
Abstract
The control of tetrahedral carbon stereocentres remains a focus of modern synthetic chemistry and is enabled by their configurational stability. By contrast, trisubstituted nitrogen1, phosphorus2 and sulfur compounds3 undergo pyramidal inversion, a fundamental and well-recognized stereochemical phenomenon that is widely exploited4. However, the stereochemistry of oxonium ions—compounds bearing three substituents on a positively charged oxygen atom—is poorly developed and there are few applications of oxonium ions in synthesis beyond their existence as reactive intermediates5,6. There are no examples of configurationally stable oxonium ions in which the oxygen atom is the sole stereogenic centre, probably owing to the low barrier to oxygen pyramidal inversion7 and the perception that all oxonium ions are highly reactive. Here we describe the design, synthesis and characterization of a helically chiral triaryloxonium ion in which inversion of the oxygen lone pair is prevented through geometric restriction to enable it to function as a determinant of configuration. A combined synthesis and quantum calculation approach delineates design principles that enable configurationally stable and room-temperature isolable salts to be generated. We show that the barrier to inversion is greater than 110 kJ mol−1 and outline processes for resolution. This constitutes, to our knowledge, the only example of a chiral non-racemic and configurationally stable molecule in which the oxygen atom is the sole stereogenic centre.
Similar content being viewed by others
Main
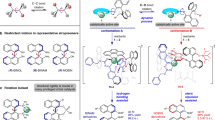
The pyramidal inversion of trisubstituted amines, phosphines and sulfonium salts is a fundamental stereochemical process that has been intensively studied, leading to an understanding of the factors that affect the magnitude of their inversion barriers1. This has enabled the generation of a myriad of configurationally stable chiral phosphines (which possess barriers to inversion of 120–185 kJ mol−1)3 and sulfonium salts (with barriers of 100–130 kJ mol−1)4. The inversion of the nitrogen atom in a tertiary amine has a relatively low barrier (20–25 kJ mol−1) and there is a substantial contribution from quantum tunnelling to the observed rate2; this means that the preparation of configurationally stable tertiary amines bearing a stereogenic nitrogen atom is particularly challenging. However, there are numerous examples of bicyclic amines such as Tröger’s base 18,9,10, cinchona alkaloids11 and others12,13 in which nitrogen inversion is restricted by virtue of conformational rigidity, enabling access to compounds bearing a stereogenic nitrogen (Fig. 1a)14. By contrast, the study of oxonium ions is much less developed15,16. The only empirical measurement of the inversion barrier for a trivalent oxygen compound is for the epoxide-derived oxonium salt 2 that has a barrier to inversion of 42 kJ mol−1, which is substantially below that required to maintain configurational stability at ambient temperature7. Consequently, there are no examples of the isolation of enantioenriched oxonium ions in which the oxygen atom17,18 is the sole stereogenic centre (Fig. 1b).

a, Pyramidal inversion of common trisubstituted heteroatoms occurs at different rates, leading to different configurational stabilities for tertiary amines, sulfonium salts and phosphines. b, Inversion of oxonium ions is rapid unless prevented by geometrical restriction: there are no examples of room-temperature configurationally stable oxonium ions bearing oxygen as the sole stereogenic centre. c, Synthesis and characterization of configurationally stable oxonium ions bearing a stereogenic oxygen. Counterions are not shown for clarity.ΔG‡inv = Gibbs free energy of activation for the inversion process.
