
Abstract
Modern humans have populated Europe for more than 45,000 years1,2. Our knowledge of the genetic relatedness and structure of ancient hunter-gatherers is however limited, owing to the scarceness and poor molecular preservation of human remains from that period3. Here we analyse 356 ancient hunter-gatherer genomes, including new genomic data for 116 individuals from 14 countries in western and central Eurasia, spanning between 35,000 and 5,000 years ago. We identify a genetic ancestry profile in individuals associated with Upper Palaeolithic Gravettian assemblages from western Europe that is distinct from contemporaneous groups related to this archaeological culture in central and southern Europe4, but resembles that of preceding individuals associated with the Aurignacian culture. This ancestry profile survived during the Last Glacial Maximum (25,000 to 19,000 years ago) in human populations from southwestern Europe associated with the Solutrean culture, and with the following Magdalenian culture that re-expanded northeastward after the Last Glacial Maximum. Conversely, we reveal a genetic turnover in southern Europe suggesting a local replacement of human groups around the time of the Last Glacial Maximum, accompanied by a north-to-south dispersal of populations associated with the Epigravettian culture. From at least 14,000 years ago, an ancestry related to this culture spread from the south across the rest of Europe, largely replacing the Magdalenian-associated gene pool. After a period of limited admixture that spanned the beginning of the Mesolithic, we find genetic interactions between western and eastern European hunter-gatherers, who were also characterized by marked differences in phenotypically relevant variants.
Similar content being viewed by others

Main
Modern humans left sub-Saharan Africa at least 60 thousand years ago (ka), and during their initial expansion into Eurasia, they genetically mixed with Neanderthals, resulting in 2–3% Neanderthal ancestry in the majority of present-day non-African populations5. Genomic data have shown that modern humans were present in western Eurasia1,2 at least 45 ka. Some of those early groups from more than 40 ka further admixed with Neanderthals, as shown by signals of recent introgression in individuals from Bacho Kiro in Bulgaria—associated with an Initial Upper Palaeolithic (IUP) archaeological culture—and from Peştera cu Oase in Romania2,6. Other individuals from that period, such as Zlatý kůň from Czechia and Ust’Ishim from Russia, do not carry significantly more Neanderthal ancestry than other non-African groups1,7, indicating differential interactions between Neanderthals and early modern humans during their initial expansions across Eurasia. Surprisingly, however, none of those pre-40 ka individuals left substantial traces in the genetic makeup of present-day Eurasian populations1,2,6,7. The oldest genomes carrying ancestries that derive primarily from the lineage leading to present-day Europeans are Kostenki 14 (from 37 ka, with uncertain archaeological association from western Russia), Goyet Q116-1 (35 ka, Aurignacian-associated from Belgium) and Bacho Kiro 1653 (35 ka, probably Aurignacian-associated from Bulgaria)2,4,8. These data suggest that the genetic ancestries identified in the pre-40 ka individuals analysed so far went largely extinct or were assimilated by subsequent expansions1,9. The Kostenki genetic signature (related to the Kostenki 14 genome, and hereafter referred to as the Kostenki cluster or ancestry) contributed to the later Věstonice genetic cluster (hereafter, Věstonice cluster or ancestry), named after the Dolní Věstonice site in Czechia4. This genetic signature is shared among individuals associated with the archaeologically defined Gravettian culture (33–26 ka) in central and southern Europe and seemingly disappeared after the Last Glacial Maximum4 (LGM). However, the genetic profile of contemporaneous Gravettian-associated individuals from western Europe remains unknown, as is their contribution to populations after the LGM. Known to have been the coldest phase of the last Ice Age, the LGM is considered to have caused a demographic decline in large parts of Europe10, with populations retracting to southern latitudes as attested—for example—by the contemporaneity of the Solutrean culture (24–19 ka) in the Iberian peninsula and southern France. Other proposed climatic refugia for human survival during this period are the Italian peninsula, the Balkans and the southeastern European Plain, but the actual genetic contribution of populations from these regions to post-LGM Europeans is highly debated11,12,13.
After the LGM, a genetic component distantly linked to the Goyet Q116-1 individual from Belgium dated to 35 ka—named GoyetQ2 ancestry (hereafter, GoyetQ2 cluster or ancestry)—reappeared in individuals from southwestern and central Europe associated with the Magdalenian culture (19–14 ka from Iberia to eastern Europe across central Europe) and in an admixed form in subsequent Final Palaeolithic and Mesolithic hunter-gatherers4,14, but the geographic extension of this ancestry is still unclear. Instead, in southern Europe, a distinct hunter-gatherer genetic profile was found as early as 17 ka in individuals associated with the Epigravettian culture15 (24–12 ka, from the Italian peninsula to the southeastern European Plain across the Balkans). This ‘Villabruna’ ancestry (hereafter, Villabruna cluster or ancestry) showed connections to ancient and present-day Near Eastern populations4,16, but the mode and tempo of its expansion into the Italian peninsula remain unexplored. The Villabruna ancestry later appeared in central Europe and it is thought to have largely replaced groups related to the GoyetQ2 ancestry4. However, its formation, diffusion and interaction with contemporaneous hunter-gatherers from eastern Europe and their interplay with later expansions of Neolithic farmers from southeastern Europe are not well characterized.
In this study, we analyse 356 ancient hunter-gatherer genomes including new genomic data of 116 individuals dated to 35–5 ka alongside a novel contamination-estimation method based on runs of homozygosity. We provide a systematic description of the genomic transformations that hunter-gatherer groups experienced from the early Upper Palaeolithic onwards across western and central Eurasia and how those are possibly linked to cultural and climatic changes.
Ancient DNA data generation
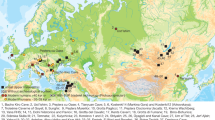
We generated genome-wide sequencing data for 102 newly reported hunter-gatherers, and increased coverage for 14 previously published individuals4. These data cover a time span of around 30,000 years from the Upper Palaeolithic to the Late Neolithic (defined here by the presence of pottery rather than by farming subsistence economy if not indicated), derive from multiple prehistoric cultural contexts, and originate from 54 archaeological sites in 14 countries: 1 Aurignacian-associated individual from Belgium and 1 culturally unassigned individual from Romania (35–33 ka), 15 Gravettian-associated individuals from Spain, France, Belgium, Czechia and Italy (31–26 ka), 2 Solutrean-associated individuals from Spain and France (23–21 ka), 9 Magdalenian-associated individuals from France, Germany, and Poland (18–15 ka), 4 Epigravettian-associated individuals from Italy (17–13 ka), 2 Federmesser-associated individuals from Germany (14 ka), and 81 Mesolithic to Neolithic foragers from across western Eurasia (11–5 ka), together with 1 central Eurasian Neolithic individual from Tajikistan (8 ka) (Fig. 1, Extended Data Table 1, Supplementary Data 1.A, Supplementary Information, section 1 and Supplementary Fig. 1).

a, Geographic locations of newly reported individuals (filled symbols with black outline) and representative previously published individuals (outlined stars). Dotted lines delimit geographic regions described in the text. b, Calibrated radiocarbon dates of individuals plotted in a. The y axis shows the average of calibrated radiocarbon dates in thousands of years (kyr) (Supplementary Data 1.A). The horizontal dashed line marks the boundary between Late Pleistocene and Holocene. c, MDS plot of European hunter-gatherers based on 1 − f3(Mbuti; pop1, pop2). The dimensions are calculated using newly reported and previously published hunter-gatherer groups or individuals with more than 30,000 SNPs. The detailed grouping of individuals shown with empty coloured circles is described in Supplementary Data 1.I.
We built 1 to 8 single- and double-stranded genetic libraries for each individual and enriched them for human DNA on 1.24 million single nucleotide polymorphisms6 (SNPs), which were then sequenced and yielded 0.04- to 7.64-fold coverage on average over the targeted SNPs. Genetic sexing revealed 78 male individuals and 38 female individuals (Supplementary Fig. 12). The levels of contamination from modern human DNA were estimated on the basis of mitochondrial DNA (mtDNA), X chromosome and autosomal DNA, and with a haplotype copying model that is extended here to autosomal data in runs of homozygosity (ROH) (Methods, Supplementary Information, sections 2 and 3, Supplementary Figs. 2–11 and Supplementary Table 1). Substantially contaminated libraries as well as marginally contaminated libraries of individually analysed genomes were filtered to maintain reads showing postmortem DNA damage (Methods and Supplementary Figs. 10 and 11). Pseudo-haploid genotypes were called on the targeted SNPs by randomly sampling a single allele at each position, resulting in individuals with 6,600 to 1.07 million SNPs covered on the 1.24-million-SNP panel (Extended Data Table 1 and Supplementary Data 1.A). The newly generated genotypes were merged with 240 published ancient hunter-gatherer genomes and modern worldwide populations for downstream analyses (Supplementary Data 1.G). Contrary to the proposal in Fu et al.4 but in agreement with Petr et al.17, we do not observe a substantial decrease of Neanderthal ancestry in most European hunter-gatherers through time (Supplementary Information, section 6 and Supplementary Figs. 15–17). This provides further support for the model with no long-term decline of genome-wide Neanderthal ancestry in modern humans following their introgression18.
Before the LGM
The Gravettian culture was one of the most widely distributed Upper Palaeolithic cultures across western Eurasia before the LGM19. It is often considered as a pan-European cultural mosaic with regional variations in material to symbolic productions20,21. In this debated framework, Gravettian-associated individuals have been suggested to represent a biologically homogeneous population on the basis of craniometric and genomic data4,22. However, published Gravettian-associated genomes originate from central and southern Europe, leaving the genetic profile of Gravettian-associated human groups from western and southwestern Europe undescribed.
To gain an overview of the genomic background of European hunter-gatherers before the LGM, we used multidimensional scaling (MDS) to plot a dissimilarity matrix of pairwise outgroup f3-statistics in the form 1 − f3(Mbuti; pop1, pop2) (Fig. 2a). This plot reveals the presence of three distinct groupings: (1) a pre-40 ka group with individuals from the Ust’Ishim, Bacho Kiro, Zlatý kůň and Peştera cu Oase sites, (2) a Věstonice cluster including Gravettian-associated individuals from central–eastern and southern European sites (Dolní Věstonice, Pavlov, Krems-Wachtberg, Paglicci and Ostuni), and (3) a Fournol cluster (hereafter, Fournol cluster or ancestry) comprising Gravettian-associated individuals from western and southwestern European sites (Ormesson, La Rochette, Fournol and two Serinyà cave sites (Mollet III and Reclau Viver)). The previously described Věstonice cluster, including a newly reported 29,000-year-old individual from Paglicci cave (Paglicci 12) in southern Italy, is closely related to the previously published genomes from Sunghir and Kostenki 12 in western Russia, which are dated to 34 ka and 32 ka, respectively4,23. The newly defined Fournol cluster is closely related to Aurignacian-associated individuals from Belgium dated to 35 ka (Goyet Q116-1 and the newly reported Goyet Q376-3 individual). Notably, and contrary to the report by Fu et al.4, another Gravettian-associated population from central–western Europe (Goyet in Belgium, n = 6 individuals) is both geographically and genetically intermediate between the Věstonice and Fournol clusters. The similarity between Goyet Q116-1 and Goyet Q376-3 and the Fournol cluster is also observed at the mtDNA level, with both groups including individuals who carried mtDNA haplogroup M, which has not been found in European individuals from after the LGM24 (Extended Data Figs. 1 and 2).

