
Abstract
Accurate prediction of antigen presentation by human leukocyte antigen (HLA) class II molecules would be valuable for vaccine development and cancer immunotherapies. Current computational methods trained on in vitro binding data are limited by insufficient training data and algorithmic constraints. Here we describe MARIA (major histocompatibility complex analysis with recurrent integrated architecture; https://maria.stanford.edu/), a multimodal recurrent neural network for predicting the likelihood of antigen presentation from a gene of interest in the context of specific HLA class II alleles. In addition to in vitro binding measurements, MARIA is trained on peptide HLA ligand sequences identified by mass spectrometry, expression levels of antigen genes and protease cleavage signatures. Because it leverages these diverse training data and our improved machine learning framework, MARIA (area under the curve = 0.89–0.92) outperformed existing methods in validation datasets. Across independent cancer neoantigen studies, peptides with high MARIA scores are more likely to elicit strong CD4+ T cell responses. MARIA allows identification of immunogenic epitopes in diverse cancers and autoimmune disease.
Similar content being viewed by others

Main
Major histocompatibility complex class II (MHC-II) is a glycoprotein complex on the surface of professional antigen-presenting cells that displays short antigen peptides to CD4+ helper T cells. Human antigen-presenting cells, such as dendritic cells and B cells, rely in large part on HLA class II (HLA-II) for the presentation of antigens to CD4+ T cells. This human form of MHC-II can also be conditionally expressed by many other human cell types, including tumor cells. Antigen presentation by these HLA-II molecules on human cells involves three loci on chromosome 6 (DR, DQ and DP) which encode the corresponding heterodimeric proteins through combinations of alpha and beta chains.
Such HLA-II presentation of endogenous and exogenous antigenic peptides is essential for robust immune responses against diverse pathogens, and is also of major significance for autoimmunity and antitumor immunity1. For example, recent mass spectrometry (MS)-based studies have shown that lymphoma and melanoma cells present somatically mutated cancer peptides (neoantigens) in the context of HLA-II2,3. CD4+ T cell recognition of neoantigens is commonly observed across diverse human tumor types and in animal models2,4,5,6,7, which underscores the potential clinical relevance of HLA-II-restricted neoantigens for cancer immunotherapy. Furthermore, neoantigens presented by HLA-II elicit potent antitumor responses in T cells from immunized patients8,9. Reliably identifying presentation by HLA-II would allow us to prioritize vaccine candidates and potentially identify likely responders to immune therapies10,11,12.
Owing to the high cost and technical challenge of experimentally testing all possible peptide candidates, researchers have attempted to computationally identify HLA-II peptides with machine-learning algorithms13. However, nearly all current HLA-II prediction methods rely on in vitro binding affinities of recombinant HLA-II molecules as surrogates, and therefore ignore other contributing factors including gene expression and protease cleavage preferences14,15. When combined with the remarkably variable length of HLA-II peptides and their binding promiscuity, this deficiency makes HLA-II antigen presentation prediction task especially challenging12,16. For example, the latest benchmarks report average receiver operating characteristic area under the curve (ROC-AUC or AUC) of ~0.83 for current prevailing HLA-II prediction models, even when validated on in vitro binding data15,17.
In this study, we present MARIA, a deep neural network trained to accurately predict the likelihood of a peptide being presented by HLA-II complexes. Rather than relying on in vitro binding affinities alone, MARIA is trained on naturally presented HLA-II peptides (ligands) identified from human samples profiled by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Despite some inherent limitations of MS methods, peptide ligand sequences identified by antigen presentation profiling currently provide the closest sample population to the true presented ligands3,18,19,20. Such training data could enable new prediction models to consider multiple relevant features including expression and binding affinities. Here we show that MARIA allows robust and more accurate HLA-II prediction, and that its performance gains are achieved by combining these improved training data with a new supervised machine learning model using a multimodal recurrent neural network (RNN).
Results
Performance of binding-based HLA-II peptide prediction methods
Immunoprecipitation of MHC molecules followed by peptide elution and LC–MS/MS analysis enables direct recovery of peptides presented by HLA-II in primary cells2,3. In comparison to traditional in vitro binding affinity assays, MS-based profiling methods can rapidly yield large datasets of peptides actively presented by cells or tissues. The Immune Epitope Database (IEDB), the largest public depository of results of HLA binding assays, contains quantitative HLA-DR binding affinities for ~12,000 non-redundant peptide sequences21 (as of December 2018). By comparison, two studies employing HLA-DR immunoprecipitation and MS analysis2,3 identified >23,000 and >16,000 non-redundant peptide sequences, respectively (Fig. 1a).

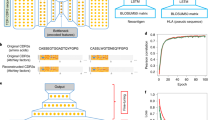
a, Comparison of the number of unique HLA-DR ligands identified within two antigen presentation profiling studies2,3, each exceeding all HLA-DR in vitro quantitative binding measurements from 239 previous studies within the IEDB (as of December 2018)21. b, Performance of NetMHCIIpan for discrimination of decoys from bona fide HLA-II ligands recovered by antigen presentation profiling. For each patient2, NetMHCIIpan-predicted affinities and ranks were separately evaluated (x axis), and performance measured by ROC-AUC (y axis, dotted lines represent the median). NetMHCIIpan ranks (mean AUC = 0.68) slightly outperformed NetMHCIIpan binding affinities (mean AUC = 0.65, n = 18; two-tailed paired t test, P = 0.003; Supplementary Table 1). c, Limited sensitivity of NetMHCIIpan for classification of HLA-DR ligands. Depicted is the distribution of NetMHCIIpan ranks for all 6,063 peptides identified from the JeKo-1 cell line, where 22% of HLA-II ligands identified by MS had predicted values worse than the recommended NetMHCIIpan rank cut-off for binding (10%). d, In vitro binding assay results for HLA-II peptide ligands identified by MS but predicted by NetMHCIIpan not to bind HLA-II. Among ten such peptides predicted by NetMHCIIpan not to bind, nine were nevertheless confirmed to bind cognate HLA-DR alleles (04:03 and/or 07:01) by two independent flow cytometry experiments. Scatter plots depict binding of two exemplar FITC-conjugated peptides (x axis) to APC-conjugated HLA-DR proteins (y axis); remaining peptides are separately depicted in Supplementary Fig. 1. e, Training and evaluation scheme of MARIA, as a new machine learning framework for more accurate prediction of HLA-II ligands. Positive examples are HLA-II ligand peptide sequences directly identified by antigen presentation profiling of human cells and tissues by immunoprecipitation (i.p.) and MS, and negative examples are length-matched random human peptides (decoys). The model separately considers binding affinities estimated using in vitro binding data. Patient HLA-II allele or genotype and gene expression information are obtained from next-generation sequencing. A RNN integrates information and produces a predictor for HLA-II ligand presentation by minimizing training errors. Independent test sets determine the final performance of the model. See Supplementary Fig. 2 for detailed machine learning schemes.
We tested the performance of the HLA binding affinity prediction tool trained on in vitro binding data to identify HLA-DR ligands presented by human antigen-presenting cells. We applied NetMHCIIpan16,22, a widely used HLA-II binding prediction method, to predict the binding affinity of HLA-DR ligands experimentally identified from 18 mantle cell lymphomas (MCLs)2 representing 16 HLA-DR alleles (Supplementary Table 1). We assessed the AUC of NetMHCIIpan using MS-identified ligands as true positives and randomly selected length-matched human peptide sequences (decoys) as negative examples. For each HLA allele and peptide sequence pair, NetMHCIIpan generates a binding affinity and binding ranks. Separately using these two values, we obtained average AUCs of 0.64 and 0.68 for NetMHCIIpan binding affinities and ranks, respectively (Fig. 1b). Consistent with previous studies15, binding ranks showed better prediction performance for ligand presentation (P = 0.003), but nevertheless had mediocre accuracy in predicting true HLA-II ligands.
We next tested the performance of NetMHCIIpan on >6,000 HLA-DR ligands discovered through deep profiling of the MCL-derived JeKo-1 cell line2. When using the recommended threshold of binding affinities (top 10% ranks)22, NetMHCIIpan labeled only ~22% of the ligands as positive (Fig. 1c). To confirm that the MS-identified peptides were indeed true ligands despite their weak NetMHCIIpan-predicted binding affinities, we experimentally validated binding of a subset of peptides with weak NetMHCIIpan scores along with positive and negative controls (Supplementary Fig. 1 and Supplementary Table 11). Remarkably, nine of ten synthesized peptides strongly bound to one or more cognate HLA-DR alleles in vitro, confirming the fidelity of the underlying MS data (Fig. 1c and Supplementary Fig. 1d). Thus, NetMHCIIpan, the top HLA-II peptide prediction binding algorithm15 has limited accuracy for predicting antigen presentation when applied to MS-based datasets.
Development of MARIA
We aimed to improve upon previous HLA-II prediction methods by developing a tool predicting active HLA-II presentation in vivo, rather than from in vitro binding affinities. We therefore examined whether HLA-II prediction could be refined by learning directly from MS-based antigen presentation profiling datasets, in addition to traditional HLA binding affinity data. Additionally, we tested whether gene expression and protease cleavage signatures also have utility in predicting HLA-II peptide presentation. We trained MARIA using the HLA-II ligands identified by MS-based antigen presentation profiling, along with empirical in vitro HLA binding measurements, and gene expression levels (Fig. 1e and Supplementary Fig. 2). Given the challenges associated with the high variability in the length of HLA-II peptide ligands (8–26 amino acids), we used a recurrent neural network (RNN) framework, a form of deep learning that excels at handling variable-length sequence data (Fig. 1e). To prevent model overfitting owing to similarities in the training and validation sequences, we filtered out any peptides in the cross-validation set that were a substring or highly similar to a training peptide. We evaluated the performance of the full model, as well as other models trained on each possible combination of biological features (Supplementary Table 2).
Impact of gene expression levels
We observed that gene expression levels of recovered HLA-DR ligands were significantly higher than both non-presented genes and random genes (Fig. 2a and Supplementary Fig. 3). Nevertheless, 8.4% of peptide ligands were encoded by genes with extremely low RNA expression levels in tumor cells (<0.1 transcripts per million (TPM)). Consistent with the known role of HLA-II molecules in sampling and presenting extracellular antigens, the presented ligands from these outliers were highly enriched for extracellular proteins and blood microparticles (P < 2 × 10−14, FDR-corrected hypergeometrical test; Fig. 2a, Supplementary Fig. 3 and Supplementary Table 3). We therefore included gene expression levels in our model and applied a correction to address presentation of extracellular proteins or blood particles (Supplementary Fig. 3b). When considering gene expression levels alone, we achieved an AUC of 0.81 when differentiating presented ligands from random human peptides (as detailed below). Not surprisingly, gene expression values had much weaker discriminatory power after removing lowly expressed genes in negative decoys (Supplementary Fig. 3d).

a, Comparison of gene expression levels of HLA-DR ligands and non-ligands. Gene expression was estimated by RNA-seq for HLA-DR-presented genes, all protein-coding genes and non-presented protein-coding genes, respectively. HLA-DR ligand genes have significantly higher gene expression levels than the set of all protein-coding genes (n = 34,049, 23,165 and 19,464, respectively; **P < 1 × 10−5, Mann–Whitney U test). Some HLA-DR ligands (8.4%) had undetectable levels of expression; those in this set were enriched for extracellular protein (GO enrichment; Fisher’s exact test, P < 1 × 10−17). Violin curves represent the probability distribution function of gene expression, black boxes represent middle two quartiles and white dots represent the median. See Supplementary Fig. 3 for detailed analysis on HLA-I ligands and the predictive power of gene expression levels. b, Cleavage signature analysis for HLA-DR ligands. Frequencies of 20 amino acids at 6 positions upstream (−6 to −1) and downstream (+1 to +6) of HLA-DR ligands (n = 12,150) are compared to the background distribution (n = 23,218) to determine amino acid enrichment and depletion surrounding HLA-DR ligands. Colors of the heat map and sizes of the logo plot letters indicate fold change. The logo plot only includes statistically significant enrichment (P < 0.001, two-tailed independent t test by IceLogo60). The minus symbol in the top row of the heat map indicates presented peptides that are located at the beginning or end of source protein sequences. See Supplementary Fig. 4 for cleavage signatures across different cell types. c, Workflow of MARIA for predicting HLA-DR ligand presentation score. Two separate models first calculate HLA-DR peptide binding scores and peptide cleavage scores. The neural network further integrates peptide sequence and estimated gene expression level with two scores, via a recurrent layer and merge layers, to generate a presentation score indicating likelihood of HLA-II presentation. d, Performance of MARIA and four alternative predictors on 10% of the held-out validation set (true MCL HLA-II ligands, n = 3,300; random human decoy peptides, n = 10,000; the same sample set is used in e and f). MARIA scores incorporating gene expression levels, peptide sequence, binding scores and cleavage scores outperformed methods using each of these features individually (DeLong test, P < 1 × 10−5; AUC = 0.92). See Supplementary Fig. 5 for detailed training data source and cross-validation performance. e, Comparison of model precision and specificity across a range of presented MCL HLA-DR peptide prevalences. Sensitivity for each model was controlled at 30% for all calculations, with corresponding specificity denoted adjacent to inset legend. The shaded areas represent the 95% confidence interval around the mean value, on the basis of tenfold cross-validation. f, Comparison of precision and recall for different models for predicting HLA-DR ligands using various types of training data. Precision was calculated assuming 1% prevalence of presented HLA-DR ligands. The shaded areas represent 95% confidence interval around the mean value (line), based on tenfold cross-validation.
