
Abstract
Mutations in a diverse set of driver genes increase the fitness of haematopoietic stem cells (HSCs), leading to clonal haematopoiesis1. These lesions are precursors for blood cancers2,3,4,5,6, but the basis of their fitness advantage remains largely unknown, partly owing to a paucity of large cohorts in which the clonal expansion rate has been assessed by longitudinal sampling. Here, to circumvent this limitation, we developed a method to infer the expansion rate from data from a single time point. We applied this method to 5,071 people with clonal haematopoiesis. A genome-wide association study revealed that a common inherited polymorphism in the TCL1A promoter was associated with a slower expansion rate in clonal haematopoiesis overall, but the effect varied by driver gene. Those carrying this protective allele exhibited markedly reduced growth rates or prevalence of clones with driver mutations in TET2, ASXL1, SF3B1 and SRSF2, but this effect was not seen in clones with driver mutations in DNMT3A. TCL1A was not expressed in normal or DNMT3A-mutated HSCs, but the introduction of mutations in TET2 or ASXL1 led to the expression of TCL1A protein and the expansion of HSCs in vitro. The protective allele restricted TCL1A expression and expansion of mutant HSCs, as did experimental knockdown of TCL1A expression. Forced expression of TCL1A promoted the expansion of human HSCs in vitro and mouse HSCs in vivo. Our results indicate that the fitness advantage of several commonly mutated driver genes in clonal haematopoiesis may be mediated by TCL1A activation.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Individual whole-genome sequence data for TOPMed whole genomes, individual-level harmonized phenotypes and the CHIP variant call sets used in this analysis are available through restricted access via the dbGaP TOPMed Exchange Area available to TOPMed investigators. Controlled-access release to the general scientific community via dbGaP is ongoing. dbGaP accession numbers are included in the Supplementary Tables 19 and 20. GWAS summary statistics have been deposited to dbGaP at accession phs001974. CHIP amplicon sequencing data from WHI have been deposited in dbGaP (parent study phs000200.v12.p3 and sub-study phs003206.v1). Data from scRNA-seq and ATAC-seq generated for this study are deposited under Gene Expression Omnibus (GEO) accession GSE205637. Source data are provided with this paper.
Code availability
Code developed for this study is available at: Re-analysis of Fabre et al.16 data https://github.com/weinstockj/longitudinal_clonal_expansion_analysis; Simulation of mutation counts in HSCs https://github.com/weinstockj/hsc_simulation; Rust binary used to call U2AF1 mutations https://github.com/weinstockj/pileup_region; Passenger count variant calling pipeline https://github.com/weinstockj/passenger_count_variant_calling; Analyses using PACER estimates https://github.com/weinstockj/PACER_analyses; Analysis code for TCL1A over-expression CITE-seq data https://github.com/jkgopa/HSC_TCL1A_overexpression_scRNAseq; Mutect2 WDL pipeline c https://dockstore.org/workflows/github.com/broadinstitute/gatk/mutect2:4.1.8.1?tab=info; Zenodo archives https://doi.org/10.5281/zenodo.7474678 and https://doi.org/10.5281/zenodo.7474719.
References
Steensma, D. P. et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 26, 2488–2498 (2014).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Abelson, S. et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018).
Desai, P. et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 24, 1015–1023 (2018).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Bick Alexander, G. et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 (2020).
Young, A. L., Challen, G. A., Birmann, B. M. & Druley, T. E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 12484 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590, 290–299 (2021).
Bick, A. G. et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020).
Osorio, F. G. et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Reports 25, 2308–2316.e4 (2018).
Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022).
Williams, N. et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 602, 162–168 (2022).
Lee-Six, H. et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018).
Fabre, M. A. et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 606, 335–342 (2022).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Zink, F. et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017).
Watson, C. J. et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449–1454 (2020).
Deuren, R. C. V. et al. Clone expansion of mutation-driven clonal hematopoiesis is associated with aging and metabolic dysfunction in individuals with obesity. Preprint at bioRxiv https://doi.org/10.1101/2021.05.12.443095 (2021).
