
Abstract
Plasma-derived polyclonal antibody therapeutics, such as intravenous immunoglobulin, have multiple drawbacks, including low potency, impurities, insufficient supply and batch-to-batch variation. Here we describe a microfluidics and molecular genomics strategy for capturing diverse mammalian antibody repertoires to create recombinant multivalent hyperimmune globulins. Our method generates of diverse mixtures of thousands of recombinant antibodies, enriched for specificity and activity against therapeutic targets. Each hyperimmune globulin product comprised thousands to tens of thousands of antibodies derived from convalescent or vaccinated human donors or from immunized mice. Using this approach, we generated hyperimmune globulins with potent neutralizing activity against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) in under 3 months, Fc-engineered hyperimmune globulins specific for Zika virus that lacked antibody-dependent enhancement of disease, and hyperimmune globulins specific for lung pathogens present in patients with primary immune deficiency. To address the limitations of rabbit-derived anti-thymocyte globulin, we generated a recombinant human version and demonstrated its efficacy in mice against graft-versus-host disease.
Similar content being viewed by others

Main
Many diseases, such as those caused by infectious viruses or bacteria with many variants or serotypes, are best treated by drugs that target multiple epitopes. An established therapeutic modality is multispecific (multivalent) antibodies derived from human or animal plasma, such as intravenous immunoglobulin (IVIG)1. Polyclonal antibody drugs with higher potency, known as hyperimmune globulins, are often derived from the plasma of recently vaccinated human donors, for example, HepaGam B against hepatitis B virus (HBV)2 and BabyBIG against infant botulism3. In diseases for which human vaccination is not possible, hyperimmune globulins can be generated by immunizing animals, for example, rabbit-derived thymoglobulin (‘rabbit-ATG’) against human thymocytes for transplant tolerance4. For rapid response to emerging pathogens with poorly characterized neutralizing epitopes, many groups have developed hyperimmune globulins derived from immunized animal plasma or convalescent human serum, for example, Zika virus hyperimmune globulin5 or severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2)6,7.
Plasma-derived antibody therapeutics have substantial drawbacks. First, demand for normal and convalescent donor plasma often outstrips supply8. Plasma-derived drugs have suffered from impurities, including infectious viruses and clotting factors, that have resulted in serious adverse events9,10. Antibody drugs derived from animal plasma occasionally cause allergic reactions11, lead to antidrug antibodies and have suboptimal effector properties12. Because they are derived from naturally occurring proteins, plasma-derived drugs are not easily engineered; for example, it is not possible to modify Fc sequences to improve mechanism of action or drug half-life. Finally, each batch of plasma-derived drug is usually derived from a different cohort of human donors or animals, resulting in batch-to-batch variation13,14,15.
Many of these problems could be solved by generating multivalent hyperimmune globulins using recombinant DNA technology. However, this strategy presents substantial technical hurdles. Most important, a recombinant hyperimmune globulin technology would have to isolate significant numbers of B cells from donors or animals, natively pair heavy and light chain immunoglobulin at a single-cell level, and then clone the sequences into recombinant expression libraries for manufacturing. Conventionally, most production cell lines for recombinant antibody drugs are generated by random integration of expression constructs into mammalian cell genomes16. To prevent mispairing between heavy and light chain immunoglobulin, a recombinant polyclonal hyperimmune globulin technology would require a single genome-integration site. Pioneering work used 96-well plates to capture antibody sequences from B cells isolated from human donors immunized with Rho(D)+ erythrocytes and then engineer multivalent recombinant antibodies17, but this approach produced drug candidates with <30 antibodies, complicating broad application and reducing potential for polyvalence.
The technology described here generates recombinant hyperimmune globulins for diverse conditions through high-throughput microfluidics, genomics and mammalian cell engineering. B cells from human donors or mice are run through a microfluidic platform, heavy and light chain immunoglobulin nucleic acid sequences are fused on a single-cell level to create antibody repertoires18, antibody repertoires are engineered into full-length expression constructs en masse, and then the full-length antibody expression constructs are stably introduced en masse into Chinese hamster ovary (CHO) cells in a site-directed manner. We applied our technology to develop 103 to 104 diverse recombinant hyperimmune globulin drug candidates to address unmet clinical needs for the COVID-19 pandemic, Zika virus disease, primary immune deficiency (PID) and transplant tolerance. We validated a drug candidate in vivo and/or in vitro for each of the four clinical applications.
Results
Capturing diverse antibody repertoires as CHO libraries
Mammalian antibody repertoires are extremely diverse, comprising as many as 107 antibody clonotypes19. Advanced molecular technology is required to capture a substantial fraction of a mammalian donor’s diverse antibody repertoire. We reported methods for generating millions-diverse libraries of natively paired heavy and light chain immunoglobulin sequences in yeast18. That method used microfluidics to isolate millions of single B cells per hour into picoliter droplets for lysis, followed by overlap extension–reverse transcriptase–polymerase chain reaction (OE–RT–PCR), to generate libraries of natively paired single chain variable fragments (scFv).
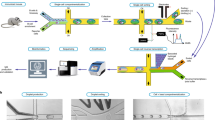
Because antibody repertoires often contain many antibodies not directed against the target(s) of interest, we used a variety of enrichment methods (Fig. 1). For ATG, Zika virus, Haemophilus influenzae b (Hib) and Streptococcus pneumoniae (pneumococcus), we administered immunogens to human donors or humanized mice before sampling antibody-producing cells. For SARS-CoV-2, we recruited convalescent donors who recently tested positive for COVID-19, made yeast display scFv libraries from donor B cells and sorted the libraries derived from these donors to enrich for antibodies directed against SARS-CoV-2 antigen. In all cases, the output was a library of thousands to tens of thousands of natively paired scFv DNAs, enriched for activity against their respective target(s).

a, B cells were isolated from human donors (vaccinated or convalescent) or immunized humanized mice. b, Droplet microfluidics were used to capture natively paired antibody sequences from millions of single cells. c, An optional yeast scFv display system was used to enrich for binders to a soluble antigen. d, A two-step Gibson assembly process converted the scFv fragment to full-length antibody expression constructs, which were then stably integrated into CHO cells following electroporation and selection. e, After bioproduction, the libraries were characterized in many ways including deep sequencing, in vitro binding and efficacy assays, and in vivo mouse efficacy studies.
Next, we used each library of scFv DNAs to produce natively paired full-length antibody expression constructs, which were then engineered into mammalian cells for production of recombinant hyperimmune globulins (Fig. 1). Cloning into full-length antibody expression constructs was performed en masse, that is, we performed all molecular steps on full libraries rather than individual clones. Briefly, the protocol involved a series of two Gibson assemblies20, which we termed Gibson assembly 1 (GA1) and Gibson assembly 2 (GA2) (Supplementary Figs. 1 and 2). In GA1, the scFv library was inserted into a vector backbone that contained a promoter, a fragment of the IgG1 constant domain and a poly(A) signal. In GA2, we linearized the GA1 plasmid, and subcloned it into a DNA fragment that contained a fragment of the IgK constant domain, a second poly(A) signal and a second promoter.
Production cell lines for monoclonal antibodies are typically produced by randomly inserting expression constructs into the CHO genome16. This method produces cell lines with genomic insertion of multiple copies of the expression construct. If we randomly inserted our polyclonal antibody construct libraries into the CHO genome, because each cell might contain several inserted transgenes, many clones would express multiple antibodies, which would result in frequent nonnative pairing between heavy and light chain immunoglobulin. Additionally, different genome locations have different transcriptional activity levels21, which could result in heterogeneous, inconsistent and/or unstable bioproduction. We therefore used CHO cell lines engineered with a Flp recombinase recognition target landing pad (Supplementary Fig. 3). We then used these cell lines for stable expression of recombinant hyperimmune globulins in polyclonal cell banks.