We were able to obtain RNA sequencing (RNA-seq) gene expression profiles for six patients with MCL2. MARIA AUCs did not differ significantly between using patient-specific RNA-seq and an external RNA-seq profile (Supplementary Fig. 3e). Furthermore, we observed only a modest degradation in prediction performance when using tissue-mismatched gene expression values from a generalized reference database as compared to tissue-matched gene expression data (Supplementary Fig. 3f; change of AUC < 1%, not significant). Supplementary Note 1 contains a detailed discussion for the predictive power of gene expression for HLA-II presentation.
Impact of cleavage signatures
We next assessed whether information from the flanking residues of a peptide could further improve predictive performance. We observed both significant enrichment and depletion of certain amino acids at specific residues immediately upstream of the ligand N terminus or downstream of its C terminus (Fig. 2b). For example, tyrosines were significantly enriched in sequences immediately flanking both termini of presented ligands (P < 0.001), whereas histidines and prolines were generally absent from these regions (P < 0.001). Among peptides presented by HLA-II, we observed significant enrichment of those derived from the C termini of the mature proteins (indicated as ‘–’ at +1 to +6 position; average fold change >150%; P < 1 × 10−5). As these flanking sequences are not directly involved in HLA complex binding of peptide ligands, the observed enrichments likely reflect the cleavage preferences of proteases involved in processing proteins for presentation. Of note, these cleavage signatures were distinct for HLA-I and HLA-II ligands (Supplementary Fig. 4a–c), consistent with their distinct cleavage and processing pathways1,20. Therefore, to capture the added predictive information from flanking residues similar to Paul et al.23, we developed a dedicated neural network for assigning HLA-DR cleavage scores from a given peptide sequence (Supplementary Figs. 2b and 4d).
MARIA data integration framework
On the basis of the findings above, we developed an integrative strategy to better predict HLA-II presentation called MARIA. The model takes in three input values: the query peptide sequence, the patient or cell HLA-DR allele(s) and the corresponding gene name (Fig. 2c). As an intermediate step, MARIA calculates HLA-DR binding scores and cleavage scores using two pretrained neural network models (Supplementary Fig. 2). Gene expression values are estimated by either tissue-matched external RNA-seq or patient-specific RNA-seq results (Supplementary Fig. 5d–f). MARIA then generates presentation scores for a potential antigen by integrating all available information including peptide sequences with a merge layer (Fig. 2c). To process variable length peptide sequence inputs, MARIA includes a recurrent layer with long short-term memory (LSTM)24. We tested MARIA and models with all possible feature combinations using tenfold cross-validation (Supplementary Figs. 2e and 5b, and Supplementary Table 2). When considering average AUCs, MARIA outperformed an RNN model trained on peptide sequences alone with an AUC value of 0.92 versus 0.87 (Fig. 2d and Supplementary Fig. 5d; P < 1 × 10−5). By contrast, a logistic regression model trained using binding scores, gene expression levels and cleavage scores achieved a lower AUC value of 0.82 (Supplementary Fig. 5e). MARIA provided higher precision (positive predictive values) as compared to alternative models across a broad range of HLA-DR ligand prevalences (0.1–10%; Fig. 2e). Assuming 1% prevalence of HLA-II antigens (Supplementary Note 2), MARIA achieved 99.5% specificity and 38.7% precision while identifying 30% of positive peptides (Fig. 2e–f).
MARIA benchmarking
To systematically compare the performance of MARIA with alternative methods, we applied antigen presentation profiling2 to directly identify HLA-DR ligands from a human cell line (K562) expressing single HLA-DR alleles (DRB1*01:01, DRB1*04:04). Given the myeloid hematopoietic origin of this cell line (in contrast to the lymphoid tumors using for MARIA training), this allowed us to assess both the effects of HLA-II allelic variation and the cell-of-origin on performance (Supplementary Fig. 6). We identified ~3,600 non-redundant peptide ligands from these two alleles. When allowing substring matching, ~31% of ligands were shared (Fig. 3a) and MEME25 identified 15 shared potential binding motifs (Supplementary Table 4). This is consistent with known promiscuity of HLA-II binding and presentation.

a, Overlap and sequence motifs of two HLA-DR ligand sets identified from two monoallelic K562 cell lines. A proportion (31%) of peptides appeared in both the HLA-DRB1*01:01 (n = 2,430) and HLA-DRB1*04:04 (n = 2,072) ligand sets when considering substring matches. The sequence motifs with highest statistical significance (P < 1 × 10−7, multiple hypergeometric test implemented by MEME) are shown. For full potential motifs, see Supplementary Table 4. b, Performance of MARIA and six alternative methods when differentiating 1,361 K562 HLA-DRB1*01:01 ligands from 1,361 human decoys. MARIA outperformed the second-best method (SMM Align; DeLong test, P < 1 × 10−5). Limited by the IEDB Concensus3 package, only ligand sequences ≥15 amino acids are included in this comparison. c, Performance of MARIA and four alternative methods differentiating 2,032 K562 DRB1*04:04 ligands from 2,032 human decoys. MARIA achieved an AUC of 0.89 AUC as compared to an AUC of 0.56 for NetMHCIIpan. RNN and SNN trained on MCL ligands obtained AUC values of 0.83 and 0.78, respectively.
We selected DRB1*01:01 for initial testing in this system, as it has the most abundant training data for existing binding prediction methods, including NetMHCIIpan3.122, SMM Align26, NN Align27, Sturniolo28, CombLib29 and IEDB Consensus330. We benchmarked the performance of MARIA and these six methods when predicting the presentation of 1,331 DRB1*01:01 ligands empirically and distinguishing them from length-matched decoys (Fig. 3b and Supplementary Table 5). When compared to the previous MHC-II prediction tools, MARIA outperformed the second-best method (SMM Align) by a significant margin (AUC 0.89 versus 0.64; P < 1 × 10−5). We also tested performance when predicting presentation by HLA-II alleles not present in the training data. Specifically, we directly profiled the HLA-DR ligands from a second monoallelic K562 isogenic line engineered to express HLA-DRB1*04:04 (Supplementary Table 6), an allele absent from the individuals considered for MARIA training. MARIA again outperformed other methods with an AUC 0.89 (Fig. 3c).
We also explored the influence of neural network structure on prediction performance. Using the same MCL dataset of HLA-DR ligands used for training MARIA, we trained a shallow neural network (SNN) similar to NetMHCIIpan with a single hidden layer, as well a deep RNN model (Supplementary Fig. 2c). These two models only considered peptide sequences, yet both outperformed NetMHCIIpan on external validation data from K562 (Fig. 3c). This is consistent with our hypothesis that directly learning from MS-identified HLA ligands substantially boosts prediction power. Importantly, when trained on the same data, deep neural networks outperformed single-layer architectures (Fig. 3c and Supplementary Fig. 5f).
Extension of MARIA to the HLA-DQ locus
HLA-DQ2.2 (DQA1*02:01 and DQB1*02:02) and HLA-DQ2.5 (DQA1*0501 and DQB1*0201) are known to present wheat gluten peptides and to predispose patients to celiac disease31. To test our prediction framework in the context of the HLA-DQ locus, we trained MARIA on 11,482 HLA-DQ2.2 human peptide ligands identified from previously profiled32 cell lines (Fig. 4a and Supplementary Fig. 7a). Similar to the HLA-DR alleles profiled above, HLA-DQ2.2 and HLA-DQ2.5 had a large number of shared peptide ligands (65%; Fig. 4a) and associated sequence motifs (40; Supplementary Table 4). After cross-validation (Supplementary Fig. 7b), we first tested MARIA on an independent set of 650 held-out human DQ ligands (positives) and 650 length-matched decoys (negatives) and observed an AUC of 0.89 (Fig. 4b). To allow comparison between prediction methods on HLA-DQ, we normalized both raw MARIA and NetMHCIIpan scores as percentiles, where higher scores reflect better predicted binding (Fig. 4b; Methods). Within this comparison on an identical test set of HLA-DQ ligands, NetMHCIIpan achieved an AUC of 0.68. Therefore, MARIA provides advantages over existing methods across HLA-II loci.

a, Overlap and sequence motifs of two HLA-DQ ligand sets. A majority (65%) of peptides were present in both HLA-DQ2.2 (n = 7,374) and HLA-DQ2.5 (n = 4,249) ligand sets when considering substring matches. The sequence motifs with highest statistical significance (P < 1 × 10−7, multiple hypergeometric test implemented by MEME) are shown. Full potential motifs are included in Supplementary Table 4. b, Performance of MARIA trained on HLA-DQ2.2 ligand sequences and tested on a held-out human HLA-DQ2.2 peptide set (n = 650). MARIA was trained on 90% of the HLA-DQ2.2-associated peptide sequences shown in a. MARIA achieves an AUC of 0.89 when differentiating DQ2.2 ligands from length-matched decoys. By comparison, NetMHCIIpan percentiles obtained an AUC of 0.68. Dashed red lines indicate the 90th percentile, the default cut-off for NetMHCIIpan. See Supplementary Fig. 7 for detailed training schemes of MARIA for HLA-DQ2.2. c, Performance of MARIA and NetMHCIIpan when identifying immunogenic gluten peptide fragments (n = 69). MARIA trained on human DQ2.2 ligands identified 49% of HLA-DQ2.2-binding gluten peptides with 92% specificity. By comparison, NetMHCIIpan had 6% sensitivity and 88% specificity. Dashed red lines indicate the 90th percentile, the default cut-off for NetMHCIIpan. The x axes in b and c capture the percentiles for depicted bins, where higher percentiles reflect higher likelihood of presentation, by defining the percentiles as 100% minus the absolute rank reported by each method.
In addition to the presentation of human peptides described above, the presentation of diverse gluten peptides by DQ2.2 has also previously been profiled by MS33. We scored 69 presented wheat peptides and 69 decoys with both NetMHCIIpan and MARIA, which was exclusively trained on human peptides. NetMHCIIpan identified 6% of positive gluten peptides with 88% specificity at the recommended cut-off (90th percentile). By comparison, MARIA identified 49% of positive gluten peptides with 92% specificity (Fig. 4c and Supplementary Table 7) with the same cut-off. MARIA also assigned significantly higher presentation scores to deamidated gluten peptides (Supplementary Fig. 7c), a result which is consistent with increased immunogenicity of gluten peptides upon deamination31.
We also identified a small number of reported natural HLA-DP ligands34,35,36 (n = 20; Supplementary Table 12) and a new dataset of pan-HLA-II ligands37. We trained a pan-HLA-II model with the same framework of our HLA-DQ model and demonstrated the utility of our framework for differentiating them from random human peptides (AUC = 0.82; Supplementary Fig. 5g–h). Overall, these results demonstrate that MARIA trained on human peptides can predict presentation of exogenous antigenic peptides by distinct HLA-II alleles.
MARIA identifies diverse cancer neoantigens
Finally, we explored the ability of MARIA to identify immunogenic neoantigens in cancer. Ideal antitumor neoantigen candidates should be both presentable by HLA complexes and capable of inducing proinflammatory responses by interacting with T cell receptors38. Most current cancer vaccine platforms prioritize candidate neoantigens for vaccine production by selecting only highly expressed candidates with high predicted binding affinity for self-HLA alleles8,9,39,40. Yet, many vaccine peptides do not elicit T cell responses upon vaccination, despite rigorous candidate selection. We therefore tested whether MARIA could better select those neoantigens that were most likely to induce a corresponding T cell response upon vaccination.