a, MDS plot of pre-LGM individuals. The pre-40 ka group and the Fournol and Věstonice clusters are marked as shaded areas in different colours. Individuals and groups are plotted with the same colours and symbols as in Fig. 1 and names are indicated next to the symbols. b, Gravettian-associated individuals form two distinct groups, with central-eastern and southern European individuals as part of the Věstonice cluster and western and southwestern European individuals as part of the Fournol cluster. In central-western Europe, Gravettian-associated individuals from Goyet show affinity to both clusters. Error bars show 1× s.e.m. (black) or 3× s.e.m. (grey) of the f4 values estimated from 5 cM-block jackknife analysis. c, Admixture graph modelling of the main pre-LGM European hunter-gatherer lineages created using qpGraph.
We further validated the genetic distinction between the Věstonice and Fournol clusters observed in the MDS plot with a series of f4-statistics (Supplementary Data 2.B). All individuals belonging to the Fournol cluster show higher affinity to Goyet Q116-1 than to the Sunghir group (n = 4), and the Věstonice-cluster individuals show higher affinity to the Sunghir group than to Goyet Q116-1 (Extended Data Fig. 3). These f4-statistics also confirm that Goyet Q376-3 carries a similar ancestry to Goyet Q116-1 and Kostenki 12 carries a similar ancestry to the Sunghir group, whereas Bacho Kiro 1653 (35 ka) from Bulgaria, Muierii 1 (34 ka) and Cioclovina 1 (32 ka) from Romania, and Paglicci 133 (33 ka) from southern Italy are equally related to Goyet Q116-1 and Sunghir. We further tested whether individuals included in the Věstonice and Fournol clusters share similar allele frequencies with the main representatives of those two clusters. With the statistics f4(Mbuti, Fournol 85; Věstonice, test) and f4(Mbuti, Věstonice; Fournol 85, test), we show that all Věstonice-cluster individuals are significantly closer (|Z|>3) to the Věstonice group (n = 5) and the Fournol-cluster individuals are closer to Fournol 85, whereas the geographically intermediate Gravettian-associated Goyet group shows extra affinity to both clusters (Fig. 2b).
We further modelled the genetic profile of pre-LGM individuals with qpGraph (Supplementary Information, section 10 and Supplementary Figs. 19–25). The admixture graph shows that the Bacho Kiro IUP group (n = 3) shares ancestry with multiple early modern human lineages2 (Supplementary Information, section 7), and that the more than 45,000-year-old Zlatý kůň genome1 is the most deeply divergent non-African lineage sequenced to date (Extended Data Fig. 4). This is also validated by f4-statistics of the form f4(Mbuti, Zlatý kůň; test1, test2), which are consistent with zero for all other pre-LGM hunter-gatherers (Supplementary Data 2.C), indicating an equidistant relationship of Zlatý kůň to the tested groups. When Gravettian-associated individuals are included in an admixture graph also featuring Kostenki 14, we find that Fournol 85 fits best as a sister lineage of Goyet Q116-1, whereas the Věstonice group is modelled as a mixture between a lineage related to the Sunghir group and one related to the Goyet Q116-1–Fournol 85 branch (Fig. 2c). This is also supported by f4-statistics of the form f4(Mbuti, Fournol 85; Sunghir, test), which are significantly positive for all the individuals included in the Věstonice cluster (Supplementary Data 2.B). Therefore, as previously reported2, the Věstonice cluster itself results from admixture between western and eastern lineages, which might contribute to the observed homogeneity in cranial morphology among Gravettian-associated individuals22.
These results show that some, but not all, of the genomic ancestries present in Europe between around 40 ka and 30 ka survived in the Gravettian-associated populations studied so far. The Kostenki (and Sunghir group) ancestry contributed to the previously described Věstonice cluster represented by Gravettian-associated individuals from central-eastern and southern Europe4. By contrast, the Goyet Q116-1 genetic profile gave rise to the newly described Fournol cluster identified in Gravettian-associated individuals from western and southwestern Europe. Notably, this genetic distinction coincides with dissimilarities in mortuary practice among genetically analysed Gravettian-associated individuals from different parts of Europe. Individuals in western and southwestern Europe related to the Fournol cluster are consistently deposited in cave sites and occasionally exhibit anthropogenic marks whereas individuals related to the Věstonice cluster are buried with grave goods and/or personal ornaments and ochre in open air or cave sites in central-eastern and southern Europe, respectively (Supplementary Figs. 29–32 and Supplementary Table 4). The oldest individual in the Fournol cluster is Ormesson 2988 from northeastern France (31 ka, Early/Middle Gravettian), whereas a Gravettian group from Goyet in Belgium (27 ka, Late Gravettian) is found to be a mixture between the Věstonice and Fournol clusters. This suggests that between the Early/Middle and Late Gravettian there was an east-to-west expansion of the Věstonice-associated ancestry that reached central-western Europe and created a longitudinal admixture cline between those two genetically distinct pre-LGM populations.
LGM in southwestern and western Europe
The Solutrean culture is temporally intermediate between the Gravettian and the Magdalenian (or the Badegoulian) cultures, and is found in southwestern and western Europe, which are considered to have been climatic refugia for human populations during the LGM25,26. However, the extent to which groups associated with the Solutrean culture are in genetic continuity with earlier and later populations from the same region is unknown because no genomic data from Solutrean-associated individuals have been reported previously. Both newly sequenced genomes from Solutrean-associated individuals (Le Piage II (23 ka) from southwestern France and La Riera (level 14, 21 ka) from northern Spain) show a generalized affinity with members of the Fournol and GoyetQ2 clusters in outgroup f3-statistics (Supplementary Data 2.A). In the MDS plot, the Le Piage II individual falls particularly close to individuals belonging to the Fournol cluster, suggesting a local genetic continuity of this ancestry during the LGM (Supplementary Fig. 13). F4-statistics further support this view, revealing that Le Piage II is more closely related to the Fournol cluster than the Věstonice cluster (f4(Mbuti, Le Piage II; Věstonice, Fournol 85) ≫ 0, Z = 6.58). We also compared its affinity to El Mirón (northern Spain), the oldest Magdalenian-associated individual sequenced to date (19 ka). F-statistics suggest that Le Piage II is genetically intermediate between Fournol 85 and El Mirón (Supplementary Data 2.D). Moreover, previous studies have shown that El Mirón carries a genetic contribution from the Villabruna cluster, which is found in Epigravettian-associated individuals from Italy4,15. El Mirón has a significantly higher similarity to the Villabruna cluster than Fournol 85 and Le Piage II, while the affinity to the Villabruna cluster in Le Piage II is not significantly higher than in Fournol 85 (Supplementary Data 2.D). Overall, the Solutrean-associated Le Piage II individual links the preceding Fournol ancestry with the succeeding ancestry found in El Mirón, providing direct evidence for genetic continuity throughout the LGM in southwestern and western Europe. These European regions, therefore, constitute climatic refugia where human populations survived during the LGM.
Post-LGM in the Italian peninsula
After the LGM, the Epigravettian culture was widespread in southern and southeastern Europe. In spite of growing discussions about its nature27,28, the Epigravettian culture has been traditionally assumed to be the result of a transition from the preceding local Gravettian29. However, the level of genetic continuity between individuals associated with these cultures and the population structure among Epigravettian-associated individuals have not been fully explored. Here, we report genomic data from 4 individuals, including 3 approximately 13,000-year-old genomes from northeastern Italy (Pradis 1), northwestern Italy (Arene Candide 16) and Sicily (San Teodoro 2), as well as increased genome-wide coverage from Tagliente 215 dated to 17 ka.
In the MDS plot, we find that all of the newly and previously reported Epigravettian-associated individuals fall within the Villabruna cluster4 (Fig. 1c). A series of f4-symmetry statistics confirm that all the Epigravettian-associated individuals are cladal, and do not share excess affinity with any local (Paglicci 12) or non-local preceding ancestries (Goyet Q116-1, Kostenki 14, Mal’ta 1 or Věstonice) (Supplementary Data 2.F). Moreover, none of the Epigravettian-associated individuals have more affinity to southern European than to central-eastern European Gravettian-associated groups, as shown by f4(Mbuti, Epigravettian-associated individual/group; Věstonice, Paglicci 12) that is consistent with 0 (Supplementary Data 2.G).
Next, we investigated the genetic relationships between Epigravettian-associated individuals across the Italian peninsula, by reconstructing a phylogeny based on a matrix of pairwise f2 genetic distances (Fig. 3a and Supplementary Fig. 9) and testing the relative affinity among them using f4-statistics in the form f4(Mbuti, Epigravettian A; Epigravettian B, Epigravettian C) (Supplementary Data 2.E). The inferred topology reveals a phylogeographic pattern irrespective of individual ages. In particular, the 13 ka Pradis 1 individual from northeastern Italy represents the most basal lineage compared to all other Epigravettian-associated individuals, including the older Tagliente 2 and Villabruna genomes from northern Italy (group 1). Individuals from northwestern Italy (Arene Candide 16), central Italy (Continenza) and Sicily fall on a phylogenetically more derived branch (group 2), which further diversified into a branch composed of Sicilian hunter-gatherers only (group 3). Within Sicily, the 14 ka Oriente C individual shows higher affinity with the much younger but geographically closer 10 ka Uzzo group30 (n = 2) than with the almost contemporaneous San Teodoro 2 individual from eastern Sicily.

a, Population structure among Epigravettian-associated populations revealed by a neighbour-joining tree based on pairwise f2 genetic distances. Branch labels show unique drift lengths; black dots refer to individuals with newly generated data and white dots refer to previously published genomes; the position of each node does not imply the location where the split took place. b, Population diversity shown by PMR between individuals in different groups. The grouping of Epigravettian-associated populations is shown with the same colour in a. The black-outlined diamond marked out in the Gravettian group shows the PMR between the two Gravettian-associated individuals from southern Italy (Paglicci 12 and Ostuni 1). In the box plot the centre line is the median, box bounds delineate the interquartile range and whiskers extend to maximum and minimum values, excluding outliers. The sample size of individual pairs included in each group is reported in Supplementary Data 3.A.