Robertson, N. A. et al. Longitudinal dynamics of clonal hematopoiesis identifies gene-specific fitness effects. Nat. Med. 28, 1439–1446 (2022).
van Zeventer, I. A. et al. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood 135, 1161–1170 (2020).
Zerbino, D. R., Wilder, S. P., Johnson, N., Juettemann, T. & Flicek, P. R. The ensembl regulatory build. Genome Biol. 16, 56 (2015).
Carvalho-Silva, D. et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065 (2019).
Narducci, M. G. et al. TCL1 is overexpressed in patients affected by adult T-cell leukemias. Cancer Res. 57, 5452–5456 (1997).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017).
Thompson, D. J. et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657 (2019).
Malcovati, L. et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 129, 3371–3378 (2017).
The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Regev, A. et al. The Human Cell Atlas. eLife 6, e27041 (2017).
Velten, L. et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat. Commun. 12, 1366 (2021).
Psaila, B. et al. Single-cell analyses reveal megakaryocyte-biased hematopoiesis in myelofibrosis and identify mutant clone-specific targets. Mol. Cell 78, 477–492.e8 (2020).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Pietras, E. M. et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46 (2015).
Trapnell, C. et al. Pseudo-temporal ordering of individual cells reveals dynamics and regulators of cell fate decisions. Nat. Biotechnol. 32, 381–386 (2014).
Laine, J., Künstle, G., Obata, T., Sha, M. & Noguchi, M. The protooncogene TCL1 is an akt kinase coactivator. Mol. Cell 6, 395–407 (2000).
Brunet, A. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 (1999).
Eijkelenboom, A. & Burgering, B. M. T. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 (2013).
Kakiuchi, N. & Ogawa, S. Clonal expansion in non-cancer tissues. Nat. Rev. Cancer 21, 239–256 (2021).
Martincorena, I. et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Martincorena, I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Regier, A. A. et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 9, 4038 (2018).
Jun, G., Wing, M. K., Abecasis, G. R. & Kang, H. M. An efficient and scalable analysis framework for variant extraction and refinement from population scale DNA sequence data. Genome Res. 25, 918–925 (2015).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Voss, K., Gentry, J. & Van der Auwera, G. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000 Research https://doi.org/10.7490/f1000research.1114631.1 (2017).
Beauchamp, E. M. et al. ZBTB33 is mutated in clonal hematopoiesis and myelodysplastic syndromes and impacts RNA splicing. Blood Cancer Discov. 2, 500–517 (2021).
Miller, C. A. et al. Failure to detect mutations in U2AF1 due to changes in the GRCh38 reference sequence. J. Mol. Diagn. 24, 219–223 (2022).
Pedersen, B. S. & Quinlan, A. R. cyvcf2: fast, flexible variant analysis with Python. Bioinformatics 33, 1867–1869 (2017).
Asuni, N. & Wilder, S. VariantKey: a reversible numerical representation of human genetic variants. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/473744v3 (2019).
Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015).
Stan Modeling Language Users Guide and Reference Manual version 2.17 (Stan Development Team, 2020).
Stan Development Team. RStan: The R interface to Stan v.2.21.5. https://mc-stan.org/ (2020).
Bezanson, J., Edelman, A., Karpinski, S. & Shah, V. B. Julia: A fresh approach to numerical computing v.1.4 (2017).
Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S (Springer-Verlag, 2002).
Hiatt, J. B., Pritchard, C. C., Salipante, S. J., O’Roak, B. J. & Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 23, 843–854 (2013).
mimips. https://github.com/kitzmanlab/mimips (2020).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Uddin, M. M. et al. Longitudinal profiling of clonal hematopoiesis provides insight into clonal dynamics. Immun. Ageing 19, 23 (2022).
Ma, C., Blackwell, T., Boehnke, M. & Scott, L. J. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet. Epidemiol. 37, 539–550 (2013).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B 82, 1273–1300 (2020).
Li, Z. et al. Dynamic scan procedure for detecting rare-variant association regions in whole-genome sequencing studies. Am. J. Hum. Genet. 104, 802–814 (2019).
Bates, D. et al. Matrix: Sparse and Dense Matrix Classes and Methods v.1.4-1 (2019).
