
Abstract
Disease resistance (R) genes from wild relatives could be used to engineer broad-spectrum resistance in domesticated crops. We combined association genetics with R gene enrichment sequencing (AgRenSeq) to exploit pan-genome variation in wild diploid wheat and rapidly clone four stem rust resistance genes. AgRenSeq enables R gene cloning in any crop that has a diverse germplasm panel.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
Code availability
The programs, scripts, and bait library sequences used in this analysis are on Github (https://github.com/steuernb/AgRenSeq, https://github.com/kgaurav1208/AgRenSeq_GLM and https://github.com/arorasanu/KASPTree).
Data availability
Sequence reads were deposited in the European Nucleotide Archive (ENA) under project number PRJEB23912. The Sr46 and SrTA1662 loci were deposited at the National Center for Biotechnology Information under accession numbers MG851023 and MG763911. Ae. tauschii accessions with the GRU accession numbers in Supplementary Table 2 are available from the Germplasm Resources Unit, John Innes Centre, Norwich, UK (https://www.jic.ac.uk/germplasm/).
References
Jones, J. D. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Dangl, J. L. & Jones, J. D. Plant pathogens and integrated defence responses to infection. Nature 411, 826–833 (2001).
Knott, D. R. The genetic nature of mutations of a gene for yellow pigment linked to Lr19 in ‘Agatha’ wheat. Can. J. Genet. Cytol. 26, 392–393 (1984).
Niu, Z. et al. Development and characterization of wheat lines carrying stem rust resistance gene Sr43 derived from Thinopyrum ponticum. Theor. Appl. Genet. 127, 969–980 (2014).
McDonald, B. A. & Linde, C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379 (2002).
Dangl, J. L., Horvath, D. M. & Staskawicz, B. J. Pivoting the plant immune system from dissection to deployment. Science 341, 746–751 (2013).
Steuernagel, B. et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 34, 652–655 (2016).
Sánchez-Martín, J. et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 17, 221 (2016).
Periyannan, S. et al. The gene Sr33, an ortholog of barley Mla genes, encodes resistance to wheat stem rust race Ug99. Science 341, 786–788 (2013).
Saintenac, C. et al. Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341, 783–786 (2013).
Witek, K. et al. Accelerated cloning of a potato late blight-resistance gene using RenSeq and SMRT sequencing. Nat. Biotechnol. 34, 656–660 (2016).
Zhao, B. et al. A maize resistance gene functions against bacterial streak disease in rice. Proc. Natl Acad. Sci. USA 102, 15383–15388 (2005).
Thind, A. K. et al. Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat. Biotechnol. 35, 793–796 (2017).
Rahman, A., Hallgrímsdóttir, I., Eisen, M. & Pachter, L. Association mapping from sequencing reads using k-mers. eLife 7, e32920 (2018).
Audano, P. A., Ravishankar, S. & Vannberg, F. O. Mapping-free variant calling using haplotype reconstruction from k-mer frequencies. Bioinformatics 34, 1659–1665 (2018).
Lees, J. A. et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat. Commun. 7, 12797 (2016).
Jones, H. et al. Strategy for exploiting exotic germplasm using genetic, morphological, and environmental diversity: the Aegilops tauschii Coss. example. Theor. Appl. Genet. 126, 1793–1808 (2013).
Steuernagel, B., Witek, K., Jones, J. D. G. & Wulff, B. B. H. MutRenSeq: a method for rapid cloning of plant disease resistance genes. Methods Mol. Biol. 1659, 215–229 (2017).
Jupe, F. et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 76, 530–544 (2013).
Steuernagel, B., Jupe, F., Witek, K., Jones, J. D. & Wulff, B. B. NLR-parser: rapid annotation of plant NLR complements. Bioinformatics 31, 1665–1667 (2015).
Rouse, M. N., Olson, E. L., Gill, B. S., Pumphrey, M. O. & Jin, Y. Stem rust resistance in Aegilops tauschii germplasm. Crop Sci. 51, 2074–2078 (2011).
Yu, G. et al. Identification and mapping of Sr46 from Aegilops tauschii accession CIae 25 conferring resistance to race TTKSK (Ug99) of wheat stem rust pathogen. Theor. Appl. Genet. 128, 431–443 (2015).
Olson, E. L. et al. Simultaneous transfer, introgression, and genomic localization of genes for resistance to stem rust race TTKSK (Ug99) from Aegilops tauschii to wheat. Theor. Appl. Genet. 126, 1179–1188 (2013).