Finally, we estimated the genetic diversity of Epigravettian-associated individuals in the dataset by calculating both pairwise mismatch rates (PMR) on pseudo-haploid genotypes and individual heterozygosity levels on pseudo-diploid genotypes (Supplementary Data 3.A). Compared with the genetic diversity observed among all analysed Gravettian-associated groups, Epigravettian-associated individuals show significantly lower amounts of genetic diversity (two-tailed t-test, P < 0.001) (Fig. 3b). Moreover, we reveal a north-to-south decrease in genetic diversity among the Epigravettian-associated groups, with the highest PMR and heterozygosity values found in northern Italian individuals (group 1), intermediate in western and central Italian individuals (group 2) and the lowest in Sicilian individuals (group 3) (Fig. 3b). A similar pattern is observed through the analysis of ROH segments (Extended Data Fig. 5 and Supplementary Information, section 9). We detect the highest amount of ROHs in Epigravettian-associated individuals from Sicily, who carry an extreme amount of more than 200 cM of short ROHs (4–8 cM). This suggests a very small recent effective population size, estimated to be in the order of around 70 individuals (Supplementary Table 2), causing the low genetic diversity in Sicilian Epigravettian hunter-gatherers.
To summarize, our results highlight a genetic turnover in the Italian peninsula of the Gravettian-associated Věstonice cluster by the Epigravettian-associated Villabruna cluster that might correlate with discontinuities observed in the archaeological record31. We show that all analysed Epigravettian-associated individuals carry a homogeneous Villabruna ancestry, with the intra-group genetic structure mainly determined by their geographical, and not temporal, distribution. The phylogenetic reconstruction of Epigravettian-associated genomes, with Pradis 1 diverging more deeply than all others, indicates that the turnover took place much earlier than 17 ka—the date of the more derived Tagliente 2 genome. This, together with the evidence of Villabruna ancestry in El Mirón 19 ka, further suggests that this genetic discontinuity could be the result of palaeogeographic and palaeoecological transformations connected to the LGM32, rather than to the Bølling–Allerød warming period4,15 (14.7–12.9 ka). In addition, our phylogeographic analysis points to northeastern Italy as the possible entry point of the Epigravettian-associated gene pool in the Italian peninsula. This finding, in conjunction with the genetic affinity of the Villabruna cluster to ancient and present-day Near Eastern ancestries4,15,16 (Supplementary Information, section 8, Supplementary Fig. 18 and Supplementary Data 2.O), suggests the Balkans as a source of the incoming Epigravettian-associated population. The LGM could thus have created a corridor south of the Alps for east-to-west human movements that genetically connected hunter-gatherer populations from the Balkans to Iberia, possibly also via dispersals along existing lower-sea-level coasts32.
Post-LGM in western and central Europe
The Magdalenian culture was widely distributed in southwestern, western and central Europe after the LGM33. Despite this wide geographical range, it is not clear whether different groups associated with this culture originated from a common source population and how those groups were genetically related to each other. Previous studies identified two different genetic compositions in Magdalenian-associated individuals—the GoyetQ2 cluster including central-western European genomes dated to around 15 ka (from France, Belgium and Germany), and the ancestry of the El Mirón individual from Spain4,14 from around 19 ka. Both of these ancestries carry a genetic component distantly related to the Goyet Q116-1 individual dated to 35 ka, with the Iberian individual also showing an affinity to the Villabruna cluster4,14. By co-analysing previously published data with our newly reported genomes associated with the Magdalenian from La Marche (18 ka) and Pincevent (15 ka) in western and northern France, respectively, and Maszycka (18–16 ka) in southern Poland, we confirm that the Goyet Q116-1 ancestry survived in all studied Magdalenian-associated genomes besides in Gravettian and Solutrean-associated individuals from southwestern and western Europe (Fig. 1). Notably, the Fournol ancestry provides a better proxy than Goyet Q116-1 for the genetic component found in the GoyetQ2 cluster and in El Mirón (Supplementary Data 2.H). However, using f4-statistics, we show that all Magdalenian-associated individuals, and not only El Mirón, carry Villabruna-related ancestry when compared to the Fournol cluster (Supplementary Data 2.H). This affinity is even stronger towards Epigravettian-associated individuals from western and central Italy and Sicily (group 2 and group 3, respectively) than to those from northern Italy (group 1) (Supplementary Data 2.F).
We thus modelled individuals belonging to the GoyetQ2 cluster and El Mirón as a mixture between the Fournol 85 and Arene Candide 16 genomes as proxies to represent the Fournol and Villabruna ancestries, respectively, in Magdalenian-associated groups (Fig. 4a). Besides El Mirón, who has around 43% Villabruna ancestry, all other Magdalenian-associated individuals have a lower proportion of this component (19–29%) and can thus be assigned to the GoyetQ2 cluster (Fig. 4a and Supplementary Data 3.C). This is further validated by f4-statistics of the form f4(Mbuti, Arene Candide 16; Goyet Q-2, Magdalenian-associated individuals), which is significantly positive only for El Mirón, whereas all other tested individuals and Goyet Q-2 are symmetrically related with respect to Arene Candide 16 (Supplementary Fig. 26 and Supplementary Data 2.H).

a,b, The ancestries of individuals in the GoyetQ-2 cluster and Iberian hunter-gatherers (HGs) (a) and individuals in the Oberkassel cluster (b) were modelled using qpAdm, with Fournol 85 and Arene Candide 16 representing the Fournol and Villabruna ancestries, respectively. The length of the colour bar shows the proportion of each ancestry. The error bar shows the s.e.m. of estimates from 5-cM-block jackknife analysis. Details of the modelling are provided in Supplementary Data 3.C.
Our analyses demonstrate that the Fournol cluster is a better source for Magdalenian-associated genomes than Goyet Q116-1. Therefore, most of the ancestry found in these post-LGM individuals probably traced back to Gravettian-associated groups from western and southwestern Europe. The genetic affinity to the Villabruna ancestry is present in El Mirón and in Magdalenian-associated individuals from western and central Europe. This suggests that genetic links between southern and southwestern European hunter-gatherers around the time of the LGM extended north of the Pyrenees. The resulting GoyetQ2 cluster includes individuals spanning from western France to Poland in the period between 18 and 15 ka. Therefore, contrary to previous suggestions34, this demonstrates that the post-LGM diffusion of the Magdalenian was indeed associated with northward and northeastward population expansions from western Europe35.
Post-14 ka to Neolithic
Previous studies have shown that two main hunter-gatherer ancestries were predominant across most parts of Europe after around 14 ka—that is, the western hunter-gatherer (WHG) ancestry, related to the Villabruna cluster, and the eastern hunter-gatherer (EHG) ancestry, showing affinity to both the Villabruna and the ancient north Eurasian (ANE) ancestry found in Upper Palaeolithic Siberian individuals4,36. Hunter-gatherers carrying an admixed WHG/EHG genetic profile have been sequenced from various regions of northern and eastern Europe, raising the question of how these two types of ancestries formed and interacted with each other through time and space37,38,39,40.
In the MDS plot (Fig. 1c) and a west Eurasian principal component analysis (PCA) (Extended Data Fig. 6 and Supplementary Fig. 14), most post-14 ka individuals from western and central Europe fall close to the WHG cluster and those from eastern Europe close to the EHG cluster, whereas the Tutkaul 1 individual from central Asia falls close to the ANE-related group. The two 14 ka Oberkassel individuals mark the earliest presence of WHG ancestry north of the Alps, which we therefore rename the Oberkassel cluster (hereafter, Oberkassel cluster or ancestry), using the name of the oldest reported individual to date carrying such ancestry with more than one-fold coverage, for consistency4. On the basis of f4-statistics, we find that individuals assigned to the Oberkassel cluster are closer to the Arene Candide 16 genome than any other Epigravettian-associated group from Italy (Supplementary Data 2.F). Moreover, the Oberkassel cluster carries both Villabruna ancestry and a contribution from GoyetQ2 ancestry (Supplementary Data 2.J). This was confirmed with qpAdm, in which we could model all individuals from the Oberkassel cluster as a broadly constant mixture of approximately 75% Arene Candide 16 and 25% Goyet Q-2 (or 90% Arene Candide 16 and 10% Fournol 85) (Fig. 4b and Supplementary Data 3.C). The observation that post-14 ka individuals from western and central Europe and also from Britain41 carry a homogeneous genetic makeup instead of displaying repeated local admixtures with GoyetQ2 ancestry implies that the Oberkassel-ancestry profile was already largely formed before its dispersal. This is in sharp contrast to the genetic history of Iberian hunter-gatherers, where the spread of the Villabruna/Oberkassel ancestry involved multiple local admixture events with groups carrying high proportions of GoyetQ2 ancestry14 (Fig. 4 and Supplementary Data 3.C). The long-lasting genetic continuity in Iberia is also reflected in the preservation until the Mesolithic of Y-chromosome haplogroup C, which was predominant in pre-LGM groups but rarely found after the LGM in other parts of Europe (Extended Data Figs. 1 and 2).
Using f4-statistics and qpAdm, we confirm that EHG populations in eastern Europe are a mixture of Villabruna/Oberkassel and ANE ancestries (Supplementary Information, section 11 and Supplementary Data 2.K). F4-statistics also show that the approximately 8.2 ka Yuzhniy Oleniy Ostrov group from Karelia in western Russia formed by 19 genomes has comparable or lower affinity to Villabruna ancestry than all the other EHG groups (Supplementary Data 2.K). The oldest individual revealing an indistinguishable genetic profile from the Yuzhniy Oleniy Ostrov group is the 11 ka Sidelkino individual from Samara in western Russia42. For consistency with the previously discussed nomenclature, we rename the EHG ancestry as the Sidelkino cluster (hereafter, Sidelkino cluster or ancestry). The genetic distinction between the Oberkassel and Sidelkino clusters is also clearly noticeable in the diversity of uniparentally inherited markers, as the Oberkassel cluster is dominated by mtDNA haplogroup U5 and Y-chromosome haplogroup I, whereas individuals from the Sidelkino cluster show a higher frequency of mtDNA haplogroups U2, U4 and R1b, and carry uniquely Y-chromosome haplogroups Q, R and J (Extended Data Figs. 1 and 2).