Using antigen presentation profiling, we recently reported hotspots within specific immunoglobulin (Ig) regions that are presented by HLA-DR and associated with antitumor CD4+ T cell responses to lymphoma neoantigens2. We therefore tested whether MARIA could accurately identify potential Ig antigens as potential lymphoma-specific targets for immunotherapy. For this test, we intentionally excluded all Ig-derived peptides for the training. We then applied this Ig-naive version of MARIA to predict presentation of Ig sequences in the tumors. The resulting MARIA-predicted presentation scores were significantly correlated with MS-identified HLA-DR ligand frequencies across the full-length heavy and light chains (Fig. 5a; Spearman’s ρ of 0.65 and 0.55). By comparison, NetMHCIIpan-predicted hotspots had weaker correlation to observed presentation of peptides (Spearman’s ρ of 0.1 and 0.48; Supplementary Fig. 8). MARIA also outperformed NetMHCIIpan in precision and recall analysis (Supplementary Fig. 8b). Importantly, consistent with our previous findings from antigen presentation profiling, MARIA identified framework region 3 (FR3) as a presentation hotspot for the heavy chain variable region (Fig. 5b; P < 1 × 10−5). We stimulated patient peripheral blood leukocytes with the corresponding patient-specific Ig neoantigens identified by MARIA, and measured induction of T cell surface CD137, a previously validated marker for T cell activation2,41,42. We observed evidence of ex vivo CD4+ T cell activation upon antigen–peptide simulation in two of three patients (Supplementary Fig. 8c–d).

a, Correlation of MARIA-predicted and experimentally identified HLA-DR-presented immunoglobulin antigens. Eighteen MCL immunoglobulin sequences were analyzed by a version of MARIA trained on non-immunoglobulin HLA-DR ligands to determine the presentation hotspots (left, blue). The same 18 MCL samples were profiled with LC–MS/MS to determine the regions of immunoglobulin presented by HLA-DR (right, orange). Predicted and observed presentation hotspots were significantly correlated on both heavy chains and light chains (Spearman’s ρ of 0.63 and 0.55, P = 1 × 10−65 and 7.5 × 10−19; n = 1,015 and 311, respectively). MARIA-predicted ligand numbers were normalized with the MS-identified maximum ligand numbers for visualization purposes. See Supplementary Fig. 8 for the presentation heat map predicted by NetMHCIIpan. b, MARIA identified HLA-DR presentation hotspots in the immunoglobulin heavy chain variable region (IGHV). MARIA-predicted HLA-DR-presented peptides from IGHV FR3 regions more than the other six regions across patients (P < 1 × 10−5, Mann–Whitney U test), consistent with MS findings (P < 1 × 10−5, Mann–Whitney U test). Each dot represents predicted or experimentally identified ligand coverage in a 15-amino-acid sliding window on the aligned IGHV sequence (n = 38 for the FR3 region and n = 87 for the non-FR3 regions).
Immunoglobulin heavy chain variable regions represent challenging test examples as most HLA ligand prediction algorithms including MARIA were trained on wild-type peptides. To further address the utility of MARIA for predicting presentation of mutated peptides, we tested MARIA predictions of HLA-II intrinsic ligand (CLIP) with and without specific point mutations. For diverse CLIP variants, MARIA scores consistently correlated with stabilizing versus destabilizing structural changes, while NetMHCIIpan did not (Supplementary Note 3 and Supplementary Fig. 9).
Personalized protein-coding somatic mutations are attractive cancer vaccine candidates in melanoma owing to the high mutation burden of patients with melanoma. We tested whether MARIA could help prioritize vaccine candidates for melanoma. We first analyzed 10,513 melanoma self-antigens identified by MS3, generated from two bulk melanoma tumors with distinct HLA-DR alleles (Mel15, DRB1*03:01 and DRB1*07:01; Mel16, DRB1*13:01 and DRB1*08:03). We scored each melanoma-presented ligand or decoy using both NetMHCIIpan and MARIA (trained on lymphoma data). Even without patient-specific gene expression data (Methods), MARIA outperformed NetMHCIIpan when differentiating melanoma HLA-II ligands from decoys (Fig. 6a and Supplementary Fig. 10e; AUC of 0.89 versus 0.64; P < 1 × 10−5).

a, Performance of MARIA on an independent melanoma HLA-II ligand set3. MARIA trained on MCL ligands achieved an AUC of 0.89 when differentiating patient melanoma HLA-II peptides from length-matched decoys, as compared to NetMHCIIpan with an AUC of 0.64. Shuffling correct training labels diminished the prediction performance of MARIA, reducing its AUC to 0.53. b, Neoantigen gene expression in patients with melanoma is not associated with post-vaccination CD4+ T cell responses. Personalized gene expression values were obtained from tumor RNA-seq in two personalized melanoma vaccine trials8,9. In both trials, there is no difference in gene expression values between positive and negative vaccine candidates for their ex vivo CD4 cytokine release tests (n = 127 and 97; P = 0.49 and 0.50, two-tailed unpaired t test). NS, not significant. c,d, Post-vaccination CD4+ T cell responses are associated with MARIA scores. Peptide sequences from the same two clinical trials were scored with MARIA (c, n = 127 for Sahin et al.8, and d, n = 97 for Ott et al.9). Each candidate was stratified into three categories on the basis of the highest MARIA percentile scores among 15-amino-acid oligomer sliding windows: low (<95th), medium (95–99.5th) and high (>99.5th). Dashed red lines indicate average response rates of the whole cohort. c, A majority (73%) of peptides in the MARIA high category elicited positive CD4+ T cell responses after vaccinations as compared to 26% in the low category and 47% in the medium category (χ2 test, 2 degrees of freedom, P = 0.019). d, A higher proportion (38%) of peptides in the MARIA high category elicited positive CD4+ T cell responses after vaccinations as compared to the low category (8.1%) and the medium category (23%) (χ2 test, 2 degrees of freedom, P = 0.023). See Supplementary Fig. 10 for detailed analysis on melanoma cancer vaccines. e, Relationship between MARIA percentile scores and CD4+ T cell responses to tumor-associated antigens across cancer types and studies. When considering seven different studies of CD4+ T cell immune responses to cancer-associated antigens (including this one)6,7,42,43,44,45 (rows), we identified immunogenic (positive; n = 27, rows 1–7) and non-immunogenic (negative; n = 494, row 8) peptides across diverse tumor types. Each of these 521 peptides (dots) were then tested by MARIA, allowing comparison of percentile scores (x axis, right) with immunogenicity (blue, immunogenic; green, non-immunogenic). As depicted by the summarized inset table, 74% of immunogenic peptides (20 of 27, blue) scored above the 95th MARIA percentile threshold. Teff, effector T cells.
After confirming the performance of MARIA in non-hematopoietic tissue, such as melanoma, we used MARIA to analyze two sets of personalized melanoma vaccine neoantigens with corresponding immune response data8,9 (ex vivo CD4+ T ell enzyme-linked immunospot (ELISPOT) test; n = 121 and 96). Gene expression levels of neoantigens alone for T cell reactive and non-reactive neoantigen candidates were largely indistinguishable (Fig. 6b; P > 0.4). MARIA assigned each peptide a percentile score by comparing the raw score to scores of 20,000 random human peptides as described (Methods; Supplementary Table 8). In each of these independent cohorts, the majority of selected neoantigens for vaccination (81% and 62.5%) were scored in the 95th percentile or above of MARIA scores (Fig. 6c,d), consistent with the authors’ attempts to select the best HLA binders. Neoantigens with lower than 95th percentile MARIA scores (Fig. 6c,d) were less likely to successfully induce a T cell response upon vaccination. Specifically, only 26% and 8.3% of such neoantigens resulted in successful ex vivo CD4+ T cell responses upon vaccination.
Conversely, those peptides with highest MARIA scores (>99.5% MARIA percentiles) were more likely to elicit a T cell response upon vaccination (Fig. 6c,d; 73% and 38%; P = 0.019 and P = 0.023). This stringent MARIA cut-off achieved a high positive predictive value (PPV) in both trials, and MARIA showed higher than baseline PPVs across a range of cut-offs (Supplementary Fig. 10a,b). Of note, ~7% of all melanoma somatic mutations in the Ott et al. clinical trial cohort9 scored higher than 99.5% MARIA percentiles (Supplementary Fig. 10c), suggesting the availability of many more vaccine candidates for effective immunization than were tested. With HLA-DR alleles available in the same trial, we also scored each candidate with NetMHCIIpan. NetMHCIIpan scores were weakly correlated with T cell responses but did not reach statistical significance (Supplementary Fig. 10d).
To further validate MARIA performance for predicting immune responses, we identified seven additional cancer-related CD4+ T cell response studies6,7,42,43,44,45. Each of these studies individually identified a small number of cancer-associated CD4+ T cell epitopes across diverse cancer types. Using a 95% MARIA percentile cut-off (Fig. 6c,d), we identified 74% of CD4 epitopes with 67% specificity (Fig. 6e and Supplementary Tables 13–15). Therefore, while MARIA was not trained on T cell response data, MARIA scores show promise for prioritizing HLA-II neoantigens most likely to induce corresponding CD4+ T cell responses.
Discussion
Historically, tools for prediction of HLA-II interactions have largely relied on in vitro peptide-binding data for training15,16,21. However, when using predicted HLA-II binding affinities alone, we observed suboptimal prediction performance for identifying naturally presented ligands from MS (Fig. 1b and Supplementary Fig. 5c). One potential reason for this observation might be the sparsity of binding data for adequate training15, especially for human peptide HLA-II ligands (Fig. 1a). Furthermore, as peptide binding by HLA-II is known to be more promiscuous than HLA-I28,46, this could make identifying binding motifs more challenging. Finally, binding-based methods ignore factors such as levels of gene expression and protease cleavage signatures, which play an important role in HLA-II presentation (Fig. 2). Given these limitations, tools trained on naturally identified peptide ligands might better reflect how prediction scores correlate with physiological HLA-II presentation, and the associated benchmarks could help to refine the accuracy of such predictions.
HLA-I and HLA-II both play central roles in antigen recognition and adaptive immune responses. Our HLA-II gene expression analysis in the Cancer Genomic Atlas (TCGA) cohorts suggests abundance of antigen-presenting cells or tumor HLA-II presentation in various cancer types (Supplementary Fig. 11). Historically, HLA-I ligand prediction algorithms have superior performance as compared to HLA-II16,47. Recent studies in HLA-I have shown that prediction accuracy can be improved by learning directly from naturally identified ligands and considering non-sequence features20,46,48,49,50,51,52. However, the variable length of HLA-II peptide ligands as well as the heterogeneity of other useful features have made translating the same framework for HLA-II challenging within conventional neural networks. MARIA represents a tool to tackle these two challenges by using multimodal RNNs39,53, which are capable of integrating heterogeneous features and variable length sequences. Our results suggest that using deep learning methods are superior to shallow neural networks (SNNs) for HLA-II prediction (Fig. 3c and Supplementary Fig. 5f). This is likely due to the ability of RNNs to consider multiple binding motifs, as SNNs typically rely on a single nine-amino-acid binding core27.
The SysteMHC Atlas51 was recently constructed to consolidate HLA-I and HLA-II ligand sequences from a diverse set of studies. We designed MARIA to be capable of integrating additional training from emerging sources such as the SysteMHC Atlas and expect its performance to improve as such ligand datasets continue to grow. For example, we observed recurrent patterns in HLA-II cleavage signatures similar to previous studies20,23,54, including the enrichment of ligand flanking sequences for tyrosines and their depletion for prolines. However, cleavage signatures from different cell types showed subtle variation in motif sequences (Supplementary Fig. 4d–i). Accordingly, additional HLA-II ligand data in the SysteMHC Atlas can provide a window to systematically investigate cleavage signatures in each cell type, thus allowing better MARIA predictions for distinct tissues. Separately, as MARIA was not trained on presented non-human peptides, emerging microbial datasets55 can be used to further refine MARIA for predictions relevant to infectious disease.