Recombinant hyperimmune globulins for SARS-CoV-2
To address the urgent unmet clinical need of the COVID-19 pandemic, we used our technology to build recombinant hyperimmune globulins against SARS-CoV-2, which we call recombinant coronavirus-2 immune globulin, or rCIG. In March 2020, we recruited 50 human donors from a single clinic in Louisiana who either had tested positive for SARS-CoV-2 by nasal swab PCR testing or had shown symptoms of COVID-19 around the time of a major local outbreak. First, we assessed anti-SARS-CoV-2 plasma titer for each of the donors using the S1 and receptor binding domain (RBD) regions of SARS-CoV-2 spike glycoprotein (Fig. 2a and Supplementary Table 1). We observed a wide range of half-maximum effective concentration (EC50) values among patients who tested positive for COVID-19 (range 0.0056–9.94 mg ml−1). We selected 16 donors with high plasma antibody titers and used our technology to build yeast scFv display libraries from pools of two donors, for a total of eight libraries. The libraries comprised a median of 70,940 antibodies (range 54,986–156,592, Supplementary Table 2).

a, ELISA of individual human plasma donors against SARS-CoV-2 S1 antigen (top) or RBD antigen (bottom). Dark blue indicates donors used in rCIG. Each data point represents a single measurement at a single test article dilution in a single experiment. b, Example FACS enrichment of scFv against CoV-2 RBD from library 1 using yeast display. The x axis measures presence of a C-terminal c-Myc tag, indicating expression of an scFv on the surface of the cell. The y axis measures binding of antigen to the scFv-expressing cells. The gates used for yeast selection (double positive) are indicated, with the percentage of scFv-expressed antigen binders in red. Each plot summarizes a single FACS experiment with one yeast scFv library. c, Clonal cluster analysis of rCIG antibodies. Each node represents an antibody clone (full-length heavy chain). The color of the nodes indicates the sorted scFv library from which the CHO antibody clones were derived. The size of the nodes reflects the frequency of the clones in the final CHO cell bank (only clones ≥0.01% are plotted). We computed the total number of amino acid differences between each pairwise alignment, and edges indicate ≤5 amino acid differences. d, ELISA of the indicated samples against SARS-CoV-2 S1 antigen (top) or RBD antigen (bottom). Each data point represents a single measurement at a single test article dilution, in a single experiment. e, ELISA of the indicated samples (indicated by the color) against the indicated antigens (different shapes). For rCIG, no binding was observed against MERS CoV S1. For the CoV-2 mAb (SAD-S35), no binding was observed against MERS CoV S1 and SARS-CoV RBD. Each data point represents a single measurement at a single test article dilution, in a single experiment. f, Live virus neutralization. Individual dots are separate test articles that represent the minimum antibody concentration that achieved neutralization. Bars represent median measurements for each test article category. Each test article was run in duplicate using different aliquots of cells and virus, in a single experiment, with the same result observed for each replicate. No neutralization was seen for IVIG. A Wilcoxon rank sum test was used to compare the minimum concentration to achieve SARS-CoV-2 live virus neutralization between convalescent plasma measurements (n = 16) and rCIG measurements (n = 2).
We used flow sorting to enrich for anti-SARS-CoV-2 antibodies in the eight yeast scFv libraries (Fig. 2b, Supplementary Fig. 4 and Supplementary Table 2). One round of flow sorting suggested that a median of 0.99% of antibodies (range 0.42–2.29%) were directed against SARS-CoV-2. After two rounds of sorting, a median of 62.7% of unsorted antibody sequences were human IgG1 subtype (range 51.5–83.4%), whereas in the sorted libraries a median of 82.4% of antibody sequences were human IgG1 subtype (range 63.6–92.2%), suggesting that the COVID-19 antibody response was generally dominated by IgG1 antibodies. Next, we used our technology to make full-length polyclonal antibody preparations from each of the eight scFv libraries. The antibodies were formatted as human IgG1, regardless of the initial IgG subtype. We used anti-SARS-CoV-2 enzyme-linked immunosorbent assay (ELISA), spike:ACE2 blocking assays and pseudotype and live virus neutralization assays to assess the relative activity of each of the eight antibody libraries (Fig. 2f, Supplementary Figs. 5 and 7 and Supplementary Table 2). We pooled the eight scFv-sorted CHO cell banks in a way that sought to balance high antibody diversity with high anti-SARS-CoV-2 pseudotype neutralization titer (Supplementary Table 3) and used the combined cell bank to generate rCIG protein product (Supplementary Fig. 8). In preparation for manufacturing rCIG for clinical trials, a comprehensive polishing strategy was developed. Stress testing showed that the polished protein quality and function was highly stable, suggesting that rCIG was amenable to large-scale manufacturing (Supplementary Fig. 9). We completed this entire process, from delivery of the first donor sample to laboratory-scale generation of the rCIG protein product, in less than 3 months.
Antibody RNA sequencing of the final CHO cell bank indicated that the rCIG drug candidate comprised a diverse set of 12,500 antibodies (Fig. 2c and Supplementary Table 4). Additional repertoire analysis of the linked scFv and CHO cell bank libraries for rCIG was performed, including variable gene usage frequency, divergence from germline, CDR3H length distribution and sequence logos of the most abundant clonal clusters (Supplementary Figs. 10 and 11). Anti-SARS-CoV-2 ELISA suggested that the binding titer of rCIG was between 99- and 747-fold higher than corresponding plasma (Fig. 2d, Supplementary Fig. 5 and Supplementary Tables 2 and 4). ELISAs with several natural variants of SARS-CoV-2 and antigens from related viruses, including SARS-CoV and Middle East respiratory syndrome (MERS) CoV, showed that rCIG bound a broader variety of antigen targets than IVIG or a neutralizing CoV-2 monoclonal antibody (mAb; Fig. 2e, Supplementary Fig. 12 and Supplementary Table 4). Finally, spike:ACE2 blocking assays, pseudotype virus neutralization assays and live SARS-CoV-2 neutralization assays suggested that the neutralizing titer of rCIG was between 44- and 1,767-fold higher than corresponding convalescent plasma (Fig. 2f, Supplementary Figs. 6 and 7 and Supplementary Tables 2 and 4). Antibody RNA sequencing of the CHO cells and SARS-CoV-2 ELISA binding and SARS-CoV-2 pseudotype neutralization of rCIG protein generated from replicate 3-l bioreactor runs did not show significant batch-to-batch variation in antibody sequence content (Wilcoxon rank sum test, P > 0.05) or in vitro pseudotype neutralization (Feltz and Miller’s asymptotic test, P > 0.05; Supplementary Fig. 13).
Recombinant hyperimmune globulin for Zika virus
To address the Zika pandemic, we used our technology to build recombinant hyperimmune globulins against Zika virus, which we termed recombinant Zika immune globulin, or rZIG. Although convalescent Zika-infected donors may have been available internationally, we decided to use Zika as a test case to show how recombinant hyperimmune globulins could be built against an emerging pathogen in the absence of any human donors. Therefore, to create rZIG, we used human-transgenic mice (Trianni) that expressed a complete repertoire of human antibody sequences. The mice were immunized with Zika virus antigens (Supplementary Fig. 14). To explore our ability to engineer an rZIG that would not exhibit antibody-dependent enhancement (ADE), a safety concern for anti-Zika therapeutic antibodies, we additionally boosted with four inactivated dengue virus serotypes.
We used B cells from the immunized animals and our microfluidics technology to create an scFv library of natively paired IgGs. The resulting scFv library comprised approximately 119,700 IgG–IgK clonotypes (Supplementary Table 5). Because enrichment by flow sorting is time-consuming and makes (possibly inappropriate) choices about viral epitope targets, we decided to assess the potency of an rZIG product produced without enrichment by flow sorting. To this end, we used the unsorted scFv library and our CHO engineering technology to create rZIG CHO cell banks with a wild type human IgG1 isotype (rZIG–IgG1) or a mutated human IgG1 with abrogated Fc receptor (FcR) binding (rZIG–LALA)22. Antibody RNA sequencing of IgG sequences in the rZIG cell banks suggested that the rZIG–IgG1 comprised 33,642 antibodies and rZIG–LALA comprised 26,708 antibodies (Fig. 3a and Supplementary Table 6). A Morisita overlap of 86% and a Jaccard overlap of 58% between the rZIG–IgG1 and rZIG–LALA libraries suggested that the cell banks comprised substantially similar antibody repertoires. Additional repertoire analysis of the linked scFv and CHO cell bank libraries for rZIG was performed, including variable gene usage frequency, divergence from germline, CDR3 length distribution and sequence logos of the most abundant clonal clusters (Supplementary Fig. 15). We used these CHO cell banks to produce rZIG–IgG1 and rZIG–LALA hyperimmune globulins at laboratory scale (Supplementary Figs. 16 and 17).