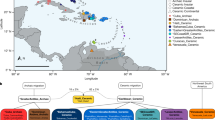
We then attempted to model 250 published and newly reported hunter-gatherers dated to 14–5 ka using qpAdm as a mixture of Oberkassel, Sidelkino, GoyetQ2 ancestries, and an ancestry maximized in Anatolian Neolithic farmers (ANF), as a considerable portion of the sequenced hunter-gatherer genomes date after around 8 ka, when ANF ancestry started spreading across Europe. Our results show that the contact zone and the admixture patterns between the Oberkassel and Sidelkino ancestries changed over time (Fig. 5). Between 14 and 8 ka, all hunter-gatherers in western and central Europe carried only Oberkassel ancestry, with no detectable contribution from the Sidelkino cluster. Further north and east, individuals from the Baltics (Baltic HG), Scandinavia (SHG), the Balkans (Iron Gates HG) and Ukraine (Ukraine HG) already carried an Oberkassel/Sidelkino admixed ancestry38,40 before 8 ka. In addition, those groups also carry affinity to ANF suggesting more complex genetic processes behind their demographic history16. Moreover, two of the oldest published groups from western Russia belonging to the Sidelkino cluster—Peschanitsa (13 ka)43 and the newly reported Minino individuals (11 ka)—showed extra affinity to the Oberkassel cluster, possibly owing to variability in this ancestry proportion during the initial formation phase of the Sidelkino-ancestry profile. Using DATES software, we estimated the admixture between Villabruna/Oberkassel and ANE ancestries in these old Sidelkino-cluster-related individuals to around 15–13 ka (Extended Data Fig. 7 and Supplementary Table 3), which coincides roughly with the first appearance of the Oberkassel ancestry in central Europe. This raises the possibility that the replacement by the Oberkassel cluster and the formation of the Sidelkino cluster might have been the result of population expansions influenced by the abrupt warming during the Bølling–Allerød interstadial4,24.

a, The genetic ancestry of hunter-gatherers dated between 14 ka and 5.2 ka modelled using qpAdm, with Oberkassel, Yuzhniy Oleniy Ostrov, Goyet Q-2 and Neolithic farmers from present-day Turkey (Barcın, Menteşe and Boncuklu sites) representing Oberkassel (WHG) (blue), Sidelkino (EHG) (red), GoyetQ2 (orange) and Anatolian Neolithic farmer (green) ancestries, respectively. The average calibrated date is shown, with pie charts indicating the estimated proportion of ancestry for each group or individual. Details of the modelling are provided in Supplementary Data 3.E,F. The expansion of farming by 9, 8, 7.5 and 7 ka is shown as green shades. Adapted from https://doi.org/10.5281/zenodo.5903165 (CC BY 4.0). b, Allele frequencies of different hunter-gatherer groups (coloured dots) on four SNPs related to skin colour (SLC24A5 and SLC45A2), eye colour (HERC2/OCA2) and lactase persistence (LCT). Dots are maximum likelihood estimates and error bars show 95% confidence intervals of the derived allele frequencies (n, the number of individuals in each group, is provided in Supplementary Data 3.G). Dashed lines show the frequencies estimated for the indicated present-day 1000 Genomes Project populations (CEU, Utah residents of northern and western European ancestry; GBR, British; IBS, Spanish; TSI, Tuscan )37. Details on the allele frequency estimates are provided in Supplementary Information, section 12, Supplementary Figs. 27 and 28 and Supplementary Data 3.G.
From around 8 ka, we begin to observe admixture events with Sidelkino ancestry in central Europe. This is first detected in an individual from Gross Fredenwalde in northeastern Germany and reaches around 10% in most European hunter-gatherer individuals thereafter (Extended Data Fig. 8). Soon after 8 ka, Sidelkino ancestry was absent in eastern Spain but it had already reached northern Iberia alongside an increase in Oberkassel ancestry (Fig. 5). Conversely, additional Oberkassel ancestry is identified in eastern Europe by at least 7.5 ka in newly generated genomes from Minino I and Yazykovo from the upper Volga region, whereas a 1,000-years-older individual from Minino I did not have this genetic component. Considering a freshwater reservoir signal in the upper Volga region making radiocarbon dates on human remains appear up to about 500 years older than their true age44, there could be an interval of more than 1,000 years between the first evidence of admixture in central European hunter-gatherers with Sidelkino ancestry and eastern European hunter-gatherers with Oberkassel ancestry. However, additional genomes intermediate in time and space are needed to assess whether those two admixture events were independent or part of a common demographic process.
After 7.5 ka, as ANF ancestry had reached regions north of the Alps, individuals carrying a hunter-gatherer genetic profile were primarily restricted to the northern fringes of Europe (Fig. 5). In this period, the Oberkassel-ancestry admixture spread further east, reaching Samara by around 6.5 ka, and an increase in Sidelkino ancestry was detected in hunter-gatherers from the Baltic region, which was previously associated with the transition from the Narva culture to the Comb Ceramic culture38,39 (Extended Data Fig. 8). In central Europe, admixture with ANF ancestry became highly common but not ubiquitous, indicating the co-existence of hunter-gatherer and farmer societies without admixing for several hundred years. The youngest individual carrying large portions of hunter-gatherer ancestry in the analysed dataset is from Ostorf in northern Germany, dated to around 5.2 ka (>90% Oberkassel cluster plus Sidelkino-cluster components) (Supplementary Data 3.F). Individuals at this site might mark one of the last occurrences of such high levels of hunter-gatherer-related ancestries, just centuries before the emerging European Bronze Age.
On the basis of PCA and outgroup f3-statistics, the Neolithic Tutkaul 1 individual from Tajikistan is closely related to Upper Palaeolithic individuals from south-central Siberia (Afontova Gora 3 (AG3) and Mal’ta 1), and roughly contemporaneous West Siberian hunter-gatherers (Tyumen and Sosnoviy), both carrying high proportions of ANE ancestry45 (Fig. 1c and Extended Data Fig. 6). We tested the affinity of Tutkaul 1 to worldwide ancient and modern populations relative to AG3. Contrary to West Siberian hunter-gatherers, Tutkaul 1 does not carry an extra eastern Eurasian ancestry, but shows affinity to Iranian Neolithic farmers and some younger populations from Iran and the Turan region (Supplementary Data 2.L). Conversely, individuals in the Sidelkino cluster are genetically closer to AG3 than Tutkaul 1. This suggests that the newly reported Neolithic individual from central Asia carries an ancestry that might be a good proxy for the ANE-related contribution to Iran and the Turan region45 from around 5.5 ka but not to roughly contemporaneous hunter-gatherers from eastern Europe.
In sum, we describe the formation and interaction between the Oberkassel and Sidelkino clusters, the two main hunter-gatherer ancestries present in Europe from 14 ka onwards. The genomic similarity of the Oberkassel cluster to Arene Candide 16 in northwestern Italy might imply that Epigravettian-associated ancestry spread from the south to central Europe passing through the western side of the Alpine region. The Sidelkino ancestry also emerged around 14 ka with its first direct evidence in eastern Europe43 dated to 13 ka. The increasing level of admixture between distinct hunter-gatherer populations from around 8 ka onwards indicates an intensified mobility of those forager groups. This might have been in part triggered by the concomitant expansion of Neolithic farmers across Europe and/or by environmental factors, such as the climatic event around 8.2 ka, the largest abrupt cooling in the northern hemisphere during the Holocene epoch46,47.
Phenotypically relevant variants
Leveraging the substantially increased sample size, we investigated genetically distinct hunter-gatherer groups for allele frequencies at selected loci that are known to be associated with specific phenotypic traits in present-day Europeans (Fig. 5b and Supplementary Figs. 27 and 28). Consistent with previous findings, none of the analysed groups show the derived allele at SNP rs4988235 on the LCT gene, which is responsible for lactase persistence. As previously hinted37, we find a large frequency variation in alleles related to skin and eye pigmentation among post-LGM hunter-gatherer groups. For the SNP associated with light eye colour (HERC2/OCA2 (rs12913832)), individuals from the Villabruna cluster, Oberkassel cluster, Baltic HG and SHG groups show high frequencies of the derived allele (>90%), which is responsible for the green or blue eye phenotype, whereas Sidelkino cluster, Ukraine HG and Iron Gates HG groups show low occurrence of this allele (10–25%). Instead, for the two SNPs associated with skin colour (SLC24A5 (rs1426654) and SLC45A2 (rs16891982)), Sidelkino cluster and Ukraine HG groups show a higher frequency (>90% for SLC24A5 and 29–61% for SLC45A2) of the derived alleles related to light skin colour, compared with Oberkassel and Villabruna clusters, where those alleles are almost completely absent (<1%). On the basis of the genetic variation of present-day Europeans, this could imply phenotypic differences between post-14 ka hunter-gatherer populations across Europe, with individuals in the Oberkassel cluster possibly exhibiting darker skin and lighter eyes, and individuals in the Sidelkino cluster possibly lighter skin and darker eye colour.
Discussion and conclusions
The data generated in this study enabled us to investigate genomic transformations of and interactions between Eurasian hunter-gatherers at high resolution (Extended Data Fig. 9). We provide five novel insights into the genomic history of hunter-gatherer populations over a time span of 30,000 years from the Upper Palaeolithic to the Neolithic.
First, we show that individuals associated with the Gravettian culture across Europe were not a biologically homogeneous population. Culturally, however, we see both widespread general tendencies, such as weaponry and some portable art48, and other aspects that have a more regional character, such as mortuary practices (Supplementary Information, section 13), various originalities in lithic and hard organic materials tool kits and adornments20,21. The ancestry found in individuals associated with the preceding Aurignacian culture from central Europe (GoyetQ116-1 ancestry) gave rise to Gravettian-associated individuals from western and southwestern Europe. This derived ancestry—the Fournol cluster—survived during the LGM in Solutrean-associated individuals, possibly within the Franco-Cantabrian climatic refugium25, leading to later populations associated with the Magdalenian culture (GoyetQ2 cluster and El Mirón). Conversely, the ancestry found in pre-30 ka eastern European individuals (Kostenki cluster and Sunghir group) contributed to Gravettian-associated individuals from central and southern Europe (Věstonice cluster), the latter without descendants retrieved in post-LGM populations from those regions.
Second, the ancestry of individuals associated with the Epigravettian culture (Villabruna cluster), which was found to genetically connect European and Near Eastern hunter-gatherers, reached southern Europe well before the transition between the Early and Late Epigravettian4,15 and possibly as early as the Gravettian–Epigravettian transition. A phylogeographic reconstruction of different lineages carrying this ancestry further suggests its entry point into northeastern Italy from the Balkans followed by a north-to-south expansion into the Italian peninsula alongside a population decline through sequential bottlenecks.