Similar to existing tools, MARIA relies on predicted binding affinities to allow specificity for different HLA alleles (Fig. 2c, Supplementary Fig. 2d and Supplementary Table 9). We therefore do not expect MARIA to achieve significantly higher discriminatory power for resolving binding by different HLA-II alleles than current methods that are based on binding affinity (Supplementary Fig. 5c). While the limited availability of allele-specific datasets has resulted in validation of MARIA on a relatively small number of HLA-DP and HLA-DQ specific ligands (Supplementary Figs. 5h and 7), we expect that the method will become more comprehensive as additional allele-specific data become available across HLA loci. This can be achieved using high-throughput technologies, such as peptide-binding arrays56,57, or by profiling a diverse set of HLA-II monoallelic cell lines for their presented ligands20.
Our results also demonstrate how MARIA might allow researchers to better identify immunogens relevant to autoimmunity and to antitumor immunity. Given the inherent challenges limiting the accuracy of previous methods for characterizing tumor-derived HLA-II ligands, MARIA should allow researchers to explore less well-studied HLA-II neoantigens. Specifically, we expect that MARIA will be useful for directly identifying and prioritizing cancer vaccine candidates from patient sequencing data12,51. Nevertheless, many peptides with high MARIA presentation scores did not induce CD4+ T cell responses when used as cancer vaccines (Figs. 6c–e). This limitation is consistent with the fact that presentation of antigens is essential but not sufficient for induction of robust T cell responses51,55,58 (Supplementary Note 2). Therefore, by combining deep learning59 and large-scale T cell response data, we envision that a future method will provide refined predictions for the immunogenicity of HLA ligands, whether autoantigens relevant for autoimmunity, alloantigens relevant to transplantation or as vaccine candidates relevant for diverse applications.
Methods
In vitro binding testing for validation of HLA-DR binding
Candidate peptides were synthesized with N-terminal 2,4-dinitrophenyl (DNP) tags joined by a 6-aminohexanoic acid linker (Sigma). Biotinylated HLA-DR recombinant proteins (HLA-DRB1*04:03 and HLA-DRB1*07:01) molecules were provided by the NIH tetramer core. Intrinsic CLIP peptide was cleaved from the HLA-DR molecules with human rhinovirus 3C protease. DNP-tagged peptides were supplied in molar excess to encourage efficient exchange of binders and incubated overnight at 32°C or 37°C (pH 4.5). Exchange reactions were then neutralized with 1 M Tris, pH 8.0 and biotinylated HLA-DR molecules were bound to streptavidin microspheres (Polyscience). Microspheres were washed and stained with allophycocyanin (APC)-labeled anti-HLA-DR (clone L243; BD Biosciences, 340549) and anti-DNP (clone 2-9(4); Abcam, ab6306) followed by rat anti-mouse IgE FITC secondary antibody (clone R35-72; BD Biosciences, 553415). Microspheres that were positive for HLA-DR and DNP-tagged peptide were detected by flow cytometry. Peptides were considered to be binders if both HLA-DR and DNP signals were detectable above an HLA-DR unexchanged control (Supplementary Fig. 1). Supplementary Fig. 1d shows full benchmarking with reported binders and non-binders.
Development of K562 cells expressing single HLA-DRB1 alleles
Cell lines expressing single HLA-DR alleles were prepared from K562 cells, which do not express surface class I or II HLA, by lentiviral transduction. Sequences for the DR α-chain and the relevant β-chain alleles (DRB1*01:01 and DRB1*04:04) separated by a 2A peptide sequence were encoded in the N103 lentiviral vector backbone (kindly provided by J. Crabtree, Stanford University) and used to produce lentiviruses in HEK293 cells using previously published methods61. To enhance expression levels of HLA-DR in our K562 cell lines, we selected and expanded the top 1% of cell populations selected on the basis of surface HLA-DR signal with fluorescence-activated cell sorting (clone L243; BD Biosciences 347367) as described before62. Expression of HLA-DR was confirmed by flow cytometry before and after sorting (Supplementary Fig. 6). K562 cells were also monitored for surface HLA-I alleles to ensure no endogenous HLA expression was present (anti-HLA-I antibody; clone G46-2.6; BD Biosciences, 555555; Supplementary Fig. 6). Cells were maintained in DMEM medium (Sigma) supplemented with 2.0 μg ml−1 puromycin (Sigma).
Identification of K562 HLA-DR ligands
HLA-DR immunopeptidomes were extracted from the K562 HLA-DRB1*01:01 and K562 HLA-DRB1*04:04 cell lines. HLA-DR molecules were isolated and the associated peptides were extracted as previously described2,63. See Supplementary Note 4 for detailed HLA-DR immunopeptidome purification and MS analysis.
HLA-II ligand sequence data sources
Detailed ligand sequence data sources are listed in Supplementary Fig. 5a. MCL HLA-DR ligandomes were obtained from our pervious study on MCL samples2. Dendritic cell HLA-DR ligandomes were obtained from a MUTZ3 cell line study54. HLA-DQ2.2 ligandomes were obtained from a monoallelic B cell line study conducted with the anti-DQ antibody SPV-L3 (ref. 32). Melanoma HLA-II ligand sequences were obtained from a previous study on primary tissues from patients with melanoma3. HLA-DQ2.2-presented wheat peptides were downloaded from the IEDB database21, and the original sequences were identified by Dorum et al.33. Pan-HLA-II ligands were obtained from a study of B cell lines and ovarian carcinoma using HB-145 anti-HLA-II antibody37. Monoallelic HLA-I ligand sequences were obtained from a B cell line study with W6/32 pan-HLA-I antibody20. HLA-DP ligands were downloaded from the IEDB database21, and the original sequences were identified by three MS studies34,35,36.
Determination of patient HLA alleles
HLA alleles of patients with MCL2 were identified with PHLAT64 from patient tumor exome sequencing data. HLA alleles of melanoma patients3 were identified with HLAVBSeq65 from patient exome sequencing data. When patient alleles were not available (for example, the melanoma study by Sahin et al8), HLA-DRB1*07:01 and HLA-DRB1*01:01 were used as they are the most common alleles in general populations (http://www.allelefrequencies.net/).
Immunogenicity testing for immunoglobulin neoantigens
Neoantigen T cell stimulation was performed as previously described with some modifications2. All specimens were obtained with informed consent in accordance with the Declaration of Helsinki and this study was approved by Stanford University’s Administrative Panels on Human Subjects in Medical Research. Samples were collected from patients as part of a clinical trial of autologous tumor vaccination (NCT00490529). Patient leukocytes were collected by leukapheresis approximately 2 weeks after a series of autologous tumor vaccinations. Cells were cultured in a 1:1 mix of AIM-V medium and RPMI1640 (Thermo Fisher) with 10% pooled human AB sera (Gemini Bio) and 50 μM β-mercaptoethanol. Neoantigen peptides were synthesized (ElimBio) and added to a final concentration of 10 μg ml−1. In one patient (MCL052), cells were treated concurrently with two predicted neoantigen peptides. As a positive control, cells were stimulated with a mixture of pathogen-associated peptides, CEFT pool (JPT Peptide Technologies). Cells were incubated for 30 h before flow cytometry analysis. CD137 (clone 4B4-1; BD Biosciences, 561702) and CD69 (clone L78; BD Biosciences 341652) expression was assessed on live CD4+ (clone RPA-T4; BD Biosciences, 562659) T cells using a FACS Aria sorter (BD Biosciences)41,66. See Supplementary Fig. 8c for gating strategies.
Gene expression data sources
We observed only minor differences in gene expression profiles when using personalized versus inferred gene expression levels with modest impacts on MARIA prediction results (Supplementary Fig. 5e,f and Supplementary Note 1). Therefore, when personalized gene expression profiles were not available, expression profiles were estimated from the corresponding tumor type, using, for example, the median of TCGA RNA-seq results from the closest tissue type67. Gene expression profile of patients with MCL and JeKo-1 cell line (MCL origin) were obtained from RNA-seq results of a previous study68; MCL patient gene expression profiles were estimated as the median value across ten patients with MCL. Given the high correlation when comparing MCL transcriptomes from different tumors, gene expression profiles of the L128 cell line (MCL origin) were estimated from JeKo-1 cell line RNA-seq values. The gene expression profile of K562 cell lines was obtained from the ENCODE database69. Expression values were normalized into TPM to enable direct comparison of values across studies.
Gene expression profiles of melanoma in the cancer vaccine studies were provided by the original studies8,9 when analyzing how original neoantigen gene expressions correlated with responses. A fixed TPM of 50 was used as input for MARIA to evaluate how MARIA scores with post-vaccine responses as cancer vaccines made each neoantigen peptide readily available to antigen-presenting cells. A fixed TPM of 50 was also used when evaluating how HLA-DRB1*01:01 binds to CLIP. Gene expression values were not needed when MARIA analyzed HLA-DQ2.2 or HLA-DP peptides, as only peptide sequences were used during training.
Gene expression analysis of HLA-II peptides
We estimated the gene expression value (TPM) of each HLA-DR-peptide-producing gene with RNA-seq of samples from patients with MCL or MCL cell lines. We allowed a gene to be counted multiple times if multiple peptides were identified from the same gene. Each gene expression value was converted into logarithmic space with log10(X + 10−6) and represented as a violin plot (Fig. 2a and Supplementary Fig. 3). Peptide sequences not in the dictionary or genes with unknown RNA-seq gene expression values were excluded from this analysis (<8%).
Using a similar approach, we analyzed the gene expression profiles of all genes in patients with MCL and MCL cell lines as a background distribution. Each gene with a known RNA-seq gene expression value was converted into logarithmic space, and each gene was counted once. We calculated the median and conducted a Mann–Whitney U test on gene expressions between MCL HLA-DR-presented genes and the background distribution.
To investigate sources of MCL HLA-DR peptides with extremely low gene expression values, we conducted Gene Ontology (GO) term enrichment analysis70 on HLA-DR peptide genes with TPM < 0.1 (Fig. 2a, Supplementary Fig. 3c and Supplementary Table 3). To correct for the presence of extracellular matrix protein and blood protein inside of professional antigen-presenting cells (for example, MCL) when these genes were not endogenously expressed, we set gene expression values of genes under the following GO terms to 50 TPM: extracellular matrix (0031012), blood microparticle (0072562) and secretory granule lumen (0034774).
To test the ability of gene expression values to differentiate HLA-II ligands from decoys, we built a logistic regression model with gene expression values as a single feature to differentiate HLA-DR peptides from a random human peptide decoy. Decoy gene lists were generated from a Uniport reviewed human protein list71 and were enriched for expressed genes. Specifically, human peptide decoys contain roughly 40% genes with TPM > 10, 10% genes with unknown expression levels and 50% genes with TPM < 10. Predictive performance of gene expression values under different assumptions is shown in Supplementary Fig. 3d. No regularization was applied because it is a single-feature model.
Neural network implementation
See Supplementary Note 5 for an overview of neural networks considered and their implementation. Neural networks in this study were implemented with Keras 2.0.3 (https://keras.io/) using the Tensorflow backend and Python 2.7. For training, we utilized an NVIDIA Tesla K80 GPU with CUDA 7.5.18 on the Stanford XStream GUP cluster to speed up the gradient descent.
Amino acid representation
Each amino acid in a peptide sequence is represented by a 21-number one-hot vector (20 common amino acids + X) A one-hot vector is a vector of zeros with a single one that indicates the amino acid species in an arbitrary but consistent mapping (for example, position one = alanine; Supplementary Fig. 2a). Thus each peptide sequence is represented by a (21, L) two-dimensional vector, where L is the length of the peptide. BLOSUM5072 and ProtVec73 amino acid encodings were attempted but had little influence on the model performance.
MARIA model
The majority of existing HLA-II peptide prediction algorithms use data on the binding of recombinant HLA-II protein to peptides as a surrogate to rank potential HLA-II peptides presentation or HLA-II neoantigens. MARIA aims to directly predict the probability of a given peptide being presented by a HLA-II complex in a cell or patient context. Rather than in vitro measurements of the binding of HLA protein to peptides, we use HLA-II ligands identified by MS/MS peptide sequencing data from antigen-presenting cells as the ground truth.
The MARIA model takes peptide sequences (8–26 amino acids long), patient or cell HLA-DR alleles and corresponding gene names to predict peptide presentation by the HLA-DR complex (Fig. 2c). MARIA estimates HLA peptide-binding affinities and cleavage scores with two separate neural network models, which are described in the next section. MARIA also estimates the gene expression levels of each input gene with a gene expression profile dictionary that is based on external tissue-matched RNA-seq results (gene name and TPM). The MARIA model takes in gene expression values (TPM) as log10(TPM + 0.001) to prevent neurons that connect to gene expression input from dominating the optimizing gradient. Users can also upload their own gene expression profile dictionaries for each query. The influence of using patient-matched or external gene expression profile dictionaries are explored in Supplementary Fig. 3e,f.