R Core Team. R: A Language and Environment for Statistical Computing. http://www.R-project.org/ (R Foundation for Statistical Computing, 2020).
Gogarten, S. M. et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Corces, M. R. et al. Omni-ATAC-seq: improved ATAC-seq protocol. Protocol Exchange https://doi.org/10.1038/protex.2017.096 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Acknowledgements
WGS for the TOPMed programme was supported by the National Heart, Lung and Blood Institute (NHLBI). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity quality control and general study coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). The authors thank the studies and participants who provided biological samples and data for TOPMed. The full study-specific acknowledgments are included in Supplementary Note 4. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the US Department of Health and Human Services. The authors wish to acknowledge the contributions of the consortium working on the development of the NHLBI BioData Catalyst ecosystem. S.J. is supported by the Burroughs Wellcome Foundation Career Award for Medical Scientists, Foundation Leducq (TNE-18CVD04), Ludwig Center for Cancer Stem Cell Research, the American Society of Hematology Scholar Award, the NIH Director’s New Innovator Award (DP2-HL157540), and a Leukemia and Lymphoma Society Discovery Grant. A.G.B. is supported by a Burroughs Wellcome Foundation Career Award for Medical Scientists, the NIH Director’s Early Independence Award (DP5-OD029586), and the Pew-Stewart Scholar for Cancer Research award, supported by the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust. WHI CHIP amplicon sequencing was supported by the NHLBI (R01 HL148565). The Fred Hutchinson Cooperative Center of Excellence in Hematology cell collection and processing is supported by NIDDK Grant # DK106829. The authors thank R. Majeti, T. Koehnke and B. Ebert for helpful discussions.
Author information
Authors and Affiliations
Consortia
Contributions
J.S.W., A.G.B. and S.J. conceived of the study and conceived of PACER. J.S.W. performed somatic variant calling, developed the PACER implementation and performed the human genetic association analyses. J.G. and S.J. functionally characterized the TCL1A locus. J.G. performed mouse model experiments. J.S.W. and S.J. performed WHI validation analyses. J.S.W., J.G., A.G.B. and S.J. wrote the manuscript with input from all authors. B.B.B., M.M.U., N.K., J.A.B., H.B., B.D., Z.M., N.L., T.M.M., S.E.L., K.P.P., S.R.M. and A.T.S. performed additional bioinformatic analyses. J.O.K., E.A.W. and A.P.R. contributed WHI amplicon sequencing data. C.A.L., J.G.B., K.D.T., X.G., M.F.S., A.S.V., S.K., A.R.S., J.R.O., J.P.L., E.B., K.C.B., N.C., E.E.K., R.J.L., M.F., L.H., D.M.L., S.R., B.E.C., B.M.P., J.C.B., J.A.B., E.K.S., J.H.Y., D.Q., N.D.P., B.I.F., D.W.B., M.H.C., D.L.D., V.S.R., L.R.Y., L.C.B., S.K., P.A.P., J.H., M.R., P.V.H., R.K., S.R.H., N.L.S., K.L.W., D.K.A., M.R.I., H.T., M.J.C., S.K., J.B.M., A.C., L.M.R., Y.G., M.A., J.I.R., S.S.R., R.P.T., B.A.K., J.M.J., M.M.W., J.G.S., O.M., P.M.N., B.S.C., R.D., J.E.C., J.B., S.M., L.K.W., S.X., M.Y., C.C.G., Y.I.C., W.L., G.M.M., J.P.K., C.R.P., M.B.S., D.D., D.R., C.A., C.K., Y.Z., J.E.M., P.K., A.D.J., R.A.M., T.W.B., G.R.A., A.V.S., H.M.K., P.N., E.A.W., A.P.R. and the NHLBI TOPMed Consortium contributed to sequencing and phenotyping of the included NHLBI TOPMed cohorts. T.W.B., G.R.A., A.V.S. and H.M.K. contributed TOPMed computing infrastructure and bioinformatics advice.