Third, Magdalenian-associated individuals not only from Iberia but also from the rest of Europe carry Epigravettian-associated ancestry (Villabruna cluster). Genetic analyses of western European individuals associated with the preceding Badegoulian culture might provide clues on the processes that led to the formation of the GoyetQ2 cluster. As inferred from the archaeological record35, the spread of the Magdalenian across Europe is linked to southwestern to northern and northeastern post-LGM population expansions and not to movements from southeastern refugia34.
Fourth, we extend the finding of a large-scale genetic turnover as early as 14 ka in central and western European hunter-gatherers associated with multiple techno-complexes—Federmesser, Azilian and other Final Palaeolithic groups4—despite considerable technological continuity with the preceding late Magdalenian. This broadly distributed ancestry (the Oberkassel cluster (also known as WHG)) is most closely related to an Epigravettian-associated individual from northwestern Italy, suggesting that its expansion into continental Europe might have started from the west—and not the east—side of the Alps. Moreover, the almost complete genetic replacement of the Magdalenian-associated gene pool raises the hypothesis that parts of Europe were differentially populated during the abrupt climatic variation starting around 14.7 ka with the Bølling–Allerød warming period, creating areas where southern European populations could expand. This might also explain the genetic uniformity of the Oberkassel cluster across large parts of western Eurasia but genomic data from between 15 and 14 ka is needed to understand the exact dynamics of this turnover.
Fifth, the Oberkassel ancestry in western and central Europe and the Sidelkino ancestry in eastern Europe remained largely isolated for almost 6,000 years until genetic interactions were first observed—around 8 ka in northeastern Germany, possibly associated with cultural exchanges along the Baltics49 and around 7.5 ka in the upper Volga region, possibly linked to the spread of pottery in the region50.
In conclusion, our study reveals that western and southwestern Europe served as climatic refugia for the persistence of human groups during the coldest phase of the last Ice Age whereas populations in the Italian peninsula and the eastern European plain were genetically overturned, challenging the role of these regions as glacial refugia for humans. The incoming Villabruna ancestry later became the most widespread hunter-gatherer ancestry across Europe. Further palaeogenomic studies on Upper Palaeolithic individuals from the Balkans will be essential for understanding whether southeastern Europe represents the source of the Villabruna ancestry and a climatic refugium for human populations during the LGM.
Note added in proof: A companion paper51 describes genome-wide data of a 23,000-year-old Solutrean-associated individual from southern Iberia that extend the evidence of genetic continuity across the LGM in southwestern Europe.
Methods
Archaeological sampling
The ancient human specimens analysed in this work derive from multiple scientific collaborations. All remains were sampled with the approval of the institutions responsible for the analysis of archaeological material. This was achieved through collaboration with local curators and scientists from the countries where the skeletal material is preserved and who are listed among the authors of this study. The responsible co-authors for the material from each archaeological site are listed in Supplementary Information, section 1.
The analysed individuals span from the Upper Palaeolithic to the Neolithic. While terms such as lithic industry, techno-complex, prehistoric tradition, and so on might be more appropriate to refer to the various associated chrono-cultural subdivisions, they concern different levels of discussion and are not applicable to all contexts investigated here. Therefore, the broader terms ‘archaeological culture’ or simply ‘culture’ are used here to refer to archaeologically defined material cultures without implying links to modern anthropological and/or ethnographical concepts of culture.
Radiocarbon dating
We report 47 new radiocarbon dates performed on skeletal elements of 40 individuals by the Curt-Engelhorn-Zentrum Archaeometrie in Mannheim (MAMS, n = 29), Center for Isotope Research, University of Groningen (GrA and GrM, n = 5), University of Aarhus (AAR, n = 3), Beta Analytics (Beta, n = 2), Zürich (ETH, n = 3), International Chemical Analysis (ICA, n = 2), Natural History Museum in Paris (Echo Lab, n = 1) and Vilnius (FTMC, n = 2) (Supplementary Data 1.A). The dates were calibrated using OxCal 4.452 with calibration curve IntCal20 at 95.4% probability53 and when multiple dates were available for the same individual we used the function R_Combine to combine them52. We did not correct the calibrated dates for marine or freshwater reservoir effects but, when available, we report individual stable isotope values (δ15N/δ13C and C:N ratio) in Supplementary Data 1.A to evaluate the potential impact of such reservoir effects.
Ancient DNA processing
The human remains were processed in dedicated laboratories at the Max Planck Institute for the Science of Human History in Jena (Germany), University of Tübingen (Germany), University of Florence (Italy), Leiden University Medical Center (the Netherlands) and University of Tartu (Estonia). Human bones and teeth were sampled in clean room facilities to minimize the inclusion of modern human DNA contamination during this procedure. DNA was extracted from the generated bone or tooth powder following established protocols. A subset of samples (GER002 and GER003) were pre-treated with a washing step to reduce surface contamination54. A negative and cave bear positive controls were included. For the DNA lysis, a solution of 900 μl EDTA, 75 μl H2O and 25 μl proteinase K was added. In a rotator, samples were digested for at least 16 h at 37 °C, and for pre-treated samples this was followed55 by an additional hour at 56 °C. The suspension was then centrifuged and transferred into a binding buffer as previously described56. To bind DNA, silica columns for high volumes (High Pure Viral Nucleic Acid Large Volume Kit (Roche)) were used. After 2 washing steps using the manufacturer’s wash buffer, DNA was eluted in TET (10 mM Tris, 1 mM EDTA and 0.05% Tween) in two steps for a final volume of 100 μl. After DNA lysis, a subset of samples was extracted using silica-coated magnetic particles on an automated liquid handling system (Agilent Technologies Bravo NGS Workstation)57. Double-stranded DNA libraries were built from 25 μl of DNA extract, without the presence of uracil DNA glycosylase (ds_nonUDG) or in the presence of uracil DNA glycosylase (ds_halfUDG), following a double-stranded ‘UDG-half’ library preparation to reduce, but not eliminate, the amount of deamination-induced damage towards the ends of ancient DNA (aDNA) fragments58. Negative and positive controls were carried alongside each experiment. Libraries were quantified using the IS7 and IS8 primers59 in a quantification assay using a DyNAmo SYBR Green qPCR Kit (Thermo Fisher Scientific) on the LightCycler 480 (Roche). Each aDNA library was double indexed60 in 1–4 parallel 100 μl reactions using PfuTurbo DNA Polymerase (Agilent). The indexed products for each library were pooled, purified over MinElute columns (Qiagen), eluted in 50 μl TET and again quantified using the IS5 and IS6 primers59 using the quantification method described above. The purified products were amplified in multiple 100 μl reactions using Herculase II Fusion DNA Polymerase (Agilent) following the manufacturer’s specifications with 0.3 μM of the IS5/IS6 primers. After another MinElute purification, the product was quantified using the Agilent 2100 Bioanalyzer DNA 1000 chip. An equimolar pool of all libraries was then prepared for shotgun sequencing on Illumina Hiseq4000 platform using 75bp single-end reads for screening. Single-stranded DNA libraries were built from 30 μl of DNA extract in the absence of uracil DNA glycosylase (ss_nonUDG) followed by double indexing, using an automated version of the protocols described in61 on the liquid handling system mentioned before. The single-stranded library of Cuiry Les Chaudardes 1 was produced with partial UDG treatment (ss_halfUDG)62 (Supplementary Data 1.B).
DNA enrichment and sequencing
Both double-stranded and single-stranded libraries were further amplified with IS5/IS6 primers to reach a concentration of 200–400 ng/μl as measured on a NanoDrop spectrophotometer (Thermo Fisher Scientific). The libraries underwent shallow shotgun sequencing on an Illumina HiSeq 4000 instrument with 75 single-end-run cycles using the manufacturer’s protocol, to evaluate the human endogenous DNA content and quality. Samples with a percentage of human DNA in shotgun data around 0.1% or greater were enriched for a set of 1,237,207 targeted SNPs (1240k capture) across the human genome6. mtDNA capture63 was also performed for those libraries where mtDNA coverage was not high enough to assess mtDNA haplogroup and contamination. Illumina sequencing platforms were also used to sequence the 1240k and mtDNA captured libraries (Supplementary Data 1.B).
The de-multiplexed capture sequencing reads were cleaned and mapped to human reference genome hs37d5 using EAGER pipeline 1.92.5564. Within the pipeline, the adapters were removed by AdapterRemoval 2.2.065, reads were mapped with BWA 0.7.12 aln/samse algorithm66, duplications were removed by DeDup 0.12.1 (https://github.com/apeltzer/DeDup) and damage patterns of each library were checked with mapDamage 2.0.6 and 2.0.967. The deduplicated bam files were filtered using PMDtools 0.6068 with a threshold of 3, to reduce potential modern DNA contamination based on postmortem DNA deamination. For ds_halfUDG libraries, we masked 2 bp from both ends of the reads with trimBam in bamUtil 1.0.13 (https://github.com/statgen/bamUtil) to remove the damaged sites.
The mitochondrial capture sequencing reads were cleaned by AdapterRemoval 2.2.0 to remove the adapters and reads with lengths below 30 bp. Then the cleaned reads together with cleaned reads from 1240k capture sequencing were mapped to human reference mitochondrial sequence NC_012920.1 with BWA 0.7.12 aln/samse algorithm (parameters –n 0.01, –l 16500) and realigned with CircularMapper64. The mapped reads from the same individual and library set-up were merged and duplications were removed with DeDup. Reads with a mapping quality below 30 were then filtered with samtools, and the consensus sequences were generated by Schmutzi69.
Ancient DNA authentication and genotyping
The sex of each individual was determined by the ratio of sequencing coverages on sex chromosomes versus autosomes (Supplementary Data 1.C). Individuals with libraries showing signs of contamination were further tested using PMD-filtered bam files. Individuals with at least one library showing Y/Auto ratio > 0.2 were determined as male individuals, and with Y/Auto < 0.2 were determined as female individuals4 (Supplementary Fig. 12).
The nuclear DNA contamination was estimated with several methods. We applied ANGSD 0.93470 and hapCon71 for libraries from male individuals, and applied contamLD72 and a newly developed method that analyses contamination in ROH for female and male libraries (see Supplementary Information, section 2 for a detailed description). The mtDNA contamination was estimated by Schmutzi (--notusepredC --uselength)69 for all the libraries. Libraries showing a mitochondrial or nuclear contamination rate over 10% were considered substantially contaminated whereas those between 5 and 10% were considered marginally contaminated and were treated differently (details are provided in Supplementary Information, section 3).