Besides estimating binding affinities, cleavage scores and gene expression values, MARIA encodes each ligand peptide sequence using an RNN layer known as LSTM. All of these values are integrated with two fully connected dense layers to estimate the probability of a peptide–gene pair being presented in a allele- and gene-expression-specific manner (Fig. 2c). See Supplementary Note 6 for our models for predicting presentation with peptide sequences only.
We explored LSTM networks with 32, 64 and 128 neurons and from one to four layers deep with a 9:1 training:validation scheme (Supplementary Fig. 2e). An LSTM network with 64 neurons and one layer of depth gave the best performance. Dense (fixed-length) layers in MARIA use the rectified linear unit activation function74 and 32 neurons. Dropout of 40% is applied to each layer for regularization. L1 and L2 regularization were attempted but not included in the final model owing to the lack of influence on the model performance. We concatenated the LSTM and dense layers to merge their information, followed by two additional dense layers (neuron number = 32). The output layer contains two neurons representing non-presenting (F or 0) and presenting (T or 1) classes.
MARIA was trained with the Tensorflow GPU backend to enable parallel calculation of gradient. For general user applications, the CPU backend is sufficient. Ten thousand peptides take ~80 s with a 2.8 GHz Intel Core Xeon CPU or 11.3 s with one NVIDIA Tesla K80 GPU.
Binding affinity prediction
MARIA assumes the main influence on HLA-DR allele is from the change in HLA-DR–peptide binding affinity. We trained a pan-allele regression model with the publicly available IEDB data to estimate binding affinity given a peptide–allele pair21. Training data was curated by the authors of NetMHCIIpan3.0 (ref. 75) which includes 33,909 peptide–allele pairs for HLA-DR. Each HLA-DR allele is converted into a 19-amino-acid pseudopeptide sequence reflecting 19 amino acid residues in HLA-DRB1 interacting with ligand peptides in the binding groove76. Each peptide–allele pair has a corresponding binding affinity measured in half maximal inhibitory concentration (IC50, in nM). For computational efficiency, IC50 was converted into log space with the formula (1 − log50,000(IC50)). The model includes an LSTM layer followed by two dense layers and a single output neuron (Supplementary Fig. 3d). Mean squared error is used as the loss function.
To evaluate the performance of our LSTM-based binding affinity predictor with NetMHCIIpan3.1 (ref. 22), a widely used method, we tested our method on the same held-out set of in vitro binding as reported by the original author. We report similar ROC-AUC scores and Pearson’s correlation coefficients (predicted versus measured) in Supplementary Table 9.
Cleavage score estimation
To understand amino acid preference for HLA-II peptide cleavage, we compared amino acid frequency around HLA-II peptide cleavage sties with a background distribution. Cleavage sites included six amino acids upstream of the HLA-II peptide N terminus (−6 to −1) and six amino acids downstream of the HLA-II peptide C terminus (+1 to +6) assuming a N terminus to C terminus direction. We randomly generated an equal number of gene-matched and length-matched peptides on the basis of our HLA-II peptides, and the amino acid frequency from the same cleavage sites was used as the background distribution. Enrichment and depletion level were calculated as HLA-II peptide amino acid i frequency at j position divided by background distribution amino acid i frequency at j position (Fig. 2b and Supplementary Fig. 4). The analysis was done for MHC-I as well (Supplementary Fig. 4a).
We built a neural network to quantitatively estimate cleavage scores given the flanking regions of a query gene–peptide pair (Supplementary Fig. 2b). The model determines six amino acids upstream of the query peptide N terminus and six amino acids downstream of the query peptide C terminus with a human proteome dictionary, encodes these 12 amino acid sequences, processes them with hidden layers and outputs a probability score between 0 and 1 (score of the positive output neuron T). The neural network encodes amino acids using one-hot encoding as described before and contains two fully connected hidden layers of 32 neurons. To avoid overfitting when applying this model to lymphoma data, we trained the cleavage model on an independent dendritic cell line (MUTZ3) ligandome54 (Supplementary Fig. 4d).
Normalization of MARIA output scores to percentiles
Raw output of MARIA is a score between 0 and 1 indicating how likely a query peptide is to be presented by a specific HLA-II. To increase human interpretability and enable comparison across different peptide lengths, MARIA’s output can be represented as a percentile score. A percentile score is generated by comparing the raw output score to a score distribution generated from length-matched random human peptides. The higher the percentile, the more likely the peptide will be presented by a cell HLA-DR complex.
Specifically, for each peptide length of 8 to 26, we sampled 20,000 random natural peptides from the human proteome. We ran MARIA on each set of random peptides and used the output as empirical distributions for normalizing query peptides with a certain length. For example, MARIA output of a 15-amino-acid query peptide will be compared against the scores of 20,000 random 15-amino acid oligomers to obtain a percentile score. NetMHCIIpan used a similar approach to generate ranks, and NetMHCIIpan percentiles = 100% − NetMHCIIpan ranks.
MARIA model evaluation on held-out HLA-II ligand sets
To fully evaluate the performance of MARIA and related methods, we obtained a set of independent HLA-DR ligandomes from various cell types and MS instruments (Fig. 3a). Any peptides shorter than 8 amino acids or longer than 26 amino acids were excluded (<1%) owing to the setup of our RNN. Because of input limitations of existing methods, we further excluded peptides with certain length. When the evaluation involved IEDB-carried methods, we excluded peptides shorter than 15 amino acids. When the evaluation involved NetMHCIIpan3.1, we excluded peptides shorter than 9 amino acids. For negative examples, we used length-matched random human peptides with 1:3 (training) or 1:1 (validation) positive:negative ratios (Supplementary Fig. 5a). During cross-validation, we excluded any peptides in the validation set that were substrings (A is part of B or B is part of A) of any training peptides (Supplementary Figs. 2e and 5b).
Benchmarking of binding-based methods on MS-identified HLA-DR ligands
To evaluate how NetMHCIIpan performed on MS-identified MCL HLA-DR ligands, we ran NetMHCIIpan3.1 on a set of 18 MCL samples with a minimum of 200 peptides identified (Supplementary Table 1). For a patient or cell with heterozygous HLA-DRB1 alleles, the better binding score (lower ranking or higher percentile) was used as the predicted score. ROC-AUC scores were calculated to evaluate predictive performance of binding models on naturally presented peptides (Supplementary Table 1).
We also benchmarked MARIA with six commonly used HLA-II prediction algorithms on non-MCL data: NetMHCIIpan3.1 (ref. 22), SMM Align26, NN Align27, Sturniolo/TEPITOPE28, Comblib29 and IEDB Consensus30. All six algorithms were trained on in vitro recombinant protein binding data but differ in allele coverage and machine learning algorithms. Held-out data included K562 myeloid cells and primary melanoma patient samples. Because IEDB Concensus (Concensus3) is only compatible with 50 HLA-DR alleles and peptide sequences longer than 14, we compared MARIA with NetMHCIIpan in most cases.
NetMHCIIpan and NN align both use a dense neural network to scan through a given query peptide with a 9-amino-acid oligomer sliding window. The best 9-amino-acid oligomer score is reported. NetMHCIIpan incorporates important amino acid sequences on HLA-II alleles as a part of input features to train a universal algorithm for all HLA-II alleles22. NN Align learns binding patterns of each HLA-II allele separately27. SMM Align and Comblib both use a stabilized matrix method26,29. Sturniolo/TEPITOPE used a combination of expert rules and assembled matrices28. IEDB Consensus (Concensus3) is an ensemble method that is based on scores from NetMHCIIpan, NN Align, SMM Align and Sturniolo. Depending on the HLA-II allele, IEDB Consensus combines scores from one to three methods to report a ranking score30.
Predicting HLA-DQ peptide presentation
We trained a separate model for HLA-DQ2.2-presented peptide that was based on two available HLA-DQ ligandomes for HLA-DQ2.2 and HLA-DQ2.5 (ref. 32). The HLA-DQ model neural network architecture is identical to the sequence-only LSTM model for HLA-DR (Supplementary Fig. 2c), but the model was trained on different datasets (Supplementary Fig. 7a,b). The training HLA-DQ2.2 ligandome contains all HLA-DQ ligandome sequences from three DQ2.2 cell lines32 (PLH 9047, MOU 9050 and PITOUT 9051). We included HLA-DQ2.5 ligandome sequences (CD114, STEINLIN 9087 and PF04015 9088) if the peptide sequences were substrings of any known HLA-DQ2.2 ligandome peptides (Supplementary Fig. 7a). We held-out 7.5% of DQ2.2 ligandome as the validation set to determine model regularization parameters and the training stop point, and another 7.5% to determine the ability of the model to predict human HLA-DQ2.2 peptides (Supplementary Fig. 7b).
We tested the ability of our DQ model to differentiate DQ2.2-binding wheat gluten peptide33 (n = 69) from length-matched random human peptide (n = 138). We ran NetMHCIIpan3.1 on the same test set with the allele input of HLA-DQA10201 and HLA-DQB10202. We compared the performance of these two methods by measuring their sensitivity when the specificity was set to 90% (cut-off at the 90th percentile for both MARIA DQ model and NetMHCIIpan3.1; percentile = 100% − rank).
Analysis of immunoglobulin HLA-DR presentation heat maps
We estimated the HLA presentation of Ig from patients with MCL on the basis of how each amino acid position was covered by experimentally identified or predicted peptide ligands. HLA-presented Ig peptides from 18 MCL samples were obtained from our previous MS-based study2. All MS-identified peptides were aligned against a standard Ig template sequence (Supplementary Table 10) with the Biopython pairwise alignment function77. The MS-identified ligand number at each amino acid position is equal to the number of peptide ligands covering the position.
To generate predicted HLA-DR ligands, we broke down Ig sequences from 18 MCL samples into consecutive 15-amino-acid sliding windows. Each sliding window peptide was scored by NetMHCIIpan or MARIA (a version not trained on Ig sequences) with matched patient alleles. We aligned high-scoring peptides against the same Ig template sequence as described above. Scores (S) for peptide sequences were based on their predicted presentation percentiles (p) and the numbers of ligands identified from the corresponding patient samples (q):
where w is a constant to scale the predicted heat map and MS-identified heat map into a similar scale for plotting purposes. The predicted ligand number at each amino acid position is the sum of scores across 18 MCL samples.
We evaluated the correlation between the MS-identified heat map and predicted heat map with Spearman’s correlation. Subregions of immunoglobulin (for example, FR3) were determined with IMGT, the international immunogenetics information system (http://www.imgt.org/)78. We compared the presentation in FR3 regions versus non-FR3 regions with a Mann–Whitney U test. Methods for precision–recall analysis are described below.
Analyzing cancer neoantigen candidates with MARIA
To score each somatic mutation in two cancer vaccine trials with MARIA, we represented each somatic mutation by a sequence that includes 14 amino acids upstream and downstream of the mutation site, such that the typical sequence length is 29 amino acids (14 + 1 + 14). We then took 15-amino-acid sliding windows along these sequences and combined each window with the HLA alleles and gene expression values of each patient to produce a MARIA score. In therapeutic cancer vaccine studies, we used 50 TPM to reflect the high concentration of mutated peptides introduced by vaccines, otherwise median TCGA tissue-matched RNA-seq gene expression was used. We used the highest score of all sliding windows as the score for a given somatic mutation. Neoantigen examples with known unsupported alleles (for example, HLA-DQB1*06:01 (ref. 79)) were excluded from our analysis. We performed the same analysis with the new IEDB CD4 immunogenicity tool80 for comparison and results are shown in Supplementary Tables 13–15.
Predictive performance metric calculation
N annotates the number of a group of peptide ligands. Sensitivity (also known as recall) was calculated as:
Specificity was calculated as:
Positive predictive value (also known as precision) was calculated as:
Or
Negative predictive value was calculated as:
ROC-AUC scores were calculated on the basis of the area under sensitivity and 1 − specificity curves and implemented with Python scikit-learn81. To generate precision and recall curves, we first calculated pairs of recalls and specificities across a range of MARIA cut-offs (70th to 99.9th percentile). We then calculated precision for each recall using an assumption of positive peptide prevalence (1%; Supplementary Note 2).
