panc-Endorcine, pancreatic neuroendocrine tumor; Thy-Adeno, thyroid adenocarcinoma; CNS-PiloAstro, piloastrocytoma; CNS-Medullo, medulloblastoma.).
Extended Data Fig. 3 Distribution of PVs in stereotyped regions across the genome.
Distribution of PVs occurring in stereotyped regions across the genome of multiple cancer types. In this plot, the genome was divided into 1000 bp bins, and the fraction of samples of a given histology with a PV in each 1000 bp bin was calculated. Only bins that have at least a 2 percent recurrence frequency in any cancer subtype are shown.
Extended Data Fig. 4 Performance of PhasED-Seq for recovery of PVs across lymphomas.
a, Univariate scatter plot comparing the fraction of all PVs across the genome identified by WGS (n = 79) that were recovered by our previously reported lymphoma CAPP-Seq panel3 (left) compared to PhasED-Seq (right). b, Univariate scatter plot comparing the expected yield of SNVs per case identified from WGS using a previously established lymphoma CAPP-Seq panel or the PhasED-Seq panel. c, Univariate scatter comparing the expected yield of PVs per case identified from WGS using a previously established lymphoma CAPP-Seq panel or the PhasED-Seq panel. Data from three independent publicly available cohorts are shown in a-c). d,e, Plots showing the improvement in recovery of PVs by PhasED-Seq compared to CAPP-Seq in 16 patients sequenced by both assays. This includes improvement in d) two SNVs in phase (that is, 2x or ‘doublet PVs’) and e) three SNVs in phase (3x or ‘triplet PVs’). Statistical testing in panels a-e) performed by 2-sided Wilcoxon signed-rank test. f, A cartoon describing the terminology for phased variants in this manuscript. The figure shows one region of an individual’s cancer genome (300 bp). Phased variants on a single strand of DNA of DNA can occur with different numbers of SNVs, including 2 variants in phase (doublets) and 3 in phase (triplets). For the purpose of detecting ctDNA, ‘independent reporters’ are defined as PVs that will typically co-segregate on separate cfDNA molecules, resulting in independent evaluable fragments. Given the size of cfDNA molecules, these are separated in 150 bp regions. g–j, These panels show the number of SNVs and PVs identified for patients with different types of lymphomas. These panels show the number of g) SNVs, h) doublet PVs, i) triplet PVs, and j) independent PV reporters; bars represent median and interquartile range. *, P < 0.05 by two-sided Wilcoxon rank sum test; comparisons only shown for all histologies vs DLBCL. (DLBCL, diffuse large B-cell lymphoma; GCB, germinal center B-cell like DLBCL; ABC, activated B-cell like DLBCL; PMBCL, primary mediastinal B-cell lymphoma; FL, follicular lymphoma; HL, Hodgkin lymphoma; MCL, mantle cell lymphoma).
Extended Data Fig. 5 Technical aspects of PhasED-Seq by hybrid-capture sequencing.
a, Theoretical binding energy for 150-mers across the genome. Mutations were either clustered to one end (green), clustered in the middle (blue), or randomly throughout the sequence (red). Data represent the median and IQR from 10,000 in silico simulations. b, Histograms of summary metrics of the mutation rate of 151-bp windows from all patients in this study. c, The percentile of mutation rate across all mutated 151-bp windows across all patients in this study. d, Rate of background-signal in the PhasED-Seq panel for multiple variants, including SNVs (red), PVs (blue), and indels (green). Different methods of error-suppression for each variant type are shown. Bars represent median and IQR. UMIs, unique molecular identifiers; PhasED-Seq 2x, doublet PVs; PhasED-Seq 3x, triplet PVs. e) Error-rate for SNVs (left), doublet PVs (middle), and triplet PVs (right) by type of mutation. For triplet PVs, the x and y-axis represent the first and second type of base alteration in the PV. f, Error rate for doublet PVs across n = 12 healthy cfDNA samples as a function of inter-SNV distance. Data show mean and standard deviation. g, Limiting dilution series simulating cfDNA similar to Fig. 5a; cfDNA from 3 independent patient samples were used in each dilution. In this plot, PhasED-Seq is assessed without the use of UMIs. Data are presented as mean and range. *, P < 0.05; CAPP-Seq vs duplex, P = 3.2e-5; CAPP-Seq vs PhasED-Seq (2x), P = 1.6e-4; CAPP-Seq vs PhasED-Seq (3x), P = 1.9e-5; duplex vs PhasED-Seq (2x), 0.017; duplex vs PhasED-Seq (3x), 0.0046. h, Theoretical rate of detection for a sample with a given number of PV-containing regions, according to binomial sampling, assuming unique sequencing depth of 4000–6000x (shaded area; 5000x shown as line). i, Observed rate of detection given a true tumor fraction, with varying numbers of PV-containing regions. Filled-in points represent ‘wet’ experiments; open points represent in silico dilution experiments. Data represent mean and range. j, Predicted vs observed rate of detection for samples from the dilution series shown in panels h) and i). Error-bars are as described in h) and i) above (see the Supplementary Methods).