The cleaned reads with base quality and mapping quality over 30 were piled up with mpileup in SAMtools 1.373 on the 1240k targeted sites. For contaminated libraries we used the PMD-filtered bam files as the input for genotyping. Then pseudo-haploid genotypes were called using pileupCaller 1.4.0.2 (https://github.com/stschiff/sequenceTools) under random haploid calling mode. For ds_halfUDG libraries, we called genotypes on all targeted sites from 2bp-masked bam files; for ds_nonUDG libraries, we called genotypes on transversion sites only; for ss_nonUDG libraries, we called genotypes with single-strand mode, which ignores forward reads at C/T polymorphisms and reverse reads at G/A polymorphisms.
Then we merged the genotypes from different libraries of the same individual, by randomly picking alleles from available genotype calls, using a custom script. After merging, individuals with less than 6,000 SNPs on 1240k sites were excluded from further analysis because of low coverage. We also genotyped a selection of previously published individuals with the same approach (Supplementary Data 1.G)2,4,30,74,75,76,77,78. Then we combined our newly generated genotypes with published genotypes from ancient and modern individuals from AADR v42.4 (Allen Ancient DNA Resource (https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data) version 42.4) for downstream analysis1,4,7,14,16,23,36,37,38,39,40,42,43,45,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94.
For individual heterozygosity calculation, we also called pseudo-diploid genotypes from each library, using pileupCaller 1.4.0.3 under random diploid calling mode and the same strategy for different types of libraries as pseudo-haploid genotype calling.
Uniparental markers
The mitochondrial haplogroups were determined using HaploGrep 295, based on the consensus sequences generated from Schmutzi inspected for each sample at increasing quality filters (from q0 to q20). Inconsistent haplogroup assignments were manually verified as indicated24 (Supplementary Data 1.L). For phylogenetic reconstruction (Extended Data Fig. 1) we used MUSCLE (-maxiters 2)96 to create a multiple genome alignment of previously published sequences and newly reported mtDNA consensus sequences with q20 according to defined thresholds (minimum average coverage >5-fold, contamination estimate <20%, HaploGrep 2 haplogroup assignment consistent with manual assignment). We built a Maximum Parsimony tree with 103 mtDNA sequences plus an African sequence as the outgroup (not shown) after the removal of individuals younger than 6.5 ka and mtDNAs from the same site with an identical placement. The tree was calculated on 16,528 positions (partial deletion 95%) and with 500 bootstrap iterations using MEGA1097.
To determine the Y-chromosome haplogroups of male individuals, we genotyped the Y-chromosome reads using a Y-SNP list (v.15.73) from the International Society of Genetic Genealogy (ISOGG) dataset, ignoring C-to-T and G-to-A transitions on the forward and reverse reads, respectively. This procedure allowed us to manually traverse the ISOGG Y-Haplogroup Tree, checking in a semi-automatic way which positions were covered. This process allowed us to assign an ancestral or derived haplogroup for covered branches, and to make corrections to calls in cases where, for instance, a more derived haplogroup was called because of residual ancient damage (C-to-T or G-to-A mismatches) in terminal read positions at diagnostic SNPs98 (Supplementary Data 1.A). For the placement of individuals onto a Y-chromosomal phylogenetic tree (Extended Data Fig. 1), we used pathPhynder99 based on the tree from Karmin et al.100. We used the default posterior threshold of 0.01, and mapping and sequencing quality cutoffs of 30. We then removed samples with less than 0.04X coverage (calculated on the mappable, non-recombining region of the Y chromosome98) to avoid arbitrarily placing low-coverage samples at the root of major haplogroups. This results in a tree with 57 newly reported and previously published ancient individuals while present-day sequences are collapsed in the major Y-chromosome haplogroups (the most basal lineages are not shown). The tentative placements of low-coverage ancient individuals based on their haplogroup assignment (Supplementary Data 1.A) are indicated with arrows on the respective branches.
Biological relatedness and population diversity
The analysis of biological relatedness was performed by calculating relatedness coefficient (r) based on PMR on the autosomal SNPs (Supplementary Data 1.F and Supplementary Information, section 4). The baseline of each population was determined using the average heterozygosity rate of individuals estimated from pseudo-diploid genotypes (Supplementary Data 1.E).
The ROH segments in hunter-gatherer genomes were identified using hapROH101. As recommended, we analysed individuals with over 400,000 SNPs called on the 1240k panel101 and we called ROH longer than 4 cM (Supplementary Data 3.B). The effective population sizes (Ne) were then estimated using a maximum likelihood method, after filtering individuals with a signal of close-kin inbreeding (individuals with at least 50 cM of their genome in ROH spanning >20 cM) (Supplementary Information, section 9).
Population genetics analysis
The PCA was carried out by smartpca in EIGENSOFT 6.0.1102, with modern individuals used for calculation and all the ancient individuals projected on the calculated PCs. The “lsqproject: YES” parameter was used to minimize the effect of missing data in ancient individuals. The PCA was calculated with 1379 individuals from 87 western Eurasian modern populations on the 1240k_HO dataset, which was intersected between the 1240k and Human Origins datasets (Supplementary Data 1.K).
The MDS analysis showing the genetic affinity among European hunter-gatherers was based on the distance matrix derived from outgroup f3-statistics, in the form 1 − f3(Mbuti.DG; pop1, pop2) and performed with classical MDS algorithm (cmdscale) implemented in R 3.5.1. The hunter-gatherers were grouped based on their geographic origins and dates (Supplementary Information, section 5). The f3-statistics were calculated with qp3Pop 435 in ADMIXTOOLS 5.1 package103.
The pairwise f2 distance matrix of Epigravettian-associated groups was generated with qpfstats 200 in ADMIXTOOLS 7.0.2 package, with parameters “allsnps: YES, scale: NO”, and Mbuti.DG set as the outgroup. The neighbour-joining tree was then reconstructed using the neighbour-joining method implemented in Ape 5.3 package104 of R 3.5.1.
The f4-statistics were calculated by qpDstat 755 with parameter “f4 mode: YES”, with the Mbuti.DG population from Africa used as outgroup in all f-statistics analyses. The tool qpAdm 810 in ADMIXTOOLS 5.1 was applied to model the ancestries of admixed populations, with “allsnps” mode and the outgroup set selection described in Supplementary Information, section 11. Admixture graphs were reconstructed using qpGraph 6450, with allsnps mode to correct for low-coverage sample and Mbuti.DG set as the outgroup. Admixture events were dated using the ancestry covariance pattern-based DATES 753 program105, with a bin size of 0.1 cM for covariance calculation and the start of exponential fitting at d ≥ 0.5cM.
Phenotypic SNP analysis
As the coverage for most ancient samples was not sufficient for diploid genotype calling, we counted the reads covering selected phenotypic SNPs on reference or alternative alleles and computed the group-based allele frequencies following a maximum likelihood approach described in Mathieson et al.37. Details on individuals involved in the analysis, read counts processing and allele frequency computation are provided in the Supplementary Information, section 12 and Supplementary Data 1.J and 3.G.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The aligned sequences of all individuals with new genomic data reported in this study are available at the European Nucleotide Archive (ENA) under study accession number PRJEB51862. The compiled genotype file used for analyses, including re-genotyped published genomes, has been uploaded at the Edmond Data Repository of the Max Planck Society (https://edmond.mpdl.mpg.de/dataset.xhtml?persistentId=doi:10.17617/3.Y1KJMF).
Code availability
The code for the newly developed ROH based contamination estimate method is available at https://github.com/hyl317/hapROH. A user manual including an installation guide is available at https://haproh.readthedocs.io/en/latest/hapROH_with_contamination.html. The version used for this work is archived at https://edmond.mpdl.mpg.de/dataset.xhtml?persistentId=doi:10.17617/3.Y1KJMF.
Change history
22 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41586-023-05942-8
References
Prüfer, K. et al. A genome sequence from a modern human skull over 45,000 years old from Zlatý kůň in Czechia. Nat. Ecol. Evol. 5, 820–825 (2021).
Hajdinjak, M. et al. Initial Upper Palaeolithic humans in Europe had recent Neanderthal ancestry. Nature 592, 253–257 (2021).
Olalde, I. & Posth, C. Latest trends in archaeogenetic research of west Eurasians. Curr. Opin. Genet. Dev. 62, 36–43 (2020).
Fu, Q. et al. The genetic history of Ice Age Europe. Nature 534, 200–205 (2016).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Fu, Q. et al. An early modern human from Romania with a recent Neanderthal ancestor. Nature 524, 216–219 (2015).
Fu, Q. et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014).
Seguin-Orlando, A. et al. Genomic structure in Europeans dating back at least 36,200 years. Science 346, 1113–1118 (2014).
Vallini, L. et al. Genetics and material culture support repeated expansions into Paleolithic Eurasia from a population hub out of Africa. Genome Biol. Evol. 14, evac045 (2022).
Maier, A. & Zimmermann, A. Populations headed south? The Gravettian from a palaeodemographic point of view. Antiquity 91, 573–588 (2017).
Dolukhanov, P. in Cultural Transformations and Interactions in Eastern Europe (eds Chapman, J. & Dolukhanov, P.) 122–145 (Avebury, 1993).
Gamble, C., Davies, W., Pettitt, P., Hazelwood, L. & Richards, M. The archaeological and genetic foundations of the European population during the Late Glacial: implications for ‘agricultural thinking’. Cambridge Archaeol. J. 15, 193–223 (2005).
Wren, C. D. & Burke, A. Habitat suitability and the genetic structure of human populations during the Last Glacial Maximum (LGM) in Western Europe. PLoS ONE 14, e0217996 (2019).
Villalba-Mouco, V. et al. Survival of Late Pleistocene hunter-gatherer ancestry in the Iberian Peninsula. Curr. Biol. 29, 1169–1177.e7 (2019).
Bortolini, E. et al. Early Alpine occupation backdates westward human migration in Late Glacial Europe. Curr. Biol. 31, 2484–2493.e7 (2021).
Feldman, M. et al. Late Pleistocene human genome suggests a local origin for the first farmers of central Anatolia. Nat. Commun. 10, 1218 (2019).
Petr, M., Pääbo, S., Kelso, J. & Vernot, B. Limits of long-term selection against Neandertal introgression. Proc. Natl Acad. Sci. USA 116, 1639–1644 (2019).
Harris, K. & Nielsen, R. The genetic cost of Neanderthal introgression. Genetics 203, 881–891 (2016).
Kozłowski, J. K. The origin of the Gravettian. Quat. Int. 359, 3–18 (2015).
Goutas, N. in Les Gravettiens (ed. Otte, M.) 105–160 (Errance, 2013).