Corresponding authors
Ethics declarations
Competing interests
S.J. is on advisory boards for Novartis, AVRO Bio, and Roche Genentech, reports speaking fees and a honorarium from GSK, and is on the scientific advisory board of Bitterroot Bio. P.N. reports grants support from Amgen, AstraZeneca, Apple, Novartis and Boston Scientific, is a paid consultant for Apple, AstraZeneca, Novartis, Genentech, Blackstone Life Sciences and spousal employment at Vertex, all unrelated to the present work. S.J., A.G.B. and P.N. are paid consultants for Foresite Labs and co-founders, equity holders, and on the scientific advisory board of TenSixteen Bio. Stanford University has filed a patent application for the use of PACER to identify therapeutic targets on which S.J., A.G.B. and J.S.W. are inventors (US patent 63/141,333). The patent has been licensed to TenSixteen Bio. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. E.K.S. reports grant support from Bayer and GSK. J.H.Y. reports consulting fees from Bridgebio Therapeutics. M.H.C. reports grant support from Bayer and GSK, and consulting and speaking fees from Illumina and AstraZeneca. D.L.D. reports grant support from Bayer. L.M.R. is a consultant for the TOPMed Administrative Coordinating Center (through Westat). P.D. reports grant support from Janssen Research. H.M.K., G.R.A. and A.R.S. are employees of Regeneron Pharmaceuticals and recieve salary, stock and stock options as compensation. A.T.S. is a founder of Immunai and Cartography Biosciences and receives research funding from Allogene Therapeutics and Merck Research Laboratories. J.O.K. is on the scientific advisory board of MyOme Inc.
Peer review
Peer review information
Nature thanks Moritz Gerstung and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 PACER Estimates Clonal Expansion Rate.
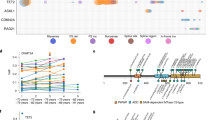
A. The passenger counts are enriched by 54% (95% CI: 51%-57%) after adjusting for age and study using a negative binomial regression. The different colors in the density plots correspond to quartiles of the marginal probability distributions. As the density estimates are smoothed, the underlying data points are indicated with hash marks. B. The distributions of passenger counts are stratified by the number of CHIP driver variants acquired. The different colors in the density plots correspond to quartiles of the marginal probability distributions. C. The observed clonal expansion rates (dVAFdT), as expressed in the change in variant allele frequency (VAF) over time (years), were associated with increased PACER fitness estimates in 55 CHIP carriers from the Women’s Health Initiative. The PACER fitness estimates have been inverse normal transformed. D. The posterior inclusion probabilities (PIP) as estimated by SuSIE63 are plotted on the y-axis, and the genomic position of a 0.8 Mb region including TCL1A is plotted on the x-axis. The linkage disequilibrium (LD) estimates are plotted on a color scale and are estimated on the genotypes used for association analyses. E. Rare variant analyses were performed using the SCANG46 rare variant scan procedure including all variants with a minor allele count less than 300. Identified rare variant windows are plotted as gray rectangles where the width corresponds to the size of the genomic region and the height corresponds to the pvalue of the SCANG64 test statistic for the window. F. Rare variant analyses were performed including the rs2887399 genotypes as covariate. Hypothesis testing was performed using the SCANG rare variant scan procedure. G. Multiz alignments across multiple species are shown for the TCL1A locus.
Extended Data Fig. 2 GWAS Implicates rs2887399 as a Modifier of Clonal Expansion Rate.
A. The distributions of the four conditions – DNMT3A and TET2 mutant clones stratified by homozygous genotype of rs2887399. The y-axis indicates the density of the distributions and the x-axis indicates the log10 founding censored passengers, which are the simulated equivalent to the singleton mutations observed in the real data analysis. Simulated DNMT3A mutations out-compete TET2 when rs2887399 is set to the protective T/T allele even though its fitness is unchanged by rs2887399. B. The top panel includes the -log10 pvalues from both the PACER GWAS and TCL1A cis-eQTLs in whole blood from GTEx v829. The GWAS p-values are estimated with SAIGE. In the bottom panel, posterior probability of colocalization from COLOC30 identifies rs2887399 as the likely shared causal variant. C. UMAP plot of scRNA-seq data from immune cells in the Human Cell Atlas31. TCL1A expression is highlighted on the bottom plot. UMAP plot was generated in the EMBL-EBI Single Cell Expression Atlas.