Statistical analyses
Sequence logo plots and amino acid frequency enrichment were generated with IceLogo60. Plotting in this study was done in matplotlib and seaborn82. We used two-tailed paired t tests in Fig. 1b for comparing AUC scores with two different methods but on the same set of patient data. We determined statistical significances difference between two AUC curves (for example, Fig. 2d) using the fast DeLong test83. Unless otherwise stated, statistically significant differences between distributions were determined by Mann–Whitney U tests. GO term enrichment analysis was conducted with ToppGene70. Except GO term enrichment, any statistical P values below 10−5 were denoted as P < 10-5 or P < 1 × 10−5.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Ligandomes are available from the PRIDE Archive under accession numbers PXD004746 and PXD005704. Data from two K562 ligandomes (Fig. 3) are provided in Supplementary Tables 5 and 6. The remaining HLA ligand datasets are publicly available from the provided references. All other data are available from the corresponding authors upon reasonable request.
Code availability
Researchers can run MARIA online at https://maria.stanford.edu/. Custom software code described in this work is available for academic research upon request from the authors or through https://maria.stanford.edu/. Commercial entities with interest in the software should contact Stanford University’s Office of Technology Licensing and reference docket S19-020.
References
Neefjes, J., Jongsma, M. L., Paul, P. & Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836 (2011).
Khodadoust, M. S. et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 543, 723–727 (2017).
Bassani-Sternberg, M. et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 7, 13404 (2016).
Toes, R. E., Ossendorp, F., Offringa, R. & Melief, C. J. CD4 T cells and their role in antitumor immune responses. J. Exp. Med. 189, 753–756 (1999).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Linnemann, C. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81 (2015).
Tran, E. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015).
The problem with neoantigen prediction. Nat. Biotechnol. 35, 97 (2017).
Khodadoust, M. S. & Alizadeh, A. A. Tumor antigen discovery through translation of the cancer genome. Immunol. Res. 58, 292–299 (2014).
Moss, D. L., Park, H. W., Mettu, R. R. & Landry, S. J. Deimmunizing substitutions in Pseudomonas exotoxin domain III perturb antigen processing without eliminating T-cell epitopes. J. Biol. Chem. 294, 4667–4681 (2019).
Andreatta, M. et al. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics 34, 1522–1528 (2017).
Marty, R., Thompson, W. K., Salem, R. M., Zanetti, M. & Carter, H. Evolutionary pressure against MHC class II binding cancer mutations. Cell 175, 416–428 (2018).
Zhao, W. & Sher, X. Systematically benchmarking peptide-MHC binding predictors: from synthetic to naturally processed epitopes. PLoS Comput. Biol. 14, e1006457 (2018).
Yadav, M. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014).
Caron, E. et al. Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry. Mol. Cell. Proteomics 14, 3105–3117 (2015).
Abelin, J. G. et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46, 315–326 (2017).
Vita, R. et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412 (2015).
Andreatta, M. et al. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 67, 641–650 (2015).
Paul, S. et al. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front. Immunol. 9, 1795 (2018).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Computation 9, 1735–1780 (1997).
Liu, H., Han, F., Zhou, H., Yan, X. & Kosik, K. S. Fast motif discovery in short sequences. In Proc. 32nd IEEE International Conference on Data Engineering 1158–1169 (IEEE, 2016).
Nielsen, M., Lundegaard, C. & Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics 8, 238 (2007).
Nielsen, M. & Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics 10, 296 (2009).
Sturniolo, T. et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat. Biotechnol. 17, 555–561 (1999).
Sidney, J. et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4, 2 (2008).
Wang, P. et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 11, 568 (2010).
Ciccocioppo, R., Di Sabatino, A. & Corazza, G. R. The immune recognition of gluten in coeliac disease. Clin. Exp. Immunol. 140, 408–416 (2005).
Bergseng, E. et al. Different binding motifs of the celiac disease-associated HLA molecules DQ2.5, DQ2.2, and DQ7.5 revealed by relative quantitative proteomics of endogenous peptide repertoires. Immunogenetics 67, 73–84 (2015).
Dorum, S. et al. HLA-DQ molecules as affinity matrix for identification of gluten T cell epitopes. J. Immunol. 193, 4497–4506 (2014).
Falk, K., Rotzschke, O., Stevanovic, S., Jung, G. & Rammensee, H. G. Pool sequencing of natural HLA-DR, DQ, and DP ligands reveals detailed peptide motifs, constraints of processing, and general rules. Immunogenetics 39, 230–242 (1994).
Chicz, R. M., Graziano, D. F., Trucco, M., Strominger, J. L. & Gorga, J. C. HLA-DP2: self peptide sequences and binding properties. J. Immunol. 159, 4935–4942 (1997).
Lorente, E. et al. Proteomics analysis reveals that structural proteins of the virion core and involved in gene expression are the main source for HLA class II ligands in vaccinia virus-infected cells. J. Proteome Res. 18, 900–911 (2019).
Chong, C. et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferongamma-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol. Cell Proteomics 17, 533–548 (2018).
Butterfield, L. H State of the art review: cancer vaccines. BMJ 350, h988 (2015).
O'Donnell, T.J. et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132 (2018).
Rubinsteyn, A., Hodes, I., Kodysh, J. & Hammerbacher, J. Vaxrank: a computational tool for designing personalized cancer vaccines. Preprint at biorXiv https://www.biorxiv.org/content/10.1101/142919v2 (2017).
Parkhurst, M. et al. Isolation of T-Cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clinical Can. Res. 23, 2491–2505 (2017).
Zacharakis, N. et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730 (2018).
Stevanovic, S. et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205 (2017).
Iiizumi, S et al. Identification of novel HLA Class II-restricted neoantigens derived from driver mutations. Cancers (Basel) 11, 266 (2019).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Muller, M., Gfeller, D., Coukos, G. & Bassani-Sternberg, M. ‘Hotspots’ of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization. Front. Immunol. 8, 1367 (2017).
Luo, H. et al. Machine learning methods for predicting HLA-peptide binding activity. Bioinform. Biol. Insights. 9, 21–29 (2015).
Bassani-Sternberg, M. et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 13, e1005725 (2017).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Bassani-Sternberg, M., Pletscher-Frankild, S., Jensen, L. J. & Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell Proteomics 14, 658–673 (2015).
Shao, W. et al. The SysteMHC Atlas project. Nucleic Acids Res. 46, D1237–D1247 (2018).
Racle, J. et al. Deep motif deconvolution of HLA-II peptidomes for robust class II epitope predictions. Preprint at biorXiv https://www.biorxiv.org/content/10.1101/539338v1 (2019).
Bhattacharya, R. et al. Prediction of peptide binding to MHC Class I proteins in the age of deep learning. Preprint at biorXiv https://www.biorxiv.org/content/10.1101/154757v1 (2017).
Mommen, G. P. et al. Sampling from the proteome to the human leukocyte antigen-DR (HLA-DR) ligandome proceeds via high specificity. Mol. Cell Proteomics 15, 1412–1423 (2016).
Graham, D. B. et al. Antigen discovery and specification of immunodominance hierarchies for MHCII-restricted epitopes. Nat. Med. 24, 1762–1772 (2018).
Forsstrom, B. et al. Proteome-wide epitope mapping of antibodies using ultra-dense peptide arrays. Mol. Cell Proteomics 13, 1585–1597 (2014).
Jorgensen, K. W., Rasmussen, M., Buus, S. & Nielsen, M. NetMHCstab—predicting stability of peptide–MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 141, 18–26 (2014).
Boelen, L. et al. BIITE: a tool to determine HLA class II epitopes from T cell ELISpot data. PLoS Comput. Biol. 12, e1004796 (2016).
Esteva, A. et al. A guide to deep learning in healthcare. Nat. Med. 25, 24–29 (2019).
Maddelein, D. et al. The IceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 43, W543–W546 (2015).
Tiscornia, G., Singer, O. & Verma, I. M. Production and purification of lentiviral vectors. Nat. Protoc. 1, 241–245 (2006).
Fujita, H. et al. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc. Natl Acad. Sci. USA 106, 21795–21800 (2009).
Hunt, D. F. et al. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 255, 1261–1263 (1992).
Bai, Y., Ni, M., Cooper, B., Wei, Y. & Fury, W. Inference of high resolution HLA types using genome-wide RNA or DNA sequencing reads. BMC Genomics 15, 325 (2014).
Nariai, N. et al. HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genomics 16, S7 (2015).
Verdegaal, E. M. et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 536, 91–95 (2016).
The Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 (2015).
Rahal, R. et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 20, 87–92 (2014).
The ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
Chen, J., Aronow, B. J. & Jegga, A. G. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics 10, 73 (2009).
The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699 (2018).
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA 89, 10915–10919 (1992).
Asgari, E. & Mofrad, M. R. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One 10, e0141287 (2015).
Nair, V. & Hinton, G. E. Rectified linear units improve restricted Boltzmann machines. In Proc. 27th International Conference on Machine Learning (Eds. Fuernkranz, J. & Joachims, T.) 807–814 (Omnipress, 2010).
Karosiene, E. et al. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013).
Nielsen, M., Justesen, S., Lund, O., Lundegaard, C. & Buus, S. NetMHCIIpan-2.0—improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 6, 9 (2010).
Cock, P. J. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Lefranc, M. P. et al. IMGT, the international ImMunoGeneTics information system 25 years on. Nucleic Acids Res. 43, D413–D422 (2015).
Tran, E. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014).
Dhanda, S. K. et al. Predicting HLA CD4 immunogenicity in human populations. Front. Immunol. 9, 1369 (2018).
Buitinck, L. et al. API design for machine learning software: experiences from the scikit-learn project. Preprint at arXiv https://arxiv.org/abs/1309.0238 (2013).
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Sun, X. & Xu, W. Fast implementation of DeLong’s algorithm for comparing the areas under correlated receiver operating characteristic curves. IEEE Signal Process. Lett. 21, 1389–1393 (2014).
Acknowledgements
This work was supported by National Institutes of Health (NIH) grant U01 CA194389 (to A.A.A.), NIH grant K08 CA207882 (to M.S.K.), NIH grant GM 102365 (to R.B.A.), NIH/Stanford MSTP training grant (to B.C.), an NSF GSF (to E.F.), an American Society of Hematology Scholar Award (to A.A.A.), the V-Foundation (to A.A.A.), a Damon Runyon-Rachleff Innovation Award (to J.E.E.), a W.M. Keck Foundation Medical Research Grant (to J.E.E.), a Conquer Cancer Foundation Young Investigator Award (to M.S.K.), the Leukemia and Lymphoma Society (to A.A.A. and M.S.K.), a Knut and Alice Wallenberg Foundation Postdoctoral Fellowship (to N.O.), a PD Soros New American Fellowship (to B.C.), the Stanford Bio-X Fellowship (to B.C.), the Virginia and D.K. Ludwig Fund for Cancer Research (to A.A.A.), the Bakewell Foundation (to M.D. and A.A.A.) and the SDW/DT and Shanahan Family Foundations (to A.A.A.). A.A.A. is a scholar of the Leukemia and Lymphoma Society. This work used the XStream computational resource, which is supported by the National Science Foundation Major Research Instrumentation program (ACI-1429830). This work used the shared FACS facility, which is supported by NIH S10 Shared Instrument Grant (S10RR027431-01). We thank the NIH Tetramer Facility for providing recombinant HLA-DR monomers for the peptide binding experiment. We thank M. Nielsen for providing insights regarding implementation of the NetMHCIIpan algorithm. We thank C. Linnemann and T. Schumacher for providing detailed peptide sequences. We thank Maria Birukova for discussions; MARIA is named in honor of and dedicated to the memory of the late Maria Birukova (1990–2016).
Author information
Authors and Affiliations
Contributions
B.C., A.A.A., M.S.K., J.E.E., M.M.D., R.L. and R.B.A. conceived the main algorithm design and validation of MARIA. B.C. and E.F. implemented and tested MARIA. B.C. performed statistical analysis. M.S.K., N.O., B.S. and L.E.W. generated training and validation data for MARIA. B.C. and C.L.L. implemented web infrastructure. Y.M. and B.C. conducted structural studies of HLA-DR complexes. B.C., M.S.K. and A.A.A. wrote the paper with input from M.S.K., J.E.E., R.L., M.D. and R.B.A. All authors commented on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
A.A.A. declares the following competing interests: stock or other ownership (CiberMed and Forty Seven); honoraria (Janssen Oncology); consulting or advisory roles (Celgene, Roche/Genentech and Gilead Sciences); research funding (Celgene); patents, royalties or other intellectual property (patent filings on immune deconvolution and circulating tumor DNA detection assigned to Stanford University); and travel, accommodations or expenses (Roche and Gilead Sciences). R.B.A. declares the following competing interests: stock or other ownership (Personalis); consulting or advisory role (Pfizer, Youscript, 23andme and WithHealth); patents, royalties or other intellectual property (royalties for patents related to genome sequencing).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 In vitro HLA-DR peptide binding assay for experimental validation and associated results.