Klaric, L., Goutas, N., Laccarière, J. & Banks, W. E. in Les Sociétés Gravettiennes du Nord-Ouest Européen: Nouveaux Sites, Nouvelles Données, Nouvelles Lectures (eds Touzé, O., Goutas, N., Salomon, H. & Noiret, P.) 323–266 (Presses Univ. de Liège, 2021).
Mounier, A. et al. Gravettian cranial morphology and human group affinities during the European Upper Palaeolithic. Sci. Rep. 10, 21931 (2020).
Sikora, M. et al. Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science 358, 659–662 (2017).
Posth, C. et al. Pleistocene mitochondrial genomes suggest a single major dispersal of non-Africans and a Late Glacial Population turnover in Europe. Curr. Biol. 26, 827–833 (2016).
Straus, L. G. The human occupation of southwestern Europe during the Last Glacial Maximum: Solutrean cultural adaptations in France and Iberia. J. Anthropol. Res. 71, 465–492 (2015).
Lécuyer, C., Hillaire-Marcel, C., Burke, A., Julien, M. A. & Hélie, J. F. Temperature and precipitation regime in LGM human refugia of southwestern Europe inferred from δ13C and δ18O of large mammal remains. Quat. Sci. Rev. 255, 106796 (2021).
Djindjian, F. Territories and economies of hunter-gatherer groups during the last glacial maximum in Europe. Quat. Int. 412, 37–43 (2016).
Ruiz-Redondo, A. et al. Mid and Late Upper Palaeolithic in the Adriatic Basin: chronology, transitions and human adaptations to a changing landscape. Quat. Sci. Rev. 276, 107319 (2022).
Laplace, G. Essai de Typologie Systématique (Annali dell’Università di Ferrara, 1964).
Yu, H. et al. Genomic and dietary discontinuities during the Mesolithic and Neolithic in Sicily. iScience 25, 104244 (2022).
Palma di Cesnola, A. Le paléolithique supérieur en Italie. Série ‘Préhistoire d’Europe’ 9 (Éditions, 2001).
Peresani, M. et al. Hunter-gatherers across the great Adriatic-Po region during the Last Glacial Maximum: environmental and cultural dynamics. Quat. Int. 581–582, 128–163 (2021).
Otte, M. Appearance, expansion and dilution of the Magdalenian civilization. Quat. Int. 272–273, 354–361 (2012).
Maier, A. in The Central European Magdalenian 81–180 https://doi.org/10.1007/978-94-017-7206-8_6 (Springer, 2015).
Kozłowski, S. K., Połtowicz-Bobak, M., Bobak, D. & Terberger, T. New information from Maszycka Cave and the Late Glacial recolonisation of Central Europe. Quat. Int. 272, 288–296 (2012).
Raghavan, M. et al. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505, 87–91 (2014).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015).
Mathieson, I. et al. The genomic history of southeastern Europe. Nature 555, 197–203 (2018).
Mittnik, A. et al. The genetic prehistory of the Baltic Sea region. Nat. Commun. 9, 442 (2018).
Günther, T. et al. Population genomics of Mesolithic Scandinavia: investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol. 16, e2003703 (2018).
Charlton, S. et al. Dual ancestries and ecologies of the Late Glacial Palaeolithic in Britain. Nat. Ecol. Evol. 6, 1658–1668 (2022).
Damgaard, P. et al. The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science 360, eaar7711 (2018).
Saag, L. et al. Genetic ancestry changes in Stone to Bronze Age transition in the East European plain. Sci. Adv. 7, eabd6535 (2021).
Wood, R. E. et al. Freshwater radiocarbon reservoir effects at the burial ground of Minino, Northwest Russia. Radiocarbon 55, 163–177 (2013).
Narasimhan, V. M. et al. The formation of human populations in South and Central Asia. Science 365, eaat7487 (2019).
Gronenborn, D. in The Spread of the Neolithic to Central Europe (RGZM, 2010).
Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 11 (2007).
Roebroeks, W., Mussi, M., Svoboda, J. & Fennema, K. Hunters of the Golden Age: The Mid Upper Palaeolithic of Eurasia, 30,000-20,000 bp (Univ. of Leiden, 2000).
Kotula, A., Piezonka, H. & Tergerger, T. The Mesolithic cemetery of Groß Fredenwalde (north-eastern Germany) and its cultural affiliations. Liet. Archeol. 46, 65–84 (2020).
Piezonka, H. et al. The emergence of hunter-gatherer pottery in the Urals and West Siberia: new dating and stable isotope evidence. J. Archaeol. Sci. 116, 105100 (2020).
Villalba-Mouco, V. et al. A 23,000-year-old southern-Iberian individual links human groups that lived in Western Europe before and after the Last Glacial Maximum. Nat. Ecol. Evol., https://doi.org/10.1038/s41559-023-01987-0 (2023)
Bronk Ramsey, C. Bayesian analysis of radiocarbon dates. Radiocarbon 51, 337–360 (2009).
Reimer, P. J. et al. The IntCal20 Northern Hemisphere radiocarbon age calibration curve (0–55 cal kbp). Radiocarbon 62, 725–757 (2020).
Korlević, P. et al. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 59, 87–93 (2015).
Rohland, N. & Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2, 1756–1762 (2007).
Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA 110, 15758–63 (2013).
Rohland, N., Glocke, I., Aximu-Petri, A. & Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 13, 2447–2461 (2018).
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Partial uracil–DNA–glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. B 370, 20130624 (2015).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010).
Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3 (2012).
Gansauge, M., Aximu-Petri, A., Nagel, S. & Meyer, M. Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA. Nat. Protoc. 15, 2279–2300 (2020).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Fu, Q. et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl Acad. Sci. USA 110, 2223–2227 (2013).
Peltzer, A. et al. EAGER: efficient ancient genome reconstruction. Genome Biol. 17, 60 (2016).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. MapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Skoglund, P. et al. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl Acad. Sci. USA 111, 2229–2234 (2014).
Renaud, G., Slon, V., Duggan, A. T. & Kelso, J. Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 16, 224 (2015).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014).
Huang, Y. & Ringbauer, H. hapCon: estimating contamination of ancient genomes by copying from reference haplotypes. Bioinformatics 38, 3768–3777 (2022).
Nakatsuka, N. et al. ContamLD: estimation of ancient nuclear DNA contamination using breakdown of linkage disequilibrium. Genome Biol. 21, 199 (2020).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Catalano, G. et al. Late Upper Palaeolithic hunter-gatherers in the Central Mediterranean: new archaeological and genetic data from the Late Epigravettian burial Oriente C (Favignana, Sicily). Quat. Int. 537, 24–32 (2020).
Jensen, T. Z. T. et al. A 5700 year-old human genome and oral microbiome from chewed birch pitch. Nat. Commun. 10, 5520–10 (2019).
Key, F. M. et al. Emergence of human-adapted Salmonella enterica is linked to the Neolithization process. Nat. Ecol. Evol. 4, 324–333 (2020).
Rivollat, M. et al. Ancient genome-wide DNA from France highlights the complexity of interactions between Mesolithic hunter-gatherers and Neolithic farmers. Sci. Adv. 6, eaaz5344 (2020).
Svensson, E. et al. Genome of Peştera Muierii skull shows high diversity and low mutational load in pre-glacial Europe. Curr. Biol. 31, 2973–2983.e9 (2021).
Antonio, M. L. et al. Ancient Rome: A genetic crossroads of Europe and the Mediterranean. Science 366, 708–714 (2019).
Brace, S. et al. Ancient genomes indicate population replacement in Early Neolithic Britain. Nat. Ecol. Evol. 3, 765–771 (2019).
Brunel, S. et al. Ancient genomes from present-day France unveil 7,000 years of its demographic history. Proc. Natl Acad. Sci. USA 117, 12791–12798 (2020).
Cassidy, L. M. et al. A dynastic elite in monumental Neolithic society. Nature 582, 384–388 (2020).
González-Fortes, G. et al. Paleogenomic evidence for multi-generational mixing between Neolithic farmers and Mesolithic hunter-gatherers in the Lower Danube Basin. Curr. Biol. 27, 1801–1810.e10 (2017).
Jones, E. R. et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Commun. 6, 8912 (2015).
Jones, E. R. et al. The Neolithic transition in the Baltic was not driven by admixture with early European farmers. Curr. Biol. 27, 576–582 (2017).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014).
Lazaridis, I. et al. Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419–424 (2016).
Lipson, M. et al. Parallel palaeogenomic transects reveal complex genetic history of early European farmers. Nature 551, 368–372 (2017).
van de Loosdrecht, M. et al. Pleistocene North African genomes link Near Eastern and sub-Saharan African human populations. Science 360, 548–552 (2018).
Olalde, I. et al. The genomic history of the Iberian Peninsula over the past 8,000 years. Science 363, 1230–1234 (2019).
Saag, L. et al. Extensive farming in Estonia started through a sex-biased migration from the steppe. Curr. Biol. 27, 2185–2193.e6 (2017).
Sikora, M. et al. The population history of northeastern Siberia since the Pleistocene. Nature 570, 182–188 (2019).
Skoglund, P. et al. Genomic diversity and admixture differs for stone-age Scandinavian foragers and farmers. Science 344, 747–750 (2014).
Yang, M. A. et al. 40,000-year-old individual from Asia provides insight into early population structure in Eurasia. Curr. Biol. 27, 3202–3208.e9 (2017).
Weissensteiner, H. et al. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63 (2016).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Rohrlach, A. B. et al. Using Y-chromosome capture enrichment to resolve haplogroup H2 shows new evidence for a two-path Neolithic expansion to Western Europe. Sci. Rep. 11, 15005 (2021).
Martiniano, R., De Sanctis, B., Hallast, P. & Durbin, R. Placing ancient DNA sequences into reference phylogenies. Mol. Biol. Evol. 39, msac017 (2022).
Karmin, M. et al. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res. 25, 459–466 (2015).
Ringbauer, H., Novembre, J. & Steinrücken, M. Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nat. Commun. 12, 5425 (2021).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Chintalapati, M., Patterson, N. & Moorjani, P. The spatiotemporal patterns of major human admixture events during the European Holocene. eLife 11, e77625 (2022).