a, Clonal cluster analysis of rZIG–IgG1 (blue) and rZIG–LALA (green) antibodies. Each node represents an antibody clone (full-length heavy chain). The size of the nodes reflects the frequency of the clones in the final CHO cell bank (only clones ≥0.01% are plotted). We computed the total number of amino acid differences between each pairwise alignment after combining both libraries together, and edges indicate ≤5 amino acid differences. b, ELISA of rZIG–IgG1 (blue), rZIG–LALA (green) and Zika/dengue+ serum control (red) for dengue serotypes 1–4 (y axis, indicated by shape) and Zika virus antigen (x axis). Each data point represents a single test article measured against a single dengue serotype. Linear regression trendline is indicated in black. Simple linear regression was used to calculate the coefficient of determination (R2) between Zika and dengue ELISA EC50 values (n = 7, in a single experiment). EC50 values for all dengue serotypes were pooled for the analysis. Significance of the regression model was determined using an F-statistic with 1 and 10 d.f. c, Pseudotype neutralization by rZIG–IgG1 (blue), rZIG–LALA (green) and Zika/dengue+ serum control (red) for dengue serotypes 1–4 (y axis, indicated by shape) and Zika virus antigen (x axis). Each data point represents a single test article measured against a single dengue serotype, in a single experiment. Linear regression trendline is indicated in black. Simple linear regression was used to calculate the coefficient of determination (R2) between Zika and dengue pseudotype neutralization IC50 values (n = 11). IC50 values for all dengue serotypes were pooled for the analysis. Significance of the regression model was determined using an F-statistic with 1 and 10 d.f. d, Zika pseudotype virus ADE assay for rZIG–IgG1 (blue), rZIG–LALA (green) and positive and negative controls. Test article concentration is on the x axis. Fold-increase infection is on the y axis, which was the infection-induced luciferase signal observed in the presence of antibody divided by the luciferase signal observed with a no-antibody control. Each data point represents a single measurement at a single test article dilution, in a single experiment.
Anti-Zika virus ELISA showed that both rZIG–LALA and rZIG–IgG1 had >75-fold higher titers against Zika virus than a human Zika positive serum sample (Supplementary Fig. 18 and Supplementary Table 6). Both rZIG–LALA and rZIG–IgG1 had anti-dengue binding activity across four serotypes, with pooled EC50 values showing strong correlation with anti-Zika EC50 values (linear regression, R2 = 0.9993, F-statistic P < 0.001; Fig. 3b, Supplementary Fig. 19 and Supplementary Table 5). In contrast, although both rZIG–LALA and rZIG–IgG1 had strong activity in a Zika pseudotype neutralization assay (Supplementary Fig. 20), there was no correlation between Zika and pooled dengue neutralization (linear regression, R2 = 0.00271, F-statistic P > 0.05; Fig. 3c and Supplementary Fig. 21). We investigated whether the abrogated Fc function of rZIG–LALA could decrease ADE in a Zika pseudotype virus assay (Supplementary Fig. 22). Both Zika+ human serum and rZIG–IgG1 showed considerable ADE, whereas rZIG–LALA showed no detectable ADE (Fig. 3d). Antibody RNA sequencing of the CHO cells and anti-Zika virus ELISA binding of rZIG–IgG1 and rZIG–LALA protein generated from replicate bioproduction runs did not show significant batch-to-batch variation in antibody sequence content (Wilcoxon rank sum test, P > 0.05), and batch-to-batch anti-Zika virus ELISA results were indistinguishable (Supplementary Fig. 23).
IVIG spike-in for patients with PID
Plasma-derived IVIG acts as antibody replacement for patients with humoral PID, who have low serum IgG titers. However, it has insufficient antipathogen activity for certain patients at-risk for PID. To address this unmet clinical need, we manufactured recombinant hyperimmune globulins directed against pneumococcus and Hib bacteria, designed as multivalent ‘spike-ins’ for plasma-derived IVIG, that is, recombinant Haemophilus immune globulin (rHIG) and recombinant pneumococcus immune globulin (rPIG). Note that rHIG and rPIG are not replacements for IVIG, but rather supplements meant to increase the efficacy of IVIG. A full recombinant replacement for IVIG would require much broader antipathogen activity.
We recruited healthy human donors and administered vaccines directed against pneumococcus or Hib. Eight to nine days after vaccination, peripheral blood mononuclear cells (PBMCs) were collected and shipped to our microfluidics processing facility. We selected B cells from the PBMCs, ran millions of cells through our microfluidics platform (Supplementary Table 7), and then used the scFv libraries and our CHO engineering technology to create IgG1 CHO cell banks for rHIG and rPIG. Heavy chain antibody RNA sequencing of the cell banks indicated that rHIG comprised 49,206 IgG sequences and rPIG comprised 17,938 IgG sequences (Fig. 4a and Supplementary Tables 8 and 9). Additional repertoire analysis of the linked scFv and CHO cell bank libraries for rHIG and rPIG was performed, including variable gene usage frequency, divergence from germline, CDR3 length distribution and sequence logos of the most abundant clonal clusters (Supplementary Figs. 24 and 25). We used these CHO cell banks to produce rHIG and rPIG hyperimmune globulins at laboratory scale (Supplementary Figs. 26 and 27).

a, Clonal cluster analysis of rHIG (green) and rPIG (blue) antibodies. Each node represents an antibody clone (full-length heavy chain). The size of the nodes reflects the frequency of the clones in the final CHO cell bank (only clones ≥0.01% are plotted). We computed the total number of amino acid differences between each pairwise alignment, and edges indicate ≤5 amino acid differences. b, Anti-Hib ELISA for rHIG (green) and IVIG (black). Each data point represents a single measurement at a single test article dilution, in a single experiment. c, Serum bactericidal assay (SBA) for rHIG (green) and IVIG (black) with the ATCC 10211 Hib strain. Percentage of no-antibody control (y axis) was computed as the number of bacterial colonies in the test sample divided by the number of bacterial colonies in a no-antibody control sample. Each data point represents a single measurement at a single test article dilution, in a single experiment. d, ELISA binding to (dark blue) or opsonophagocytosis of (light blue) the indicated pneumococcal serotype. Fold improvement in binding/activity over IVIG was computed as a mean of duplicate measurements for rPIG divided by a mean of duplicate measurements for IVIG (based on the binding concentration for ELISA and the number of bacterial colonies for opsonophagocytosis). Fold improvement over IVIG, by assay (ELISA or opsonophagocytosis) was tested using a one-sample Wilcoxon signed rank test, with the null hypothesis that the median equals 1, that is, H0 = 1. For each assay, all individual serotypes were pooled a single Wilcoxon signed rank test. Values for each individual serotype were generated by dividing the mean of duplicate rPIG measurements by the mean of duplicate IVIG measurements. e, In vivo assay with ATCC 10211 Hib strain. Each circle represents CFU Hib per ml (y axis) from either peritoneal fluid or blood from a single mouse in a given test group. Black bars represent mean of the CFU Hib per ml. Dotted lines represent the lower limit of detection for CFU quantification. Welch’s t-tests were used to compare CFU Hib per ml between test groups (n = 8 mice per group, in a single experiment). d.f. were 7.87 for IVIG + rHIG/rPIG (500 mg kg−1) and 7.13 for IVIG + rHIG/rPIG (200 mg kg−1) in peritoneal fluid. d.f. were 10.87 for IVIG + rHIG/rPIG (500 mg kg−1) and 8.03 for IVIG + rHIG/rPIG (200 mg kg−1) in blood.