Extended Data Fig. 3 Chromatin Accessibility and Transcript Expression of TCL1A.
A. Quantification of fraction of HSC/MPPs expressing TCL1A transcripts in patients with TET2 or ASXL1 driven acute myeloid leukemia (AML) or myeloproliferative neoplasm (MPN) compared to healthy donors. Data is from single-cell RNA sequencing generated in Psaila33 et al. and Velten32 et al. B. ATAC-sequencing tracks of the TCL1A locus near rs2887399 in HSCs from healthy donors (row 1-4), pre-leukemic hematopoietic stem cells (pHSCs) from patients with AML but no detected driver mutations (rows 5-7), in pHSCs with TET2 mutations (rows 8-10), and pHSCs with DNMT3A mutations (rows 11-12). Data is from Corces et al.34. Vertical dashed line indicates location of the rs2887399 SNP. C. ATAC-sequencing tracks of the TCL6-TCL1A locus in HSCs from healthy donors (row 1), pre-leukemic hematopoietic stem cells (pHSCs) from patients with AML but no detected driver mutations (rows 2-3), pHSCs with DNMT3A mutations (rows 4-5), and in pHSCs with TET2 mutations (rows 6-7). Amino acid change and variant allele fraction (VAF) for the driver mutations are shown. Data is from Corces et al.34.
Extended Data Fig. 4 Schematic of rs2887399 Effect on TET2 Clonal Expansion.
Proposed model for clonal advantage due to mutations in TET2. In cells with the rs2887399 REF/REF genotype, loss of TET2 function leads to an accessible TCL1A locus, aberrant TCL1A RNA and protein expression in hematopoietic stem cells (HSC’s) and multi-potent progenitors (MPP’s), and subsequent clonal expansion. The presence of rs2887399 ALT alleles diminishes the TET2 clonal expansion phenotype by limiting TCL1A locus accessibility and downstream protein expression. Figure created with BioRender under a paid license.
Extended Data Fig. 5 CRISPR Editing Efficiency.
A. ICE analysis of Sanger traces to determine targeted CRISPR editing efficiency. Bar plots display percent of CD34+ CD38− CD45RA− cells with indel formation in gene of interest. These cells were used for the OMNI-ATAC and intracellular TCL1A flow assays. B. ICE analysis of Sanger traces to determine targeted CRISPR editing efficiency. Bar plots display percent of CD34+ CD38− CD45RA− cells with indel formation in gene of interest. These cells were used for the 14-day expansion assay.
Extended Data Fig. 6 ATAC Sequencing Tracks of TCL1A.
A. ATAC-sequencing tracks illustrating chromatin accessibility at rs2887399 in TET2 or DNMT3A-edited HSC/MPPs cultured for 5 days from donors of the GG, GT, and TT genotypes. Red line indicates location of rs2887399. TET2 edited samples are the same as in Fig. 4, shown here for comparison. B. ATAC-sequencing tracks illustrating chromatin accessibility at rs2887399 in AAVS, TET2 or DNMT3A-edited HSC/MPPs cultured for 7 days from donors of the GG and TT genotypes, and then sorted for CD34hi CD38− CD45RA− Lin− cells prior to nuclei preparation. Red line indicates location of rs2887399.
Extended Data Fig. 7 Interaction of CHIP Mutations and rs2887399 in human HSPC phenotypes.
A. Representative intracellular flow plots of TCL1A protein expression in edited HSC/MPPs from each rs2887399 donor after 11 days in culture. B. Quantification of Lin−/lo CD34+ CD38− CD45RAlo HSPCs (CD45RAlo HSPCs) after 14 days of in vitro expansion stratified by edited gene and rs2887399 genotype. Results of a linear regression model for the effect of edited gene (referent to AAVS1), rs2887399 genotype (referent to GG), and the interaction term of edited gene with rs2887399 genotype are presented below. Unadjusted p-values from two-sided tests are reported. n = 4 for each group. C. Ratio of CD34+CD45RA- cells to CD34- cells after 14 days of in vitro expansion stratified by edited gene and rs2887399 genotype. Results of a linear regression model for the effect of edited gene (referent to AAVS1), rs2887399 genotype (referent to GG), and the interaction term of edited gene with rs2887399 genotype are presented below. The horizontal line in each box indicates the median, the tops and bottoms of the boxes indicate the interquartile range, and the top and bottom error bars indicate maxima and minima, respectively. Unadjusted p-values from two-sided tests are reported. n = 4 for each group.