Acknowledgements
The authors thank G. Marciani and O. Jöris for comments on archaeology; C. Jeong, M. Spyrou and K. Prüfer for comments on genetics; M. O’Reilly for graphical support for Fig. 5 and Extended Data Fig. 9; the entire IT and laboratory teams at the Department of Archaeogenetics of MPI-SHH for technical assistance; M. Meyer and S. Nagel for support with single-stranded library preparation; K. Post, P. van Es, J. Glimmerveen, M. Medendorp, M. Sier, S. Dikstra, M. Dikstra, R. van Eerden, D. Duineveld and A. Hoekman for providing access to human specimens from the North Sea (The Netherlands); M. D. Garralda and A. Estalrrich for providing access to human specimens from La Riera (Spain); J. Górski and M. Zając for providing access to human specimens from Maszycka cave; C. Di Patti for providing access to human specimens from San Teodoro 2 (Italy); P. Blaževičius for providing access to the Donkalnis human remains and the new radiocarbon dates; the Italian Ministry of Culture and Soprintendenza Archeologia Belle Arti e Paesaggio for the Provinces of Verona, Rovigo, and Vicenza for granting access to the human remains of Tagliente 2; F. Fontana, who carries out investigations of the Riparo Tagliente site (Italy); the Friuli Venezia Giulia Superintendency for providing access to the human tooth Pradis 1; and the Soprintendenza Archeologia Belle Arti e Paesaggio for the Provinces of Barletta-Andria-Trani and Foggia for providing access to the Paglicci human remains. This project has received funding by the European Research Council under the European Union’s Horizon 2020 research and innovation programme under grant agreements no. 803147-RESOLUTION (to S.T.), no. 771234-PALEoRIDER (to W.H.), no. 864358 (to K.M.), no. 724703 and no. 101019659 (to K.H.). K.H. is also supported by the Deutsche Forschungsgemeinschaft (DFG FOR 2237). E.A. has received funding from the Van de Kamp fonds. PACEA co-authors of this research benefited from the scientific framework of the University of Bordeaux’s IdEx Investments for the Future programme/GPR Human Past. A.G.-O. is supported by a Ramón y Cajal fellowship (RYC-2017-22558). L. Sineo, M.L. and D.C. have received funding from the Italian Ministry of University and Research (MUR) PRIN 2017 grants 20177PJ9XF and 20174BTC4R_002. H. Rougier received support from the College of Social and Behavioral Sciences of CSUN and the CSUN Competition for RSCA Awards. C.L.S. and T. Saupe received support from the European Union through the European Regional Development Fund (project no. 2014-2020.4.01.16-0030) and C.L.S. received support from the Estonian Research Council grant PUT (PRG243). S. Shnaider received support from the Russian Science Foundation (no. 19-78-10053).
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
C. Posth, H.Y., A.G., A.B.R., W.H. and J. Krause performed or supervised genetic analyses. H. Rougier and I.C. performed mortuary practices analyses. Y.H. and H. Ringbauer developed the contamination-estimation method. K.N., V.V.-M., R.R., T. Ferraz, R.T., D.G.D., M.L., A. Modi, S. Vai, T. Saupe, C.L.S., G. Catalano, L. Pagani, D.C. and E.A. performed or supervised laboratory work. H. Rougier, I.C., L.K., A. Morala, M. Rué, S.M., L.C., J.-B.C., E. Bocaege, S. Ricci, F.B., P. Bayle, B.M., F.L.B.-R., J.-G.B., G.O., E. Bortolini, O.B.-L., G.D., M.O., A.Z., V.S., E.S., L. Sineo, J.v.d.P., L. Pecqueur, G.M., G.G., J.-M.L., C.B.G., A.G.-O., M.P.-B., D.B., M.L.L., P. Storm, C.H., J. Kabaciński, T. Filimonova, S. Shnaider, N.B., B.G.-R., M.R.G.M., A.B.M.-A., B.L., C.A.-L., A.R., C. Polet, I.J., N. Cauwe, J. Soler, N. Coromina, I.R., R.C., G. Clark, L.G.S., M.-A.J., S. Rehart, D.T., S.B., M. Romandini, L.A., H. Bocherens, C.W., S. Villotte, J.F.-L.d.P., M.G.-P., M.A.E.-B., P. Bodu, L. Smits, B.S., R.J., J. Kozakaitė, C.C., H. Benthien, K.W., R.W.S., S.C.F., T. Schüler, C.T., D.G., F.L., A. Kotula, H.P., F.S., J. Svoboda, S. Sázelová, A.C., A. Khokhlov, N.J.C., F.V., K.H., P. Semal, B.J., A. Suvorov, R. Schulting, V.M., K.M., A.B. T.T., D.C. and E.A. assembled archaeological material, descriptions and interpretations. S.T. and H.F. produced a subset of the radiocarbon dates. A. Stoessel performed µCT scanning and advised on sampling strategy. C. Posth and H.Y. wrote the manuscript with input from T.T., E.A., W.H., J. Krause and all other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Marta Mirazón Lahr, Ludovic Orlando and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 MtDNA and Y-chromosome phylogenies.
Bold letters refer to (a) mtDNA and (b) Ychr haplogroups, whose boxes are coloured according to the legend in Extended Data Fig. 2. The labels in italic denote previously published individuals without new data generated in this study.
Extended Data Fig. 2 The distribution of mtDNA and Y-chromosome haplogroups among different hunter-gatherer groups.
The length of each coloured bar represents the fraction of individuals carrying the corresponding haplogroup (legend on the right of each panel). The number of individuals in each group is written to the right of each bar. We only plotted groups with more than two individuals and, for this reason, individuals from the GoyetQ116-1 cluster are included here into the Fournol cluster.
Extended Data Fig. 3 F4-statistics comparing the affinity of pre-LGM European hunter-gatherers to Goyet Q116-1 and Sunghir.
The colours correspond to the grouping of tested populations, dots refer to the f4-values and the dark and light error bars to 1*SE and 3*SE estimated from 5 cM block jackknife, respectively. This figure shows that the Gravettian-associated individuals from western Europe (Fournol cluster) are closely related to Goyet Q116-1 while the Gravettian-associated individuals from central-eastern and southern Europe (Věstonice cluster) are closely related to the Sunghir group, representative of the Kostenki cluster. Details are provided in Data S2.B.
Extended Data Fig. 4 Admixture graph modelling of pre-34 ka hunter-gatherer lineages.
In this admixture graph, the lineage related to Zlatý kůň splits more basally than the Bacho Kiro IUP group, who contributes to Tianyuan, Ust’Ishim and Goyet Q116-1 (indicated with red lines), but not to the Sunghir group.
Extended Data Fig. 5 Summary of ROH segments detected in Eurasian hunter-gatherers.
We visualize the total amount of ROH longer than 4 cM for (a) pre-LGM individuals, (b) Epigravettian- and Magdalenian-associated individuals, (c) individuals carrying high proportions of Sidelkino-related ancestry, and (d) individuals carrying high proportions of Oberkassel-related ancestry. Colour legend is shown in (e). Each bar represents one individual with the ROH grouped in four length categories (grouped by colour). The inferred pattern of short ROH (4–8 cM, visualized in blue) being common is in stark contrast to most later farmer populations, where the majority of individuals have no short ROH whatsoever (see101), and evidences small effective population sizes across West Eurasian hunter-gatherer groups. A dashed line of 50 cM total ROH is drawn in each panel to help comparison between panels with different y-axis scales. Details of the grouping and ROH segments are provided in Data S3.B.
Extended Data Fig. 6 West Eurasian PCA showing the genetic positioning of post-LGM hunter-gatherers.
Present-day individuals (gray dots) genotyped on the Human Origins dataset are used to define the PCA variation onto which ancient genomes (coloured symbols) are projected. The newly reported individuals with over 15,000 SNPs on the Human Origins dataset are shown in black-outlined and filled symbols, as illustrated in the legend on the right, while representative ancient genomes are shown in outlined symbols, as illustrated in the legend at the bottom of the PCA.
Extended Data Fig. 7 Admixture dates between Oberkassel/Villabruna and ANE ancestries in the oldest individuals from the Sidelkino cluster.
The triangles show the average calibrated dates of the tested groups and the dots show the estimated admixture dates with the software DATES105 using Oberkassel (red dots) or Villabruna (blue dots) clusters as one source and ANE-related individuals as the other source population. The generation time is set to 29 years and the error bars show the SE of the admixture date estimated from jackknife resampling (n = 22 autosomal chromosomes). Additional details are provided in Supplementary Information, section 11 and Supplementary Table 3.
Extended Data Fig. 8 Changes of Sidelkino and Oberkassel ancestry proportions in post-14 ka hunter-gatherers.
The bivariate plots show the expansion of Oberkassel and Sidelkino ancestries through time in two European areas (longitude below and above 30 degrees). The x-axis shows the average age of each tested individual/group and the y-axis shows the proportion of Sidelkino ancestry, relative to the total hunter-gatherer ancestry (Oberkassel + Sidelkino) in each group. The three squares highlight Baltic HG groups associated with the Comb Ceramic Culture (CCC) that show a marked increase in Sidelkino-related ancestry compared to older Baltic HG groups.
Extended Data Fig. 9 Graphical summary depicting the main genetic transformations in post-40 ka hunter-gatherers from Europe.
This figure shows (a) the distribution of and interaction between hunter-gatherer genetic ancestries and (b) a simplified schematic representation of major chrono-cultural subdivisions of the European Upper Palaeolithic (green blocks) followed by a grouped Mesolithic to Neolithic block (in gray). The x-axes report the geographic regions as divided in Fig. 1a, and the y-axes report time in thousand years before present (kBP). In panel a, genetic affinity between different ancestries is indicated by thick lines or shades, while admixture is indicated with arrows. In panel b, the colour code does not imply archaeological similarities.
Supplementary information
Supplementary Information
Archaeological background, method descriptions and discussions additional to the main text, including sections 1–13, Figs. 1–32, Tables 1–4 and references.
Supplementary Data 1
Metadata information of studied individuals.
Supplementary Data 2
Raw data of f-statistics analyses.
Supplementary Data 3
Raw data of other types of analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Posth, C., Yu, H., Ghalichi, A. et al. Palaeogenomics of Upper Palaeolithic to Neolithic European hunter-gatherers. Nature 615, 117–126 (2023). https://doi.org/10.1038/s41586-023-05726-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-05726-0
This article is cited by
-
Evidence from personal ornaments suggest nine distinct cultural groups between 34,000 and 24,000 years ago in Europe
Nature Human Behaviour (2024)
-
Signalling Palaeolithic identity
Nature Human Behaviour (2024)
-
The Persian plateau served as hub for Homo sapiens after the main out of Africa dispersal
Nature Communications (2024)
-
Us and Them: How to Reconcile Archaeological and Biological Data at the Middle-to-Upper Palaeolithic Transition in Europe?
Journal of Paleolithic Archaeology (2024)
-
Homo sapiens reached the higher latitudes of Europe by 45,000 years ago
Nature (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