Anti-Hib ELISA indicated that rHIG had 233-fold higher titer than plasma-derived IVIG (Fig. 4b and Supplementary Table 8). A serum bactericidal assay demonstrated that rHIG was strongly active against two different Hib strains, whereas no bactericidal activity was observed for plasma-derived IVIG (Fig. 4c, Supplementary Fig. 28 and Supplementary Table 8). An ELISA against a combination of 23 pneumococcus serotypes showed that rPIG has 85-fold higher titer than plasma-derived IVIG (Supplementary Fig. 29 and Supplementary Table 9). ELISA for individual pneumococcus serotypes showed that rPIG was at least fivefold higher titer than plasma-derived IVIG for 13 out of 16 serotypes measured, indicating broadly enriched multivalent reactivity and significantly higher than IVIG overall across all separate ELISAs combined (Wilcoxon signed rank test, P = 0.00123; Fig. 4d and Supplementary Table 9). Finally, semiquantitative serotype-specific opsonophagocytosis assays suggested that rPIG was as effective or more effective than plasma-derived IVIG at cell killing for 15 out of 16 serotypes tested, and had significantly higher activity than IVIG across all separate opsonophagocytosis assays combined (Wilcoxon signed rank test, P = 0.00251; Fig. 4d and Supplementary Table 9). Antibody RNA sequencing of the CHO cells and anti-Hib or antipneumococcal ELISA binding of rHIG or rPIG protein generated from replicate bioproduction runs, respectively, did not show significant batch-to-batch variation in antibody sequence content (Wilcoxon rank sum test, P > 0.05), and batch-to-batch antipathogen ELISA results were indistinguishable (Supplementary Figs. 30 and 31).
To simulate the potential clinical application, rHIG and rPIG were mixed in with plasma-derived IVIG (IVIG + rHIG/rPIG) at a ratio of 1:1:8 (rHIG:rPIG:IVIG), producing a product with 18.3-fold higher titer than plasma IVIG for Hib and 8.3-fold higher titer than plasma IVIG for a pool of 23 pneumococcus serotypes (Supplementary Fig. 32 and Supplementary Table 10). A Hib mouse challenge model using IVIG + rHIG/rPIG as prophylactic treatment showed significantly lower bacterial loads in the blood (Welch t-test, P < 0.001) and peritoneal fluid (Welch t-test, P < 0.001) as compared to plasma IVIG alone (Fig. 4e).
Recombinant human ATG for transplant tolerance
To encourage tolerance of grafts, transplant physicians use a variety immunosuppressive drugs23, such as rabbit-ATG, which is manufactured by injecting rabbits with human thymocytes and isolating antibodies from the rabbit serum24. However, rabbit-ATG can cause allergic reactions and other complications in humans11, and the drug shows significant variation in potency across lots15. To improve on rabbit-ATG, we made a recombinant human ATG, or rhATG, derived from transgenic mice that express human antibodies (Trianni). The mice were immunized with either human T cells or human fetal thymocytes (Supplementary Fig. 33). We used B cells from the immunized animals and our microfluidics technology to create four scFv libraries of natively paired IgGs: bone marrow cells from T-cell immunized mice, lymph node cells from T-cell immunized mice, lymph node cells from thymocyte immunized mice and spleen cells from thymocyte immunized mice. The resulting scFv libraries comprised a range of 13,314 to 34,324 IgG–IgK clonotypes (Supplementary Table 11). Additional repertoire analysis of the linked scFv and CHO cell bank libraries for rhATG was performed, including variable gene usage frequency, divergence from germline, CDR3 length distribution and sequence logos of the most abundant clonal clusters (Supplementary Fig. 34). We then used our CHO engineering technology to make cell banks from each of the four libraries.
We produced protein from each of the CHO cell banks, and then pooled the proteins in equal mass equivalents to create rhATG (Supplementary Fig. 35). Sequencing of individual libraries suggests that the pool comprised 49,885 antibodies (Fig. 5a and Supplementary Table 12). We then performed ELISA for a panel of known cell surface antigen targets for rabbit-ATG25 and observed that rhATG bound several immune cell surface targets, but only a subset of the targets bound by rabbit-ATG (Supplementary Fig. 36). To investigate further, we performed in vitro cell killing assays with human PBMCs, and showed that rhATG and rabbit-ATG were not significantly different in cell killing potency against cytotoxic T cells and helper T cells (linear mixed effects model, P > 0.05), whereas rhATG is significantly stronger than rabbit-ATG at killing B cells (linear mixed effects model, P < 0.01) but significantly weaker than rabbit-ATG at killing natural killer cells (linear mixed effects model, P < 0.01; Fig. 5b). We also performed anti-erythrocyte binding assays, which suggested that rhATG has less off-target activity than rabbit-ATG (Supplementary Fig. 37).

a, Clonal cluster analysis of rhATG antibodies. Each node represents an antibody clone (full-length heavy chain). The color of the nodes indicates the immunized library source. The shape of the nodes indicates the mouse tissue origin. The size of the nodes reflects the frequency of the clones in the final CHO cell bank (only clones ≥0.01% are plotted). We computed the total number of amino acid differences between each pairwise alignment, and edges indicate ≤5 amino acid differences. b, Cell killing assays of a dilution series of rabbit-ATG (red) and rhATG (blue) with three PBMC donors. The y axis (% cells) was determined by dividing the number of cells of the indicated cell type present after overnight incubation with the indicated amount of antibody by the number of cells of that cell type present in a no-antibody control. Each data point represents a single measurement at a single test article dilution, in a single experiment. Linear mixed effects models were used to compute P values for each of the four cell types, with group and concentration as fixed effects and PBMC donor as a random effect to account for the dependence of repeated measures. d.f. were 31 for each of the four models. NK, natural killer. c, Survival of mice (n = 8 per treatment group, in a single experiment) in the GVH study using PBMC donor 1 treated every other day with a negative vehicle control (black), rabbit-ATG (red) or rhATG (blue). Treatment days are indicated by green triangles. Kaplan–Meier survival models were fit on time to mortality and pairwise log-rank tests were performed to compare median survival between treatment groups. d, Flow cytometry was used to determine the concentration of CD45+ cells from each alive mouse on days 9, 16, 23 and 30 of the GVH study from c for negative vehicle control (black circles), rhATG (blue circles) or rabbit-ATG (red circles). Lines connect measurements from each mouse. No CD45+ cells were observed where circles intercept the x axis. Linear mixed effects models were used to compute P values for trends in CD45+ cell counts in each of the four GVH experiments (2 PBMC donors × 2 drug dosing regimens = 4 experiments) with day as a fixed effect and PBMC donor as a random effect to account for the dependence of repeated measures. A Wilcoxon rank sum test was used to compare CD45+ cell counts on day 9 for saline negative control versus rhATG and saline negative control versus rabbit-ATG, in each of the four GVH experiments (2 PBMC donors × 2 drug dosing regimens = 4 experiments).
We studied the efficacy of rhATG in vivo, using a graft-versus-host (GVH) model in which human PBMCs were grafted onto immune-incompetent mice26. We dosed animals (n = 8 per PBMC donor) with rhATG, rabbit-ATG or vehicle control, either every other day for 5 weeks starting 5 days after the PBMC graft, or only on days 5, 6 and 7 after the graft. Two different PBMC donors were tested for each dosing regimen. After 42 days, rhATG was not significantly different from rabbit-ATG for survival (log-rank pairwise tests, P > 0.05) and was superior to vehicle control for survival (log-rank pairwise tests, P < 0.001), in both dosing schemes across multiple PBMC donors (Fig. 5c and Supplementary Fig. 38). In both dosing regimens across both PBMC donors, immune cell (CD45+) expansion was not significantly different between rhATG and rabbit-ATG (linear mixed effects model, P > 0.05), whereas for the vehicle control immune cell counts were significantly higher than rhATG at day 9 (Wilcoxon rank sum tests, P < 0.01; Fig. 5d and Supplementary Fig. 39). We concluded that although rhATG and rabbit-ATG did not share identical antigen targets, the drugs had similar efficacy in vivo.