Yu, G. et al. Discovery and characterization of two new stem rust resistance genes in Aegilops sharonensis. Theor. Appl. Genet. 130, 1207–1222 (2017).
Krasileva, K. V. et al. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 14, R66 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Choulet, F. et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 345, 1249721 (2014).
The International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).
International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010).
Wilkinson, P. A. et al. CerealsDB 2.0: an integrated resource for plant breeders and scientists. BMC Bioinformatics 13, 219 (2012).
Winfield, M. O. et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant. Biotechnol. J. 14, 1195–1206 (2016).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant. Biotechnol. J. 12, 787–796 (2014).
Gardner, K. A., Wittern, L. M. & Mackay, I. J. A highly recombined, high-density, eight-founder wheat MAGIC map reveals extensive segregation distortion and genomic locations of introgression segments. Plant. Biotechnol. J. 14, 1406–1417 (2016).
Wicker, T. M. D. E. & Keller, B. TREP: a database for Triticeae repetitive elements. Trends Plant. Sci. 7, 561–562 (2002).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Talevich, E., Invergo, B. M., Cock, P. J. & Chapman, B. A. Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics 13, 209 (2012).
Stakman, E. C., Stewart, D. M. & Loegering, W. Q. Identification of Physiological Races of Puccinia graminis var. tritici E617 (United States Department of Agriculture, Agricultural Research Service, 1962).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Wilks, S. S. The large-sample distribution of the likelihood ratio for testing composite hypotheses. Ann. Math. Stat. 9, 60–62 (1938).
Brynildsrud, O., Bohlin, J., Scheffer, L. & Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17, 238 (2016).
Letunic, I. & Bork, P. Interactive tree of life (iTOL)v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Lagudah, E., MacRitchie, F. & Halloran, G. M. Influence of high-molecular-weight glutenin subunits from Triticum tauschii on flour quality of synthetic hexaploid wheat. J. Cereal Sci. 5, 129–138 (1987).
Moullet, O., Zhang, H. B. & Lagudah, E. S. Construction and characterisation of a large DNA insert library from the D genome of wheat. Theor. Appl. Genet. 99, 305–313 (1999).
Mago, R. et al. Generation of loss-of-function mutants for wheat rust disease resistance gene cloning. Methods Mol. Biol. 1659, 199–205 (2017).
Wang, M., Li, Z., Matthews, P. R., Upadhyaya, N. M. & Waterhouse, P. M. Improved vectors for Agrobacterium tumefaciens-mediated transformation of monocot plants. Acta Hortic. 461, 401–408 (1998).
Engler, C. et al. A golden gate modular cloning toolbox for plants. ACS Synth. Biol. 3, 839–843 (2014).
Patron, N. J. et al. Standards for plant synthetic biology: a common syntax for exchange of DNA parts. New Phytol. 208, 13–19 (2015).
Weber, E., Engler, C., Gruetzner, R., Werner, S. & Marillonnet, S. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 6, e16765 (2011).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7, e32253 (2012).
Broman, K. W., Wu, H., Sen, S. & Churchill, G. A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890 (2003).
Acknowledgements
We are grateful to the germplasm banks at Kansas State University, International Center for Agricultural Research in the Dry Areas, USDA-ARS National Small Grains Collection, and the N. I. Vavilov Research Institute of Plant Industry, and to our colleagues J. Raupp, J. Dvorak, and C. Hiebert for providing seed of Ae. tauschii. We thank our colleagues Y. Yue and JIC Horticultural Services for plant husbandry, J. Brown for helpful discussions, M. Ambrose and A. Meldrum for help with MTAs, K. Witek for technical assistance with RenSeq, M. Rouse and Y. Jin for PGT isolates, P. Solomon for help with bait source sequences, the International Wheat Sequencing Consortium for pre-publication access to REFSEQ v1.0, and B. McIntosh for critical reading of the manuscript. This research was supported by the NBI Computing Infrastructure for Science (CiS) group and financed by the Two Blades Foundation, USA to B.J.S. and B.B.H.W.; the Biotechnology and Biological Sciences Research Council (BBSRC) to B.B.H.W.; the BBSRC Designing Future Wheat Cross-Institute Strategic Programme to A.R.B. and B.B.H.W.; the Norwich Research Park Translational Fund to B.B.H.W.; the Lieberman-Okinow Endowment at the University of Minnesota to B.J.S.; the Grains Research and Development Corporation, Australia to E.L. and H.B.; the Gordon and Betty Moore Foundation to J.D.G.J.; and in-kind support by Arbor Biosciences.