Extended Data Fig. 8 Validation of TCL1A shRNA and Expression Lentivirus.
A. Histogram of TCL1A-DAPI in wild-type, TCL1A CRISPR knockout, and TCL1A shRNA knockdown in NALM-6 cell line. B. Histogram of TCL1A-DAPI in human HSC/MPPs transduced with TCL1A-eGFP lentivirus or TET2-edited HSC/MPPs. MFI = geometric mean fluorescence intensity.
Extended Data Fig. 9 TCL1A Expression Promotes HSC Fitness in Mice.
A. Post-hoc analysis of percent GFP+ cells in the lineage negative fraction of the input cell mixture used for transplant. B. GFP+ chimerism over 20 weeks post-transplant as a fraction of total donor white blood cells. Shown are mean percent GFP+ cells and error bars represent standard errors for each time point. Hypothesis testing was performed with a two-sided Wilcoxon rank sum test and unadjusted p-values are shown above each time point. n = 8 for each group. C. Percent GFP+ cells in donor HSC/MPP subsets at 22 weeks post-transplant. The horizontal line in each box indicates the median, the tops and bottoms of the boxes indicate the interquartile range, and the top and bottom error bars indicate maxima and minima, respectively. Unadjusted p-values obtained from two-sided Wilcoxon rank sum tests are reported. n = 8 for each group.
Extended Data Fig. 10 CITE-seq of TCL1A Expressing Human HSPCs.
A. UMAP feature plots of Antibody Derived Tags (ADTs) for cell surface markers for HSPC identification. B. UMAP clustering of HSC/MPP populations colored by cell subtype clusters next to UMAP clustering of HSC/MPP populations colored by Monocle Pseudotime values. C. Stacked bar plot of percent of cells in each cell cycle phase as determined by Seurat cell cycle scoring module for each cell cluster. D. UMAP feature plot of select stress response and FOXO target genes.
Extended Data Fig. 11 Effect of TCL1A Expression on Human HSC/MPP Phenotypes.
A. Normalized enrichment scores (NES) of REACTOME pathways upregulated in HSC/MPP cluster 4 compared to HSC/MPP cluster 1 and filtered for those with FDR < 0.1 and NES > 1. Pathways printed in blue contain interferon response genes and pathways printed in red contain FOXO response genes. B. Stacked bar plot of all clusters in each analyzed sample dataset as a percentage of total cells in that sample. G/G or T/T refers to the genotype at rs2887399 in the donor. C. Stacked bar plot of absolute counts for each HSC/MPP cluster from each sample. Counts are shown as number of output cells at Day 7 per 1000 HSC/MPPs plated at Day 0.
Supplementary information
Supplementary Information
This file contains the supplementary theoretical analysis of PACER, data analyses, discussion, TOPMed cohort acknowledgments and Supplementary Figs. 1 and 2.
Supplementary Tables
Supplementary Tables 1–28.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Weinstock, J.S., Gopakumar, J., Burugula, B.B. et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis. Nature 616, 755–763 (2023). https://doi.org/10.1038/s41586-023-05806-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-05806-1
This article is cited by
-
Clonal haematopoiesis, ageing and kidney disease
Nature Reviews Nephrology (2024)
-
Driver mutation zygosity is a critical factor in predicting clonal hematopoiesis transformation risk
Blood Cancer Journal (2024)
-
Tissue mosaicism following stem cell aging: blood as an exemplar
Nature Aging (2024)
-
Genetic variation across and within individuals
Nature Reviews Genetics (2024)
-
Ultraviolet light shapes the evolution of precancerous cells
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