Discussion
We have demonstrated the generation of multivalent, 103 to 104 diverse recombinant hyperimmune antibody drugs from convalescent human blood donors, vaccinated human blood donors and humanized mouse repertoires. We extensively validated the drug candidates using in vitro and in vivo methods, highlighting their advantages over plasma-derived incumbents. Our technology combines methods in microfluidics, genomics and mammalian cell engineering. Contrasted against previous methods for generating recombinant polyclonal antibodies17, our drug candidates had hundreds-fold higher antibody diversity and therefore represented higher fractions of antigen-reactive repertoires. Their advantages over plasma-derived products include higher potency, the ability to scale production without collecting further donors, consistency of production and the ability to modulate pharmacologic problems such as ADE. The technology is fast, producing a master cell bank against a poorly characterized virus (SARS-CoV-2) in less than 3 months. The rCIG product (GIGA-2050) has now been manufactured at a good manufacturing practice (GMP) facility. GMP production was similar to the methods described in this study, except that upstream bioproduction was performed in a single use bioreactor at the 250-liter scale and downstream purification was scaled equivalently.
Emerging viruses are a constant and unpredictable threat to human health. In the past two decades alone, the world has seen outbreaks of Ebola virus27, SARS28, MERS29, 2009 H1N1 swine flu30, Zika Virus31 and SARS-CoV-2 (ref. 32), among others. Notwithstanding recent successes in rapid development of SARS-CoV-2 prophylactic vaccines, previous vaccine development efforts required very long development timelines33. Development of broadly neutralizing monoclonal antibodies is often confounded by the difficulty of identifying broadly neutralizing epitopes27, and escape variants can emerge over time. Because of such issues, convalescent COVID-19 plasma emerged as a promising approach early in the COVID-19 pandemic6,7. However, convalescent plasma is difficult to manufacture at scale because convalescent plasma supply is constrained and each plasma donor supplies enough therapeutic for only 1–2 patients. We concluded that rCIG was a promising alternative to COVID-19 convalescent plasma due to significantly higher potency against live virus (Wilcoxon rank sum test, P = 0.02869) and the ability to scale GMP production without the need to recruit more donors.
Although Zika virus has been less widespread and less deadly than SARS-CoV-2, Zika can spread from mother to fetus in utero, resulting in birth defects such as microcephaly34. As of July 2020, there was no FDA-approved vaccine or therapy for Zika virus. Zika virus disease is complicated by ADE35,36, a phenomenon in which poorly neutralizing antibodies enhance viral infection by bringing virus particles to cells that express FcR. This problem is particularly troublesome for individuals who have been previously infected with dengue, a related flavivirus, since many anti-dengue antibodies are poor neutralizers against Zika virus and vice versa37. ADE is a safety concern in the development of plasma-derived hyperimmune globulins and vaccines38. We concluded that rZIG–LALA was a promising alternative to plasma-derived drugs, due to high neutralizing potency against Zika virus and complete abrogation of the risk of ADE.
Although plasma-derived IVIG reduces rates of serious infections in PID, many patients still suffer frequent serious infections that require hospitalization39. In particular, about 78% of serious lung infections are caused by pneumococcus and Hib bacteria40. Clinicians have improved outcomes by further increasing IVIG doses41, suggesting that, however, bacterial species are often incredibly diverse; for example, there are 90 known pneumococcus serotypes42, complicating therapeutic development. We concluded that a polyvalent IVIG + rHIG/rPIG product had strong potential to address unmet clinical needs in patients with PID through increased potency against key pathogens.
In 2019, nearly 40,000 solid organ transplants were performed in the USA alone (www.unos.org). Transplantation generally introduces at least some mismatch between the human leukocyte antigen genotypes of the donor and host. This frequently results in some host-versus-graft effects, leading to loss of the graft and other serious complications. Our work suggested that rhATG could one day address unmet clinical needs in transplant tolerance.
In the future, there are many opportunities to improve the manufacturing processes we report here. First, input linked scFv repertoires typically comprised approximately two- to fourfold more antibodies than the final CHO cell banks, due to inefficiency of mammalian cell transfection with plasmids. Our previous work to clone T-cell receptor (TCR) repertoires into Jurkat cells was much more efficient43, but that work used lentivirus rather than Flp-In site-directed integration, and therefore many Jurkat clones expressed multiple TCRs. Another issue is the inability to characterize the protein diversity of the drug candidates; rather, antibody RNA sequencing is used as a proxy for protein diversity. Advances in proteomics may help to solve this problem44. Our method relies on PCR amplification of RNA from single cells, which may suffer from amplification bias45. Although our goal was not necessarily to precisely recapitulate the input repertoires, PCR bias can result in undesired antibody ratios. Improvements in throughput and cost of DNA synthesis46 may abrogate the need for PCR amplification and allow for precise engineering of complex antibody mixtures.
The drug candidates we describe can also be improved. For example, rPIG did not bind equivalently to different serotypes. To improve this product, we could sort yeast scFv libraries for binders to specific serotypes and then mix in ratios that might be considered more clinically appropriate. Also, although rhATG functioned similarly to rabbit-ATG in our in vivo GVH model and showed less off-target binding, further in vivo work may reveal that a different multivalent mixture would be more efficacious. Such a mixture could be made by further optimizing mouse immunizations, and/or implementing a yeast scFv sorting protocol that selects for on-target specificity using cell lysate, as we have reported previously47.
Our technology for generating recombinant hyperimmune globulins combines the advantages of recombinant antibodies (purity, consistency, potency) with the advantages of plasma-derived antibodies (proven efficacy, diversity, polyvalence, in vivo affinity maturation). In this current study, we have shown how the technology can improve existing plasma-derived products such as IVIG and rabbit-ATG. Polyclonal antibodies contain drugs with different mechanisms of action, potentially increasing efficacy; for example, rCIG may contain some antibodies optimized to block binding of virus to ACE2 and other antibodies optimized to clear virus through complement fixation. In the future, our technology could be used to develop drugs with new mechanisms of action, for example, antitumor antibody mixtures or antiplasma cell mixtures to cure humoral-driven autoimmune disease. It could also be extended to develop recombinant polyclonal IgM or IgA. To strengthen readiness against future pandemics, recombinant hyperimmune globulin cell banks against the most pressing biodefense threats could be produced preemptively.
Methods
Generating paired heavy and light chain libraries
Generation of scFv libraries from antibody-producing cells18 comprises three steps: (1) poly(A) + mRNA capture, (2) multiplexed OE–RT–PCR) and (3) nested PCR. Briefly, a microfluidic device captures single cells in droplets with a mixture of lysis buffer and oligo dT beads (NEB). After the cell is lysed and messenger RNA is bound to the bead, the emulsion is broken and the mRNA-containing beads are purified. Next, an emulsion is created using OE–RT–PCR reagents including a pool of primers directed against the IgK C region, the IgG C region and all V regions (Supplementary Table 13)48, and the mRNA-bound beads as a template. The emulsion is subjected to thermal cycling, which creates complementary DNA, amplifies the IgK and IgG variable regions and links them together in an scFv format. Then the emulsion is broken and the linked scFv DNA product is extracted and purified. The purified scFv product is then amplified using nested PCR to remove artifacts and add adapter sequences. Depending on the adapter sequences, the product can be used for deep sequencing, yeast display libraries or full-length CHO expression.
To convert the scFv libraries into full-length CHO expression libraries, we first used nested outer PCR primers to add adapters with overhangs for Gibson assembly to the 5′ and 3′ ends of the scFv library (for rCIG, this was done after yeast scFv display enrichment, as described in the next section). Then NEBuilder HiFi DNA Assembly Master Mix (NEB) was used to insert the scFv library into a vector containing a single promoter, a secretory leader sequence for light chain immunoglobulin and the remainder of the IgG1 constant region, creating a cloned scFv library (GA1 backbone, Supplementary Fig. 2; GenBank accession number MW079271). This intermediate library (GA1 product, Supplementary Fig. 2; example plasmid sequence provided as GenBank accession number MW079272) was transformed into E. coli and plasmids were purified by either (1) spreading onto LB-ampicillin plates, scraping 0.5–1 million colonies and pooling or (2) inoculating directly into LB-ampicillin broth and growing overnight. Plasmid purification was performed using ZymoPURE II Plasmid Maxiprep Kits (Zymo Research). To create the full-length antibody library, we performed a second Gibson assembly by linearizing the GA1 product with BamHI-HF (rHIG) or NheI-HF (rCIG, rPIG, rhATG and rZIG) (NEB) and using it as a vector to insert a synthetic amplicon (Supplementary Fig. 2, GenBank accession number MW079275) containing a portion of the light chain immunoglobulin constant region, a poly(A) signal for light chain immunoglobulin, a promoter for the IgG gene and a secretory leader sequence for the IgG gene. The full-length library was then transformed into E. coli and spread on LB-ampicillin plates. We typically combined >0.5 million colonies and purified plasmid with a ZymoPURE II Plasmid Maxiprep Kits (Zymo Research) to make the full-length recombinant hyperimmune globulin maxiprep library for transfection (GA2 product, Supplementary Fig. 2; example plasmid sequence provided as GenBank accession number MW079273). When the transformed E. coli were inoculated directly into LB-ampicillin medium, a small volume of cells was plated to calculate the total number of transformants. In some cases, ampicillin was used for both plates and medium, whereas in other cases carbenicillin was used instead of ampicillin. Paired heavy and light chain libraries were made only once from each sample.