(a) Workflow of flow cytometry based HLA-DR peptide binding assay. Dinitrophenyl-tagged peptides were exchanged into biotin-tagged HLA-DR (alpha and beta chains arranged with a leucine zipper) molecules and loaded onto streptavidin microspheres. Bound HLA-DR peptide complexes were visualized with flow cytometry fluorescence read-outs from anti-DR antibodies (APC) and anti-dinitrophenyl antibodies (FITC). HLA-DR peptide binding strength was estimated with percentages of the microsphere population that is high in both fluorescence markers. Peptide-HLA microspheres with >50% of the population above background FITC and APC signals are considered as positive binders. (b) MS identified JeKo-1-1 cell HLA-DR ligands and negative controls were tested for binding to HLA-DR recombinant proteins (HLA-DRB1*07:01 or HLA-DRB1*04:03) at 37°C. All ligand sequences have >10% NetMHCIIpan ranks (predicted non-binders). (c) Two non-binders in the 37°C experiment were tested for binding to HLA-DR recombinant proteins (HLA-DRB1*07:01 or HLA-DRB1*04:03) under 32°C. KEFYLFPTVFEDN was able to bind to HLA-DRB1*07:01 under 32°C. (d) Two technical controls, five positive controls, five negative controls and five JeKo HLA-DR ligands were incubated with HLA-DRB1*04:03 or HLA-DRB1*07:01 protein in 37C overnight. HLA-DR-APC and anti-DNP-FITC double positive populations are considered to be positive binders. Three additional JeKo ligands bound to at least one HLA-DR allele in spite of their poor NetMHCIIpan scores. References of controls peptides are listed in Supplementary Table 11. Each peptide in (b)-(d) was tested in two independent flow cytometry experiments to confirm the result.
Supplementary Figure 2 Peptide sequence encoding and detailed individual neural network architectures.
(a) An example (GCSADQACN) of how variable length amino acid sequences (8-26AA) to be one-hot encoded for machine learning purposes. A peptide is represented by a 21x26 matrix. Each row represents 21 possible amino acids, and each column represents the true amino acid at that position (1 = true). Any positions not encoding for an amino acid due to short length of a peptide are encoded as an all-zero vector which will be ignored by the neural network masking layer. (b) The model architecture of peptide sequence cleavage scores for HLA-DR presentation. The algorithm takes in a pair of query gene and short peptide sequence, and look up human proteome sequence database to determine the upstream and downstream six amino acid sequences (flanking sequences). A two-layer conventional neural network takes in these 12 amino acid sequences and output a 0-1 cleavage score indicating likelihood of HLA-DR presentation by knowing flanking sequences only. (c) The deep RNN model for predicting HLA-DR peptide presentation based on peptide sequences only. The deep RNN model consists of one masking layer, one RNN layer and two conventional dense layers. The deep RNN model takes in hot-hot encoded peptide sequences and output presentation scores indicating likelihood of HLA-DR presentation by knowing peptide sequences only. This model was trained on naturally presented MCL HLA-DR peptide ligands. (d) The deep RNN model for predicting HLA-DR peptide in vitro binding affinities based on IEDB binding data. A pair of query HLA-DR and peptide is encoded as a single sequence consisting of HLA-DRB1 pseudosequence, a spacer (-), and query peptide sequence. A deep RNN model takes in one-hot encodes sequences and outputs estimated in vitro binding affinities (1 - log50k(nM)). This model was trained on the IEDB quantitative HLA-DR to peptide binding data, identical to the data used by NetMHCIIpan3.1. (e) Selecting training and validation data for HLA-DR presentation prediction models. ~35k naturally presented HLA-DR peptide ligands and ~105k length-matched random human peptides are randomly assigned into training (85%) and validation (15%) sets. Peptides in the validation set identical or of substring to any peptides in training set were moved to the training set to avoid overfitting. The training and cross validation repeated 10 times to determine regularization parameters and estimates predictive powers of various models. The final performance of MARIA was determined with independent test sets.
Supplementary Figure 3 Relationship between gene expression levels and HLA ligand presentation.
(a) Comparison of gene expression levels of HLA-DR ligands and non-ligands in JeKo-1 cell line. Distributions of RNA-Seq estimated gene expression level (TPM) from JeKo-1 cell line HLA-DR ligands (red, n=5720), whole transcriptome (green, n=23165), and non-ligand genes (blue, n=22000) were plotted in a logarithmic space. HLA-DR ligands have significantly higher gene expression levels than the whole transcriptome (Mann-Whitney U test, **=p<1e-5), non-ligands have significantly lower gene expression levels than the whole transcriptome (Mann-Whitney U test, **=P<1e-5). In (a)-(c), Violin curves represent probability distribution function of gene expression, black boxes represent middle two quartiles, and white dot represents median. (b) Effects of correcting blood and ECM genes in estimating ligand gene expression levels in MCL patients. We reassigned gene expression values of blood particle or extracellular (ECM) associated genes to 50 TPM. Statistical significantly lower numbers of MCL patient HLA-DR ligand genes have low expression levels (0.1 TPM) after correction of blood and ECM genes (green, Mann-Whitney U test, **=P<1e-5, n=34049). (c) Comparison of gene expression levels of HLA-I ligands and non-ligands in JeKo-1 cell line and MCL patients. Distributions of RNA-Seq estimated gene expression level (TPM) from HLA-I ligands (red, n=60169 and 5555) and random protein-coding genes (green, n=23165 and 23165) were plotted in a logarithmic space. HLA-I ligands have significantly higher gene expression levels than the random protein-coding genes in both JeKo-1 cell lines (Mann-Whitney U test, **=P<1e-5) and MCL patients (Mann-Whitney U test, **=P<1e-5). Lowly expressed HLA-I ligands (<0.1TPM) are weakly enriched for blood micro-particles (FDR-corrected hypergeometrical test, q-value < 0.05). (d) Influence of filtering decoy peptide gene expression values on predictive power of gene expression. Gene expressions alone can differentiate between presented peptides and random decoys with 0.81 AUC. However, the predictive decreases as we removes lowly expressed genes. Gene expression does not differentiate MCL HLA-DR ligands and decoy peptides with >25 TPM gene expression values (0.51 AUC, n=3300 for ligands and n=10000 for decoy peptides). (e) Performance of MARIA on MCL validation set using various gene expression profiles. For 6 MCL patients, MARIA was run with three different gene expression profile dictionaries: patient-matched RNASeq, external MCL RNASeq, and shuffled external MCL RNASeq. MARIA predictive powers are not different between using patient-matched and external MCL RNASeq dictionaries (P=0.3, n=6). MARIA predictive power decreases when using a shuffled RNASeq dictionary (P=0.0002, n=6). P-values were determined with two-tailed paired t-test. (f) Performance of MARIA using tissue-matched or tissue-mismatched gene expression profile. For predicting ligands presented by melanoma HLA-II (n=10513), using tissue-mismatched RNASeq dictionaries decreases MARIA by less than 1% AUC. However, using a shuffled RNASeq dictionary profile data decreased MARIA performance by 7% AUC. SKCM: Skin cutaneous melanoma, GBM: Glioblastoma multiforme, BRCA: Breast invasive carcinoma, LUSC: Lung squamous cell carcinoma. (g) Influence of expression level thresholds for correction of extracellular matrix (ECM) genes on model performance. Depicted is the relationship between cross-validation AUC at various correction levels (b, TPM thresholds) for genes associated with extracellular matrix (Gene Ontology Cellular Compartment accession GO:0031012). Asterisks capture significant differences in cross-validation AUCs when comparing lower TPM thresholds than the mean level (TPM~50) that we had originally selected (** indicates Mann-Whitney U test p=0.0002 for TPM0 vs. TPM50, n=10; * indicates Mann-Whitney U test P=0.001 for TPM10 vs. TPM50, n=10). Models using thresholds between 20-100 TPM yielded highly similar validation AUC scores.
Supplementary Figure 4 Analysis of HLA ligand cleavage signatures.
(a, b) Cleavage signature analysis for MCL patient HLA-I ligands. Frequencies of 20 amino acids plus protein terminal position (-) in for 6 amino acids upstream of the C-terminus of HLA-I ligand (-6 to -1) and 6 amino acids downstream of HLA-I ligands (+1 to +6) are compared to the background distribution (n=22100 and n=42906) to determine amino acid enrichment and depletion surrounding HLA-DR ligands. Colors of the heat-map (a) and sizes of the IceLogo plot (b) letters indicate fold-change. The logo plot only includes statistically significant enrichment (P<0.0001). Compared to HLA-DR, HLA-I showed preferences for presences of tryptophan in both upstream and downstream sequences. (c) Comparison of cleavage signatures distinguishing flanking sequences of HLA-I and HLA-DR ligands. The IceLogo plot only includes statistically significant enrichments/depletions (P<0.001, two-tailed independent t-test by IceLogo60). Methionines, lysines, glutamines, and histidines are notably enriched in sequences flanking HLA-I ligands (n=22100) compared to the same regions of HLA-DR ligands (n=12150). In contrast, tryptophans, glycines, and tyrosines appear depleted from HLA-I flanks relative to HLA-DR. Cleavage signatures of (d) JeKo-1 mantle cell lymphoma cell lines, (e) L128 mantle cell lymphoma cell lines, (f) patient melanoma tissues, and (g) MUTZ3 dendritic cell lines. Each cell or tissue type exhibits variable cleavage signatures, but their HLA-DR ligands consistently show enrichment for peptides at the tails of protein (-) and depletion for proline (P) and histidine (H) flanking regions. Cleavage signatures of our lymphoma HLA-I and HLA-II ligands compared to two previous studies. (h) HLA-II ligands from dendritic cell line MUTZ3 (HLA-DRB1*10, HLA-DRB1*11, n=11419)1 were profiled by Mommen et al. 2016 Amino acid frequencies at six amino acids upstream and downstream of the presented ligands were compared to their counterparts from our MCL HLA-II ligands. ICE logo plots showed statistically significant enrichment or depletion in each position (p-value cut-off = 0.001). Methionine showed enrichment in dendritic cell cleavage signatures, and prolines showed stronger depletions at the majority of positions. (i) Similarly HLA-I ligands from B-lymphoblastoid cells profiled by Abelin et al. 2017 (HLA-A*01:01, HLA-A*02:01, HLA-B*35:01, n=4857)2 were compared to our MCL HLA-I ligands. Fewer significant differences were observed. Phenylalanines were enriched in B-lymphoblastoid cleavage signatures.
Supplementary Figure 5 Training and validation data sources and feature importance for presentation models.