Extended Data Fig. 6 Comparison of ctDNA quantitation by PhasED-Seq to CAPP-Seq and clinical applications.
a, ROC curve of the performance for detection of ctDNA from SNVs (that is, CAPP-Seq) and PVs using PhasED-Seq. Positive samples are 107 pretreatment plasmas, negative samples are 40 control plasmas assessed for evidence of ctDNA using 107 personalized mutation lists for 4,280 total samples. Sensitivity and specificity at optimum point and AUC are shown. b, Quantity of ctDNA (measured as log10(haploid genome equivalents/mL)) as measured by CAPP-Seq vs. PhasED-Seq in individual samples. Samples taken prior to cycle 1 of RCHOP therapy (that is, pretreatment), prior to cycle 2, and prior to cycle 3, are shown in independent colors (blue, green, and red respectively; 277 total samples). Undetectable levels fall on the axes. Spearman correlation and P-value are shown.
Extended Data Fig. 7 Detection of ctDNA after two cycles of systemic therapy.
a, Scatterplot shows the log-fold change in ctDNA after 2 cycles of therapy measured by CAPP-Seq or PhasED-Seq for patients receiving RCHOP therapy. Dotted lines show the previously established threshold of a 2.5-log reduction in ctDNA for molecular response. Undetectable samples fall on the axes; the correlation coefficient represents a Spearman rho for the samples detected by both CAPP-Seq and PhasED-Seq. b, Detection rate of ctDNA samples after 2 cycles of therapy by PhasED-Seq vs CAPP-Seq. Patients with eventual disease progression are shown in red, while patients without eventual disease progression are shown in blue. c, ROC curve for detection of ctDNA after 2 cycles of treatment. Positive samples include 24 samples from patients with eventual disease progression, and therefore are known to have residual disease. Negative samples are from 4,280 tests on healthy controls as described in Extended Data Fig. 6a. d, Kaplan-Meier plots and two-sided log-rank test showing the event-free survival of 69 patients achieving an MMR stratified by ctDNA detection with CAPP-Seq (top) or PhasED-Seq (bottom).
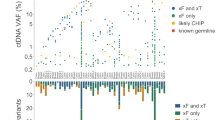
Extended Data Fig. 8 Detection of ctDNA after one cycle of systemic therapy.
a, Scatterplot showing the log-fold change in ctDNA after 1 cycle of therapy measured by CAPP-Seq or PhasED-Seq for patients receiving RCHOP therapy. Dotted lines show the previously established threshold of a 2-log reduction in ctDNA for molecular response. Undetectable samples fall on the axes; the correlation coefficient represents a Spearman rho for the samples detected by both CAPP-Seq and PhasED-Seq. b, Detection rate of ctDNA samples after 1 cycle of therapy by PhasED-Seq vs CAPP-Seq. Patients with eventual disease progression are shown in red, while patients without eventual disease progression are shown in blue. c, ROC curve for detection of ctDNA after 2 cycles of treatment. Positive samples include 22 samples from patients with eventual disease progression, and therefore are known to have residual disease. Negative samples are from 4,280 tests on healthy controls as described in Extended Data Fig. 6a. d, Waterfall plot showing the change in ctDNA levels measured by CAPP-Seq after 1 cycle of first-line therapy in patients with DLBCL. Patients with undetectable ctDNA by CAPP-Seq are shown as ‘ND’ (‘not detected’), in darker colors. The colors of the bars also indicate the eventual clinical outcomes for these patients. e, A Kaplan-Meier plot showing the event-free survival for 33 DLBCL patients with undetectable ctDNA measured by CAPP-Seq after 1 cycle of therapy. f, A Kaplan-Meier plot and two-sided log-rank test showing the event-free survival of 33 patients shown in f) (undetectable ctDNA by CAPP-Seq) stratified by ctDNA detection via PhasED-Seq at this same time-point (cycle 2, day 1). g, A Kaplan-Meier plot and two-sided log-rank test showing the event-free survival for 82 patients with DLBCL stratified by ctDNA at cycle 2, day 1 separated into 3 strata – patients failing to achieve an early molecular response (red), patients with an early molecular response who still have detectable ctDNA by PhasED-Seq and/or CAPP-Seq (grey), and patients who have a stringent molecular remission (undetectable ctDNA by PhasED-Seq and CAPP-Seq; blue).