Enrichment for antigen binders by yeast scFv display
Polyclonal COVID-19 scFv libraries were sorted18 to enrich for relevant sequences. Briefly, yeast surface display scFv libraries were generated using COVID-19 scFv DNA libraries and a custom yeast surface display vector transformed by electroporation into EBY100 yeast strain (MYA-4941, ATCC). Surface displayed scFv sequences include a C-terminal myc tag to identify scFv expression with 1 μl per sample of undiluted anti-myc primary (A21281, Thermo Fisher Scientific) and 1 μl per sample of undiluted AF488 secondary antibody (A11039; Thermo Fisher Scientific). Binding to antigen was identified by staining with soluble biotinylated SARS-CoV-2 RBD antigen (SPD-C82E9, Acro Biosystems) at 1,200 nM and APC-streptavidin (SA1005, Thermo Fisher Scientific). Stained yeast libraries were sorted on a FACSMelody (BD Biosciences, with BD fluorescent activated cell sorting (FACS) Chorus software v.1.3.3) and double positive (AF488+/APC+) cells were collected. The gating strategy is outlined in Supplementary Fig. 4. The collected cells were expanded and sorted again to further enrich the libraries. After the second round of sorting, cells were expanded a third time before plasmid isolation with a Zymoprep Yeast Plasmid Miniprep kit (Zymo Research). The plasmid libraries were then used as template for barcoding PCR and subsequent analysis by deep sequencing (Illumina). Plasmid from twice-sorted libraries was used as template for PCR toward full-length CHO antibody expression. Yeast scFv sorting was performed only once from each yeast scFv library.
Bioproduction of rHIG and rhATG
Adapted Flp-In-CHO cells stably expressing antibody libraries were grown in media consisting of 90% BalanCD CHO Growth A Medium (Irvine Scientific), 9% Ham’s F-12 (Thermo Fisher Scientific), 1% FBS (Thermo Fisher Scientific), 4 mM Glutamax (Thermo Fisher Scientific), 0.2% anticlumping agent (Irvine Scientific), 600 μg ml−1 Hygromycin-B (Gemini Bio). Protein production was performed at either small (250 ml) or medium (5 l) scale. For small-scale production, cells were seeded at 1 × 106 cells per ml into 50 ml of media in a 250 ml Erlenmeyer flask and grown at 37 °C, 5% CO2, 125 r.p.m. Cells were continually grown under these conditions and supplemented with 7.5 ml of CHO Feed 1 (Irvine Scientific) on days 2, 4 and 7 of the production run. Supernatant was harvested on days 8 or 9 by centrifugation followed by filtration through a 0.22-μm 250 ml filter bottle (MilliporeSigma) with a 1 μm prefilter (MilliporeSigma). Harvested cell culture fluid (HCCF) was stored at 4 °C (if less than 1 week) or at −80 °C (if more than 1 week) until Protein A purification. For medium-scale production, cells were grown in the same media. Cells were then seeded at 1 × 106 cells per ml in 2.3 l in a 5 l flask (in duplicate, day 0). Each flask was fed with 345 ml of CHO Feed 1 (Irvine Scientific) on days 2 and 4 of the culture. Cultures were gathered on days 8 or 9. Each of the four rhATG protein libraries were produced separately. Bioproduction was performed twice each for rHIG and rhATG.
Bioproduction of rPIG, rZIG and rCIG
CSS-1286 CHO cells stably expressing antibody libraries were grown in media without glutamine (EX-CELL CHOZN Advanced, MilliporeSigma). Protein production was performed at either small (500 ml flask) or medium (5 l flask) scale. For small-scale production, cells were seeded at 0.5 × 106 cells per ml into 100 ml media in a 500 ml Erlenmeyer flask and grown at 37 °C, 5% CO2, 125 r.p.m. Cells were continually grown under these conditions and supplemented with 15 ml of CHO Feed 1 (MilliporeSigma) on day 3, and 10 ml of CHO Feed 1 (MilliporeSigma) on days 6 and 8 of the production run. Starting on day 3, glucose was measured each day and supplemented to 6 g l−1 if below 4 g l−1. Supernatant was harvested after cell viability peak and before dropping below 70% viability between days 9 and 11, centrifuged and filtered through a 0.22-μm 250 ml filter bottle (MilliporeSigma) with a 1 μm prefilter (MilliporeSigma). HCCF was stored at 4 °C (if less than 1 week) or at −80 °C (if more than 1 week) until Protein A purification. For medium-scale production, cells were grown in the same media. Cells were seeded at 0.5 × 106 cells per ml in 2.2 l in a 5 l flask (in duplicate, day 0). Each flask was fed with 330 ml of CHO Feed 1 (MilliporeSigma) on day 3 and 220 ml of CHO Feed 1 (MilliporeSigma) on days 6 and 8 of the production run. Starting on day 3, glucose was measured each day and supplemented to 6 g l−1 if below 4 g l−1. Cultures were gathered on days 10–12. Bioproduction was performed three times for rCIG and twice each for rZIG and rPIG.
Protein production and characterization
After harvest, HCCF was purified using MabSelect PrismA (Cytiva) using 1 × PBS (Teknova) for running and wash buffer, 0.1 M Citrate, pH 3.0 (Teknova) for elution and 1 M sodium citrate pH 6.0 (Teknova) for neutralization. The protocol was 10 column volumes (CV) of equilibration, HCCF loading, 10 CV of washing and 5–10 CV of elution followed by cleaning-in-place with 1 M NaOH. HCCF was loaded with a 1-min residence time. Eluted material was neutralized to a pH of roughly 4.5 and centrifuged to remove any precipitation. This material was dialyzed into 0.2 M glycine, pH 4.5 (Teknova) using a 20K molecular weight cutoff dialysis cassette (Thermo Fisher Scientific) and optionally concentrated up to 30 mg ml−1 using a 50 kDa molecular weight cutoff spin device (MilliporeSigma). Final material was sterilized with a 0.22 μm filter and quantified by A280 (NanoDrop, Thermo Fisher Scientific). For rhATG, each of the four libraries were purified by Protein A separately and then equally pooled based on mass.
Purity of the protein was determined by size-exclusion–HPLC. Here, 20 μg of material at 1 mg ml−1 was injected over a 300 Å, 2.7 μm, 7.8 × 300 mm size-exclusion column (Agilent) using a mobile phase of 25 mM phosphate, 200 mM NaCl pH 7.0 with 10% acetonitrile at 1 ml min−1. The percentage of monomer was determined by integrating the product peaks and reporting the percent area corresponding to roughly 150 kDa. The product was further characterized by running 2 μg on a 12% SDS–PAGE gel under reduced and nonreduced buffering conditions and imaged after staining with SimplyBlue SafeStain (Thermo Fisher Scientific).
Protein production was performed once for each of the bioproduction runs.
Statistical analysis
All statistical tests were performed on nonnormalized data, two-sided without adjustments to type I error rates. We used a significance threshold of α = 0.05 for all statistical tests. All statistical analyses were conducted using R v.3.6.2.
For Fig. 2f, a Wilcoxon rank sum test was used to compare the minimum concentration to achieve SARS-CoV-2 live virus neutralization between convalescent plasma measurements (n = 16) and rCIG measurements (n = 2).