(a) Detailed HLA-II ligand data and gene expression data used in training and validation of MARIA models. (b) Distributions of minimum additive distances of validation peptide sequences to training peptide sequences. The median minimum additive distance is around 7, which indicates these validation peptides need to undergo at least seven amino acid changes to become a peptide in the training set. No identical peptides were present in both training and validation sets (minimum additive distance > 0). (c) Performance of RNN-based binding models compared to NetMHCIIpan3.1. RNN-based HLA-DR in vitro binding model was trained on the identical IEDB HLA-DR data of NetMHCIIpan3.1 and validated on naturally presented MCL HLA-DR ligands (18 MCL samples). RNN-based binding models and NetMHCIIpan3.1 got about the same predictive performance (ROC-AUC=0.64, Mann-Whitney U test P=0.34, n=18). (d) Detailed 10-fold cross validation performance on identifying naturally presented with different predictors. MARIA models considering all relevant features (peptide sequence, gene expression, predicted in vitro binding, and cleavage scores) have higher average AUC scores than the second best model (RNN with sequence only, Mann-Whitney U test P<1e-5, n=10). (e) Validation performance of logistical regression models combining gene expression, binding scores and cleavage scores. Logistical regression models were trained on training MCL HLA-DR ligand data, and the validation performance was reported as average AUCs of 10-fold cross validation. Combining gene expression, binding scores and cleavage scores moderately increases the AUC compared to gene expression alone or combined with one additional feature (AUC=0.82, DeLong test p<0.0001, n=3300 for ligand peptides and n=10,000 for decoy peptides). (f) Comparing deep RNN models and shallow neural network (NN) models on predicting HLA-DR ligands based on peptide sequences only. Trained and validated on the identical sequence data, deep RNN models achieved higher validation AUC than shallow NN models after the 6th epoch. The solid lines indicate average validation AUC of 5 independent training experiments, and the shaded areas indicate 95% confidence interval (n=3300 for ligand peptides and n=10,000 for decoy peptides). (g) Impact of training dataset size on prediction performance for pan-HLA-II MARIA models. We trained new MARIA models using varying randomly sampled levels (x-axis) of training peptide ligand examples from a pan-HLA-II dataset profiling diverse cell types, when combined with the data we originally used to train MARIA (Khodadoust et al. 2017). Validation AUCs (y-axis) were then calculated using two monoallelic HLA-DR datasets (top panel: DRB1*01:01, bottom panel: DRB1*04:04) originally shown in Fig. 3. Models with more training examples show stronger performance, but with saturating plateaus in AUC performance gains after consideration of ~20k peptides. Surprisingly, models trained using pan-HLA-II data from diverse cell types did not significantly outperform the original MARIA model trained only on HLA-DR ligands from a single tumor type (two-tailed independent t-test P=0.35, n=10). The shaded area depicts the 95% confidence interval around the mean, based on 10 independently trained models, with the mean performance depicted by the solid line. (h) Performance of pan-HLA-II models for differentiating HLA-DP ligands from random human peptides. A recurrent neural network model was trained on presented HLA II ligands identified with MS and used to scores 20 reported HLA-DP ligands (Supplementary Table 12) and 100 random human peptides. Presentation scores for HLA-DP ligands were significantly higher than those for random human peptides (Mann-Whitney U test p=3e-6) and this difference achieved an AUC of 0.82.
Supplementary Figure 6 Insert figure title here by deleting or overwriting this text; keep title to a single sentence.
(a) Comparisons of interacting residues in HLA-DRB1*01:01 and HLA-DRB1*04:04 alleles. Two alleles differ in 7 out of 19 amino acid positions which potentially interact with peptide ligands. (b) Surface HLA-DR, HLA-I, and Immunoglobulin M (IgM) densities of K562 cell lines after lentiviral transductions. Transduced K562 cell lines are HLA-DR positive and HLA-I negative. K562 cell HLA-DR densities are substantially lower than B-cell lines (JeKo-1 and HBL-1). (c) HLA-DR densities after sorting and antibiotic selection. Transduced K562 cell lines were sorted for the top 1% expression for HLA-DR densities and grown in selective media (2ug/ml puromycin). The sorted mono-allelic K562 cells for DRB1*01:01 and DRB1*04:04 have higher HLA-DR densities (~10 fold increase compared to the unsorted populations). Two flow cytometry profiling experiments were conducted for each K562 cell line in (b) and (c). (d) Overlaps of K562 DRB1*01:01 and DRB1*04:04 peptide ligand sequences. Ligands from these two alleles overlap 15% when counting identical peptide sequences only and 31% when including peptides which are substrings of each other (Fig. 3a).
Supplementary Figure 7 Training of MARIA on HLA-DQ ligands and gluten peptide deamination effects on HLA-DQ presentation.
(a) Overlap of HLA-DQ2.2 and HLA-DQ2.5 peptide ligands. Ligands from these two alleles overlap 29% when counting identical peptide sequences only. (b) Training, validation, test of MARIA models for HLA-DQ2.2 presentation. To train the MARIA DQ2.2 model, 5845 peptides shared between HLA-DQ2.2 and HLA-DQ2.5, and 2529 peptide unique to HLA-DQ2.2 were used as the positive examples; 8374 length-matched peptides were used as negative examples. Peptide sequences were assigned into training, validation, and test set. No peptides in validation and test set were substring of a training peptide, vice versa. (c) MARIA predicted presentation scores on HLA-DQ2.2 presentation of five known celiac disease related gluten peptides upon all possible Q->E or Q->K mutations. Based on MARIA-DQ ranks, deamination forms (Q->E) of gluten peptides present better compared to unmodified forms or Q -> K forms of gluten peptides (* indicates p=3e-4, ** = P<1e-5, Mann-Whitney U test, n=15, 255, 7, 31, 63).
Supplementary Figure 8 Validating MARIA performance for predicting patient IgH HLA-DR presentation and immune response.
(a) NetMHCIIpan predicted HLA-DR presentation of lymphoma immunoglobulin correlated with experimentally identified HLA-DR immunoglobulin ligands. 18 MCL immunoglobulin sequences were analyzed by NetMHCIIpan(left, blue). The same 18 MCL samples were profiled with LC-MS/MS to determine the regions of immunoglobulin presented by HLA-DR. Predicted and observed presentation hot spots were significantly correlated on light chains (Spearman rho 0.48, p=3.8e-14, n=311). NetMHCIIpan prediction correlated with observed heavy chain presentation moderately (Spearman rho 0.10, p=0.02, n=1015). NetMHCIIpan predicted ligand numbers were normalized with the MS identified maximum ligand numbers for visualization purposes. (b) Precision-Recall curves of different models for identifying immunoglobulin (Ig) HLA-DR ligands. Curves depict the comparison of the precision/PPV (y-axis) for MARIA (blue curves) versus NetMHCIIpan (green curves) when considering a range of recall/sensitivity thresholds (x-axis). At 20% recall, MARIA achieved 56% and 31% precision for predicting Ig heavy chain (left panel) and light chain (right panel) presentation, respectively. In comparison, NetMHCIIpan achieved 13% and 16% at the same recall. Prevalence of 1% was used for all calculations. (c) Gating strategies of identifying alive CD4 T-cells after peptide stimulations. Analysis (d) was based on singlet lymphocyte populations with low 7-AAD and high CD4 levels. (d) Experimental validation of CD4 immunogenicity for candidate peptide neoantigens identified by MARIA. Peripheral blood mononuclear cells (PBMCs) were isolated from 3 MCL patients after immunization with autologous tumor vaccines. Patient PBMCs were re-stimulated with MARIA identified IgH neoantigens (>99.5th percentile) or control peptides for 30hrs. Antigen-specific T-cell activation was evaluated by cell surface CD137 induction. For 2 of 3 patients (MCL005 and MCL052), neoantigens induced specific CD4 T-cell activation with CD137+ levels comparable to positive controls (Pathogen Peptide Pool). These experiments were independently repeated in three patients with no technical replicates due to limited patient samples.
Supplementary Figure 9 Comparison and structural anlaysis of MARIA and NetMHCIIpan for mutated CLIP peptides.
(a) Key amino acid residues on CLIP peptide (PVSKMRMATPLLMQALP) interacting with HLA-DRB1*01:01 complex. Based on published crystal structures (PDB ID 3PDO), seven amino acids in the natural ligand of HLA-DR (CLIP) form hydrogen bonds with either HLA-DRA1 or HLA-DRB1*01:01. (b) MARIA scores change consistently with the influence of CLIP amino acid mutations. Seven mutated CLIP HLA-DR complexes with single amino acid substitution was created in silico according to the key residues defined in a. Mutated peptide atom positions in the HLA-DR environment were optimized with FlexPepDock. M107W, L113R, and M115R have higher MARIA percentile than CLIP WT (91.60% percentile), which is consistent with their gaining of hydrogen bonds or enhanced Van der Waals interactions resulting from mutations. R108 has lower MARIA percentile than CLIP WT, which is consistent with its loss of one hydrogen bound. K106R and K106D have about the same MARIA scores as WT (91.42% and 90.48%) despite opposite charges of these two mutants. Structure analysis showed the amino acid side chain in the position 106 does not contribute to hydrogen bond forming. Six out of seven mutants have about the same NetMHCIIpan percentiles compared to WT (99.85%-99.99%). NetMHCIIpan percentiles were calculated with 100% - NetMHCIIpan rank.
Supplementary Figure 10 Performance of MARIA for predicting melanoma antigen presentation and vaccine T-cell responses.
Performance of MARIA in predicting CD4 T-cell responses to personalized vaccines. Plots depict results for two melanoma clinical trials of personalized cancer vaccines (Sahin et al. 2017 (a, top) or Ott et al. 2017 (b, bottom)), where a range of MARIA score cutoffs (x-axis) are related to the Positive predictive values (PPV), negative predictive values (NPV) and sensitivity (y-axis) for predicting post-vaccination CD4 T-cell responses. MARIA scores of 95% and 99.5% were used as cut-offs for ‘medium’ and ‘high’ confidence categories depicted in Fig. 5. (c) Potential CD4 T cell epitopes in Ott et al. cohort based on MARIA scores. Numbers of neoantigens in melanoma above MARIA-high cut-off. Each nonsynonymous mutation in 6 melanoma patients (Ott et al. 2017) was scored with MARIA on a basis of 15mer sliding windows. The best MARIA score of all potential 15mer windows was used to represent the neoantigen. ~7% of nonsynonymous mutations reached 99.5% MARIA-high cut-off. Except the patient 1, all patients had at least 20 neoantigens in the MARIA-high category (MARIA percentile >99.5th). (d) Weak association of NetMHCIIpan and CD4 T-cell post-vaccination responses. Each vaccine peptide sequence in Ott et al. was scored with NetMHCIIpan and was stratified into three categories based on the same cut-off used for MARIA (Fig. 6d): low (<95th), medium (95-99.5th) and high (>99.5th). NetMHCIIpan score categories were weakly associated with CD4 T-cell responses but did not reach statistical significance (chi-square test, P=0.3). Dashed red lines indicate average response rates of the whole cohort. (e) Precision-recall curves of MARIA and NetMHCIIpan for identifying melanoma HLA-II ligands. Curves depict the comparison of the precision (y-axis) of each of three methods (full MARIA model, NetMHCIIpan 3.1, and a ‘random’ MARIA model trained on shuffled data) when considering a range of recall/sensitivity thresholds (x-axis). At 20% recall, MARIA achieved 38% precision (PPV), assuming a 1% prevalence of true antigen presentation.
Supplementary Figure 11 Abundance of HLA-II gene expression in various tumors.
Bulk RNA-Seq values of (a) HLA-DRA, (b) HLA-DQA1, and (c) HLA-DPA of 5077 TCGA tumor samples and 6 MCL tumor samples plotted as box plots. All tumor types have higher than 100 median TPM for HLA-DRA and HLA-DPA1. Most of tumors have lower HLA-II gene expressions compared to diffuse large B-cell lymphoma (DLBC, ~1000 median TPM). Few tumor samples have lower than 10 TPM (grey dash line) for HLA-DRA and HLA-DPA1. Top and bottom lines indicate 95% confidence interval, and the box indicates the first and third quartiles. MCL: mantle cell lymphoma, n=8; PRAD: prostate adenocarcinoma, n=558; HNSC: head and neck squamous cell carcinoma, n=566; OV: ovarian serous cystadenocarcinoma, n=430; SKCM: skin cutaneous melanoma, n=473; SARC: sarcoma, n=265; BRCA: breast invasive carcinoma, n=1256; MESO: mesothelioma, n=87; GBM: glioblastoma multiforme, n=175; KIRC: kidney renal clear cell carcinoma, n=618; LUAD: lung adenocarcinoma, n=601; DLBC: diffuse large B-cell lymphoma, n=48.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11 and Supplementary Notes 1–6.
Supplementary Tables
Supplementary Tables 1–15.
Rights and permissions
About this article

Cite this article
Chen, B., Khodadoust, M.S., Olsson, N. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol 37, 1332–1343 (2019). https://doi.org/10.1038/s41587-019-0280-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-019-0280-2
This article is cited by
-
Perspective view of allogeneic IgG tumor immunotherapy
Cancer Cell International (2024)
-
Development and use of machine learning algorithms in vaccine target selection
npj Vaccines (2024)
-
Informing immunotherapy with multi-omics driven machine learning
npj Digital Medicine (2024)
-
Computational immunogenomic approaches to predict response to cancer immunotherapies
Nature Reviews Clinical Oncology (2024)
-
Reducing Immunogenicity by Design: Approaches to Minimize Immunogenicity of Monoclonal Antibodies
BioDrugs (2024)