Extended Data Fig. 9 Performance of ctDNA detection at the end of systemic therapy.
a, ROC curve for detection of ctDNA after the completion of planned systemic therapy. Positive samples include 5 samples from patients with eventual disease progression, and therefore are known to have residual disease. Negative samples are from 4,280 tests on healthy controls as described in Extended Data Fig. 6a. b, The ctDNA profile of a patient with stage 4 DLBCL undergoing systemic chemotherapy, with pretreatment PET scan shown on the left. This patient only received one cycle of EPOCH-R chemotherapy from 6 planned treatments (dashed arrows – planned therapy that was not given). Following this, the patient self-discontinued treatment. This patient was found to have cleared their ctDNA by PhasED-Seq and continues in clinical remission after > 4 years.
Extended Data Fig. 10 Extension of PhasED-Seq to solid tumors.
a, A mathematical model showing the expected total unique molecular depth (blue) and duplex molecular depth (green) from an optimized hybrid-capture workflow (Chabon et al.41; Methods). b, A comparison in projected sensitivity for ctDNA detection using PVs versus structural variants (SVs) for various histologies from the PCAWG dataset. Comparison assumes a personalized sequencing panel targeting only patient-specific variants, 64 ng of DNA input and 20 million sequencing reads, using the model of molecular recovery from a). c, A comparison in expected sensitivity for ctDNA detection using PVs versus duplex sequencing and SNVs for various histologies from the PCAWG dataset. Comparison assumes a personalized sequencing panel targeting only patient-specific variants, 64 ng of DNA input and 20 million sequencing reads, using the model of molecular recovery from a). d, Detection of ctDNA for the 6 cases of patients with solid tumors, including lung cancer (n = 5) and breast cancer (n = 1) using SNV-based detection (that is, CAPP-Seq) or PhasED-Seq with a personalized panel. Detection of ctDNA in patient plasma samples are shown in blue; samples detectable with PhasED-Seq but not SNV based approaches are in light blue. Specificity of the assay was assessed using 24 healthy control samples; detection of evidence of ctDNA by PhasED-Seq in these are shown on the right in pink across all 6 personalized panels, indicating 97% (139/144) specificity; CAPP-Seq on the same samples showed 95% (137/144) specificity. e, The ctDNA profile of a patient with stage 3 lung adenocarcinoma (LUP831) undergoing combined chemo-radiotherapy (CRT) and immunotherapy, measured by both CAPP-Seq and PhasED-Seq. The left panel shows the measured tumor fraction in the tumor biopsy sample using both methods. The right panel shows the tumor fraction from plasma DNA, including a sample detected by PhasED-Seq that is undetected by CAPP-Seq. ND: not detected.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2, table legends and Methods.
Rights and permissions
About this article

Cite this article
Kurtz, D.M., Soo, J., Co Ting Keh, L. et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol 39, 1537–1547 (2021). https://doi.org/10.1038/s41587-021-00981-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-00981-w
This article is cited by
-
Genetic and transcriptomic analyses of diffuse large B-cell lymphoma patients with poor outcomes within two years of diagnosis
Leukemia (2024)
-
Single-cell low-pass whole genome sequencing accurately detects circulating tumor cells for liquid biopsy-based multi-cancer diagnosis
npj Precision Oncology (2024)
-
Bridging horizons beyond CIRCULATE-Japan: a new paradigm in molecular residual disease detection via whole genome sequencing-based circulating tumor DNA assay
International Journal of Clinical Oncology (2024)
-
Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling
Nature (2024)
-
Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer
Nature Communications (2024)