For Fig. 3b, simple linear regression was used to calculate the coefficient of determination (R2) between Zika and dengue ELISA EC50 values. EC50 values for all dengue serotypes were pooled for the analysis. Significance of the regression model was determined using an F-statistic with 1 and 10 degrees of freedom (d.f.). All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 3c, simple linear regression was used to calculate the coefficient of determination (R2) between Zika and dengue pseudotype neutralization half-maximum inhibitory concentration (IC50) values. IC50 values for all dengue serotypes were pooled for the analysis. Significance of the regression model was determined using an F-statistic with 1 and 10 d.f. All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 4d, fold improvement over IVIG, by assay (ELISA or opsonophagocytosis) was tested using a one-sample Wilcoxon signed rank test, with the null hypothesis that the median equals 1, that is, H0 = 1. For each assay, all individual serotypes were pooled a single Wilcoxon signed rank test. Values for each individual serotype were generated by dividing the mean of duplicate rPIG measurements by the mean of duplicate IVIG measurements. All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 4e, Welch’s t-tests were used to compare colony forming units (CFU) Hib per ml between test groups and d.f. were 7.87 for IVIG + rHIG/rPIG (500 mg kg−1) and 7.13 for IVIG + rHIG/rPIG (200 mg kg−1) in peritoneal fluid, and were 10.87 for IVIG + rHIG/rPIG (500 mg kg−1) and 8.03 for IVIG + rHIG/rPIG (200 mg kg−1) in blood. All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 5b, linear mixed effects models were used to compute P values for each of the four cell types, with group and concentration as fixed effects and PBMC donor as a random effect to account for the dependence of repeated measures: the d.f. were 31 for each of the four models. All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 5c and Supplementary Fig. 38, Kaplan–Meier survival models were fit on time to mortality and pairwise log-rank tests were performed to compare median survival between treatment groups. All measurements were performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Fig. 5d and Supplementary Fig. 39, linear mixed effects models were used to compute P values for trends in CD45+ cell counts in each of the four GVH experiments (2 PBMC donors × 2 drug dosing regimens = 4 experiments) with day as a fixed effect and PBMC donor as a random effect to account for the dependence of repeated measures. A Wilcoxon rank sum test was used to compare CD45+ cell counts on day 9 for saline negative control versus rhATG and saline negative control versus rabbit-ATG, in each of the four GVH experiments (2 PBMC donors × 2 drug dosing regimens = 4 experiments). No power analysis was carried out to predetermine an appropriate sample size for this experiment.
For Supplementary Figs. 13, 23, 30 and 31, we assessed whether batch-to-batch variation was more significant than the variability inherent to the assays used to make the measurements.
For rCIG pseudotype neutralization assays (Supplementary Fig. 13), Feltz and Miller’s asymptotic test was used to determine whether the coefficient of variation of three bioproduction batch IC50 measurements (18%) was different from the coefficient of variation of eight IC50 measurements on a fourth bioproduction batch (17%).
To assess batch variation in antibody sequence content, the Wilcoxon rank sum test was used to test whether the Jaccard or Morisita indices from PCR replicates from each bioproduction batch came from the same populations as the Jaccard or Morisita indices among bioproduction batches. Sequencing was performed in a single experiment. No power analysis was carried out to predetermine an appropriate sample size for this experiment.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Plasmid and cloning insert sequences are available on GenBank (GA1 backbone, GenBank accession number MW079271; GA1 product, GenBank accession number MW079272; synthetic amplicon insert, GenBank accession number MW079275; GA2 product, example plasmid sequence provided as GenBank accession number MW079273; PMD-4681, GenBank accession number MW079274. Sequencing data are available in the Short Read Archive under project identifier PRJNA649279. All raw data (ELISAs, flow cytometry, in vitro neutralization assays and so on) can be made available on reasonable request for noncommercial use.
References
Bozzo, J. & Jorquera, J. I. Use of human immunoglobulins as an anti-infective treatment: the experience so far and their possible re-emerging role. Expert Rev. Anti Infect. Ther. 15, 585–604 (2017).
Beasley, R. P. et al. Efficacy of hepatitis B immune globulin for prevention of perinatal transmission of the hepatitis B virus carrier state: final report of a randomized double-blind, placebo-controlled trial. Hepatology. 3, 135–141 (1983).
Payne, J. R., Khouri, J. M., Jewell, N. P. & Arnon, S. S. Efficacy of human botulism immune globulin for the treatment of infant botulism: the first 12 years post licensure. J Pediatr. 193, 172–177 (2018).
Gaber, A. O. et al. Results of the double-blind, randomized, multicenter, phase III clinical trial of thymoglobulin versus atgam in the treatment of acute graft rejection episodes after renal transplantation. Transplantation. 66, 29–37 (1998).
Branche, E. et al. Human polyclonal antibodies prevent lethal Zika virus infection in mice. Sci. Rep. 9, 9857 (2019).
Bloch, E. M. et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. J. Clin. Invest. 130, 2757–2765 (2020).
Shen, C. et al. Treatment of 5 critically Ill patients with COVID-19 with convalescent plasma. JAMA 323, 1582–1589 (2020).
Boulis, A., Goold, S. & Ubel, P. A. Responding to the immunoglobulin shortage: a case study. J. Health Polit. Policy Law. 27, 977–999 (2002).
Bjøro, K., Frøland, S. S., Yun, Z., Samdal, H. H. & Haaland, T. Hepatitis C infection in patients with primary hypogammaglobulinemia after treatment with contaminated immune globulin. N. Engl. J. Med. 331, 1607–1611 (1994).
Etscheid, M. et al. Identification of kallikrein and FXIa as impurities in therapeutic immunoglobulins: implications for the safety and control of intravenous blood products. Vox Sang. 102, 40–46 (2012).
Brabant, S. et al. An avoidable cause of thymoglobulin anaphylaxis. Allergy Asthma Clin. Immunol. 13, 13 (2017).
Ober, R. J., Radu, C. G., Ghetie, V. & Ward, E. S. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 13, 1551–1559 (2001).
Simon, H. U. & Späth, P. J. IVIG–mechanisms of action. Allergy 58, 543–552 (2003).
Lejtenyi, D. & Mazer, B. Consistency of protective antibody levels across lots of intravenous immunoglobulin preparations. J. Allergy Clin. Immunol. 121, 254–255 (2008).
Popow, I. et al. Assessment of batch to batch variation in polyclonal antithymocyte globulin preparations. Transplantation 93, 32–40 (2012).
Rita Costa, A., Elisa Rodrigues, M., Henriques, M., Azeredo, J. & Oliveira, R. Guidelines to cell engineering for monoclonal antibody production. Eur. J. Pharm. Biopharm. 74, 127–138 (2010).
Frandsen, T. P. et al. Consistent manufacturing and quality control of a highly complex recombinant polyclonal antibody product for human therapeutic use. Biotechnol. Bioeng. 108, 2171–2181 (2011).
Adler, A. S. et al. Rare, high-affinity anti-pathogen antibodies from human repertoires, discovered using microfluidics and molecular genomics. MAbs 9, 1282–1296 (2017).
Soto, C. et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566, 398–402 (2019).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Kito, M., Itami, S., Fukano, Y., Yamana, K. & Shibui, T. Construction of engineered CHO strains for high-level production of recombinant proteins. Appl. Microbiol. Biotechnol. 60, 442–448 (2002).
Hessell, A. J. et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 (2007).
Hart, A. et al. OPTN/SRTR 2016 annual data report: kidney. Am. J. Transplant 18, 18–113 (2018).
Brennan, D. C., Daller, J. A., Lake, K. D., Cibrik, D. & Del Castillo, D. Thymoglobulin induction study G. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N. Engl. J. Med. 355, 1967–1977 (2006).
Popow, I. et al. A comprehensive and quantitative analysis of the major specificities in rabbit antithymocyte globulin preparations. Am. J. Transplant 13, 3103–3113 (2013).
Norelli, M., Camisa, B. & Bondanza, A. Modeling human graft-versus-host disease in immunocompromised mice. Methods Mol. Biol. 1393, 127–132 (2016).