Author information
Authors and Affiliations
Contributions
S.A., A.R.B., J.P., N.S., S.G., E.L., and B.B.H.W. configured the diversity panel. S.A. and J.C. extracted DNA and performed phylogenetic analysis. B.S., S.A., and J.E. designed and tested bait library. O.M., R.J., S.A., N.K., L.J.S., and B.J.S. phenotyped the diversity panel. J.E. prepared enriched libraries. S.A., G.Y., B.S., and B.B.H.W. performed AgRenSeq pilot studies. B.S. designed, implemented, and visualized the AgRenSeq data matrix, while K.G. and S.A. implemented the regression model and sample size power study. S.C., Y.L., S.P., N.A., H.B., M.A., S.S.X., and E.L. map-base cloned Sr46. S.P., M.A.M.H., and M.A. made Sr45 transgenics. E.O. mapped SrTA1662. B.B.H.W. conceived the study and drafted the manuscript with S.A., B.S., K.G., J.D.G.J., A.R.B., M.A., E.O., S.S.X., B.J.S., and E.L. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Patent applications filed based on this work include PCT/US2019/013430 by B.B.H.W., B.S. and S.A. and PCT/GB2019/050077 by B.B.H.W., B.S., S.A. and K.G.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Distribution of 317 KASP markers across the seven Ae. tauschii chromosome arms.
The horizontal blue bars indicate the chromosomal position of the markers. Centromeres are indicated by red bars. These markers were used to study the genetic diversity of the Ae. tauschii accessions (Figure 2b).
Supplementary Figure 2 An unweighted pair group method with an arithmetic mean (UPGMA) tree of 195 Ae. tauschii accessions.
The tree is divided into two branches representing ssp. strangulata (cyan) and ssp. tauschii (magenta) with an intermediate accession between the two subgroups (dark green). The final set of 151 nonredundant Ae. tauschii ssp. strangulata accessions used in AgRenSeq analysis are marked with black dots.
Supplementary Figure 3 Principal component analysis of the 195 Ae. tauschii lines.
SNP markers identified two different subgroups, namely ssp. strangulata (cyan; Lineage 2) and ssp. tauschii (magenta; Lineage 1) with an intermediate accession lying between the two subspecies (dark green). The PCA distribution of accessions into subgroups corresponded closely with the subgroups depicted in the UPGMA tree.
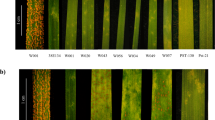
Supplementary Figure 4 Phenotypic diversity in the Ae. tauschii panel on the second leaf in response to various PGT races.
The infection types were scored using the Stakman infection type scale22 and the scores ranged from: resistant (0;, ;1-, 1), moderately resistant (1+, 2), moderately susceptible (2+, 3), to susceptible (3+, 4). The accession number and PGT race of the infected leaves from left to right are: BW_01020 (TTKSK), BW_01114 (TPMKC); BW_01020 (TTTTF); BW_01084 (TRTTF); BW_01081 (TTKSK); BW_01039 (TRTTF); BW_01065 (TTKSK); BW_01184 (TPMKC); BW_01125 (TTKSK); BW_01012 (TPMKC). The phenotype of each accession against the six PGT races used in this study and the number of replicates is provided in Supplementary Table 8.
Supplementary Figure 5 Distribution of seedling stem rust reactions across the 151 nonredundant Ae. tauschii spp. strangulata accessions.
The Ae. tauschii ssp. strangulata accessions were phenotyped with PGT races (a) QTHJC, (b) RKQQC, (c) TTKSK, (d) TRTTF, (e) TPMKC, and (f) TTTTF. The coloured scale at the bottom of each histogram indicates the variation in reaction types ranging from resistant (green), moderately resistant (olive), moderately susceptible (yellow) to susceptible (pink).
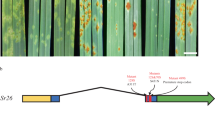
Supplementary Figure 6 Transgenic Sr45 expression provides stem rust resistance in wheat cultivar Fielder.