Iversen P. L. et al. Recent successes in therapeutics for Ebola virus disease: no time for complacency. Lancet Infect Dis. 20, 231–237 (2020).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia [published correction appears in N Engl J Med. 2013 Jul 25;369(4):394]. N. Engl. J. Med. 367, 1814–1820 (2012).
Itoh, Y. et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460, 1021–1025 (2009).
Wikan, N. & Smith, D. R. Zika virus: history of a newly emerging arbovirus. Lancet Infect Dis. 16, e119–e126 (2016).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!) [published correction appears in Lancet. 2017 Feb 4;389(10068):504] [published correction appears in Lancet. 2017 Feb 4;389(10068):504]. Lancet 389, 505–518 (2017).
Mlakar, J. et al. Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016).
Katzelnick, L. C. et al. Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017).
Priyamvada, L. et al. Human antibody responses after Dengue virus infection are highly cross-reactive to Zika virus. Proc. Natl Acad. Sci. USA 113, 7852–7857 (2016).
Stettler, K. et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016).
Langerak, T. et al. The possible role of cross-reactive dengue virus antibodies in Zika virus pathogenesis. PLoS Pathog. 15, e1007640 (2019).
Gathmann, B. & Mahlaoui, N., CEREDIH. et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 134, 116–126 (2014).
Sperlich, J. M. et al. Respiratory infections and antibiotic usage in common variable immunodeficiency. J. Allergy Clin. Immunol. Pract. 6, 159–168.e3 (2018).
Orange, J. S., Grossman, W. J., Navickis, R. J. & Wilkes, M. M. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 137, 21–30 (2010).
Geno, K. A. et al. Pneumococcal capsules and their types: past, present, and future. Clin. Microbiol. Rev. 28, 871–899 (2015).
Spindler, M. J. et al. Massively parallel interrogation and mining of natively paired human TCRαβ repertoires. Nat. Biotechnol. 38, 609–619 (2020).
Lee, J. et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat. Med. 22, 1456–1464 (2016).
Vollmers, C., Sit, R. V., Weinstein, J. A., Dekker, C. L. & Quake, S. R. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc. Natl Acad. Sci. USA 110, 13463–13468 (2013).
Plesa, C., Sidore, A. M., Lubock, N. B., Zhang, D. & Kosuri, S. Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science 359, 343–347 (2018).
Medina-Cucurella, A. V. et al. Preferential identification of agonistic OX40 antibodies by using cell lysate to pan natively paired, humanized mouse-derived yeast surface display libraries. Antibodies 8, 17 (2019).
Meijer, P. J., Nielsen, L. S., Lantto, J. & Jensen, A. Human antibody repertoires. Methods Mol. Biol. 525, 261–277 (2009).
Acknowledgements
This work was partially funded by National Institute of Allergy and Infectious Diseases (NIAID) grant nos. R44AI115892 and R44AI124901, and NSF grant no. 1230150, to D.S.J. The staff at MedPharmics and Access Biologicals were instrumental in acquiring convalescent COVID-19 donor samples under IRB-approved protocols. The staff at Waisman Biomanufacturing performed 3-l scale bioproduction of rCIG. J. Keller (GigaGen, Inc.) helped to organize collection of COVID-19, Hib and pneumococcus donor samples. Antibody Solutions provided helpful advice in design of mouse immunization work for rZIG and rhATG and performed the immunizations. Jackson Laboratories provided useful advice in design of GVH studies and performed the studies. E. Stone (GigaGen, Inc.) provided useful assistance in the field of immunology and in critical reviews of the scientific direction. C. Keller (GigaGen, Inc.) and C. Wilson provided useful strategic advice. The following reagents were obtained through BEI Resources, NIAID, NIH: vector pCAGGS containing the SARS-CoV-2, Wuhan-Hu-1 spike glycoprotein gene RBD with C-terminal hexa-histidine tag, NR-52309; vector pCAGGS containing the SARS-related coronavirus-2, Wuhan-Hu-1 spike glycoprotein gene (soluble, stabilized), NR-52394 produced under HHSN272201400008C; spike glycoprotein RBD from SARS-related coronavirus-2, Wuhan-Hu-1, recombinant from human embryonic kidney-293T cells, NR-52306.
Author information
Authors and Affiliations
Contributions
Conceptualization, S.M.K., R.A. Mizrahi, E.H.M., A.S.A., and D.S.J.; methodology, S.M.K., R.A. Mizrahi, M.A.A., K.P.C., M.J.S., J.F.S., E.K.W., N.W., C.V.L., S.M.C., B.M., C.O., C.R.A., C.R.B, A.S.A., and D.S.J.; software, R.C.E. and Y.W.L.; investigation, S.M.K., R.A.A., M.S.A., M.A.A., E.B., K.P.C., Y.C., B.K.G., A.G., J.L., R.L., E.P., V.A.M., A.V.M-C., A.R.N., J.S., J.F.S., M.J.S., K.S., B.T., E.K.W., N.W., L.R., H.R., C.V.F.C., T.H.O., A.T.R., R.A. Mosher, and M.J.W.; data curation, S.M.K., R.A. Mizrahi, M.A.A., Y.W.L., L.T., A.S.A., and D.S.J.; writing—original draft preparation, D.S.J.; writing—review and editing, S.M.K., R.A. Mizrahi, A.S.A., and D.S.J.; visualization, S.M.K., Y.W.L., A.S.A., and D.S.J.; supervision, M.J.S., C.A.R., A.S.A., and D.S.J.; project administration, S.M.K., R.A. Mizrahi, C.V.L., D.B., J.V.T., D.G., G.S., M.O.M., H.L., R.J., C.A.R.., C.R.B, A.S.A., and D.S.J.; funding acquisition, D.S.J.
Corresponding author
Ethics declarations
Competing interests
S.M.K., R.A. Mizrahi, M.S.A., M.A.A., E.B., K.P.C., Y.C., R.C.E., B.K.G., A.G., J.L., R.L., Y.W.L., V.A.M., A.V.M-C., A.R.N., J.S., J.F.S., M.J.S., K.S., B.T., E.K.W., N.W., E.H.M., A.S.A. and D.S.J., have received shares and salary from GigaGen, Inc. which was acquired by Grifols on 8 March 2021. C.V.L. receives a salary from Statens Serum Institut. D.B. and J.V.T. receive salaries from Grifols. C.V.F.C. and A.T.R. receive a salary from The University of the West Indies. L.R., E.P., H.R. and D.G. receive salary from University College London and D.G. receives support from the NIHR GOSH Biomedical Research Centre. G.S. and M.O.M. receive salary from Vitalant. L.T., S.M.C., B.M. and C.O. received consulting fees from GigaGen, Inc. T.H.O. and H.L. receive a salary from Duke University Medical Center and were paid by GigaGen, Inc. to perform SARS-CoV-2 neutralization studies. R.J. is an employee of MedPharmics, which received payments for convalescent COVID-19 samples from GigaGen, Inc. R.A. Mosher, M.J.W., C.A.R. and C.R.B are salaried employees of Waisman Biomanufacturing. Waisman Biomanufacturing received payments from GigaGen for manufacturing services. E.H.M. is a salaried employee of Stanford University Medical Center. Methods for linkage of heavy and light chain immunoglobulin in emulsion droplets are granted in patents US20200140947A1, US10787706 and EP2652155B1 to D.S.J. and E.H.M. Methods for cloning antibody libraries are granted in patents US20190256841A1, US10689641, WO2018170013A1, WO2016200577A1, EP16808004.2 and US20160362681A1 to D.S.J., A.S.A., M.J.S. and R.A. Mizrahi. Antibody library compositions are described in patent applications US63/038,470 and US62/841,097, to D.S.J., A.S.A., R.A. Mizrahi, Y.W.L., S.M.K., R.L., J.L. and M.A.A.
Additional information
Peer review information Nature Biotechnology thanks George Georgiou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods and Figs. 1–40.
Supplementary Tables
Supplementary Tables 1–13.
Rights and permissions
About this article

Cite this article
Keating, S.M., Mizrahi, R.A., Adams, M.S. et al. Generation of recombinant hyperimmune globulins from diverse B-cell repertoires. Nat Biotechnol 39, 989–999 (2021). https://doi.org/10.1038/s41587-021-00894-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-00894-8