Primary transgenic seedlings show a typical Sr45 infection phenotype when challenged with the Australian stem rust race 98–1,2,3,5 and 6. (a) PC110–1 to 12 carry a Sr45 construct with the native promoter and terminator sequences, while (b) PC147–1 carries Sr45 regulated by the Sr33 promoter and terminator sequences. Nine out of ten independent PC147 lines conferred seedling resistance typical of Sr45. (c) Controls of Chinese Spring (CS) and CS 1D substitution lines carrying Ae. tauschii Sr33 (CS1D5406) or Sr45 (CS1D5405). The phenotypes were confirmed twice in the subsequent generation.
Supplementary Figure 7 Genetic mapping of SrTA1662 in an Ae. tauschii TA1662 × T. aestivum KS05HW14 BC2F1 population.
(a) PCR amplification of a 750 bp fragment from SrTA1662 using primers R1.F3 and R1.R3. From left to right are genotypes of a resistant BC2F1 individual (U6714-B-047 (SrTA1662/srTA1662)), a susceptible BC2F1 individual (U6714-B-048 (srTA1662/srTA1662)), Ae. tauschii TA1662 (SrTA1662/SrTA1662) and the susceptible recurrent parent KS05HW14 (srTA1662/srTA1662). (b) The 750 bp PCR fragment co-segregated with SrTA1662-mediated resistance in 83 BC2F1 individuals. CentiMorgan distances are shown on left and markers are shown on right of chromosome 1D. The plants were genotyped once and the diagnostic marker was amplified in all 38 stem rust resistant individuals.
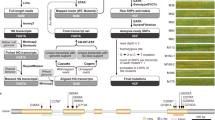
Supplementary Figure 8 Positional cloning of Sr46.
(a) Sr46 was mapped in diploid and hexaploid families resulting in the identification of a sequence tagged site marker psr649 located 0.1 cM from the resistance locus. (b) Physical mapping using BAC contigs of Ae. tauschii AL8/78 of the locus combined with (c, d) comparative genomics of the corresponding region from Brachypodium distachyon led to the identification of candidate genes in the Sr46 donor BAC library (e, f) of which unique mutations were found in the NLR gene. (g) The Sr46 candidate NLR gene was validated by transgenic studies where (g1) the wild type and sib lines without the transgene showed high infection type of 3+4, (g2) while seven independent transgenic events showed an intermediate infection type and (g3, g4) nine independent transgenic events showed a low infection type characteristic of a resistant phenotype. The transgenics were tested once each at the T0, T1 and T2 generations.
Supplementary Figure 9 The effect of sample size on the ability to identify Sr genes.
The accessions were randomly subsampled 50 times for each sample size as described in methods. The ability or power to identify the four genes SrTA1662 (dark green), Sr45 + Sr46 (pink), and Sr33 (olive green), as the top most candidate(s), is indicated with a threshold of 80% (horizontal black line). For example, 80 random accessions were enough to identify SrTA1662 in 80% of the trials.
Supplementary Figure 10 Impact of population structure on Sr33 gene identification.
Panel was subsampled to 95 accessions while retaining five Sr33 containing accessions. (a) Increase in signal (red dots) to noise ratio (black dots) when Sr33 containing accessions came from different clades. (b) Suppression of the Sr33 signal (red dots) when Sr33 containing accessions came from the same (orange) clade. (c) Phylogenetic tree of the Ae. tauschii panel (as in Supplementary Figure 2) highlighting the Sr33 containing accessions from different clades (represented with different colours). Note that the association score is defined as the negative log of p-value obtained using likelihood ratio test for nested models.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–10
Supplementary Tables
Supplementary Tables 1–13
Rights and permissions
About this article

Cite this article
Arora, S., Steuernagel, B., Gaurav, K. et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat Biotechnol 37, 139–143 (2019). https://doi.org/10.1038/s41587-018-0007-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-018-0007-9
This article is cited by
-
Are cereal grasses a single genetic system?
Nature Plants (2024)
-
Plant pangenomes for crop improvement, biodiversity and evolution
Nature Reviews Genetics (2024)
-
R gene-mediated resistance in the management of plant diseases
Journal of Plant Biochemistry and Biotechnology (2024)
-
HISS: Snakemake-based workflows for performing SMRT-RenSeq assembly, AgRenSeq and dRenSeq for the discovery of novel plant disease resistance genes
BMC Bioinformatics (2023)
-
Pan-genome of Citrullus genus highlights the extent of presence/absence variation during domestication and selection
BMC Genomics (2023)