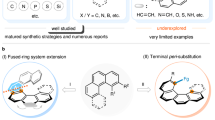
Oxonium ions are considered to be highly reactive and are invoked as transient intermediates in a range of synthetic transformations including natural product biosynthesis and total synthesis19,20,21,22,23,24. This has led to the characterization of complex trialkyloxonium ions such as 3, a proposed intermediate in the synthesis of the Laurencia family of natural products. The fused ring system in 3 prevents inversion, and hence the oxygen is stereogenic; in this case the oxonium ion is formed in a diastereoselective manner from a precursor bearing several carbon stereocenters5. Tetracyclic oxonium ion 4 possesses a stereogenic oxygen due to its non-symmetrical bridge25. The best-known oxonium ions are arguably the trialkyl tetrafluoroborates, first synthesized by Meerwein more than 80 years ago6,26. Although these are stable and isolable, they are also some of the most reactive alkylating agents used in a synthetic chemistry laboratory and react readily with weak nucleophiles including water. Other alkyl oxonium ions have been prepared and studied27,28,29,30, but species stable to nucleophiles were elusive until Mascal reported oxatriquinane oxonium ions31,32,33,34; this indicates the potential impact upon stability of embedding oxonium ions in ring structures. In contrast to most alkyl-substituted oxonium ions, simple triaryloxonium ions are almost trigonal planar about oxygen and are stable isolable solids resistant to the action of many nucleophiles35,36,37. Dibenzofuran-derived triaryloxonium ions are pyramidal about the oxygen atom38, which led to Baldridge and Siegel investigating rotation about the exocyclic C–O bond in oxonium salts such as 5 (R1 = Et, R2 = H); this showed that the barrier (estimated at more than 84 kJ mol−1) was probably relatively close to Oki’s definition of atropisomerism39. However, oxygen inversion was calculated to require substantially less energy (R1 = R2 = Me ≈ 7 kJ mol−1)40. We reasoned that joining of the aromatic rings in the triaryloxonium scaffold with linkers of various lengths and flexibilities should increase the barrier to oxygen inversion while also maintaining the stability exhibited by other triaryloxonium ions (Fig. 1c). A sufficiently high enantiomerization barrier (which we have defined as greater than 107 kJ mol−1, t1/2 rac = 7 days at 25 °C; t1/2 rac = the time during which the enantiomeric purity of a chiral sample has reduced from 100 to 50% enantiomeric excess) would effectively lock the configuration of the stereogenic oxygen lone pair on a laboratory timescale and generate helically chiral molecules41,42. We reasoned that an intramolecular O-arylation of a dibenzofuran scaffold with a diazonium salt could offer a viable route to such molecules.
We initially selected a dibenzofuran−xanthene scaffold for our investigations as the CH2 protons in the xanthene backbone could be used as a reporter group to measure inversion barriers by variable temperature 1H NMR spectroscopy. We prepared the functionalized xanthenes 6 and 7 from commercial materials (see page 3 of the Supplementary Information for details); these were cross-coupled with 4,6-dimethyl-2-bromoaniline to afford biaryls 8 and 9. Diazotization and intramolecular O-arylation with a loss of nitrogen led to the formation of pentacyclic oxonium tetrafluoroborates 10 and 11 (Fig. 2a). The structure of compound 10 was confirmed by single-crystal X-ray diffraction, which shows that the compound is pyramidal at the oxygen (sum of the angles around the oxygen θ1 + θ2 + θ3 = 338.3(3)° versus 345(1)° for the dibenzofuran parent oxonium43, and the apex height h = 0.401(2) Å, Fig. 2) with the longest C–O bond length of 1.491(2) Å. The dibenzofuran ring is planar (internal C–O–C angle 104.26 (15)°), with the central ring of the xanthene adopting a boat-like conformation that places its two adjoining aromatic rings in different planes (Fig. 2b). The xanthene C–O–C internal angle is 110.11(14)° (versus 119.3° in the parent heterocycle xanthene itself), indicative of some angle strain at the fusion with the benzofuran ring, although that does not seem to compromise the stability of the compound, which can be stored indefinitely at room temperature without special precautions. Low-temperature 1H NMR spectroscopy was used to determine the enantiomerization barrier as 58.3 kJ mol−1 (see page 23 of the Supplementary Information for further details), which is substantially lower than the estimated value of 107 kJ mol−1 required to enable configurationally stable molecules to be isolated. By contrast, oxonium ion 11 was only observed as a transient species, and we initially reasoned that this may have been a consequence of the tetrafluoroborate counterion functioning as a nucleophilic fluoride source (the Balz−Schiemann reaction). Anion exchange of the diazonium tetrafluoroborate salt derived from 9 to a less nucleophilic B(C6F5)4− counterion and ring closure afforded 12, but this compound slowly decomposed in solution at ambient temperature over 72 hours. We considered that the introduction of a second substituent onto the dibenzofuran−xanthene scaffold may have increased the strain in 11 and 12, compromising the relative stability observed in 10.

a, Reagents and conditions: (i) Pd(dppf)Cl2·CH2Cl2 (5 mol%), Na2CO3 (4 equivalents), PhMe/H2O (1:1, v/v), 95 °C, 24 h; (ii) HBF4 (48% aq, 5 equivalents), t-BuONO (5 equivalents), CH2Cl2/IPA (1:1), 0 °C, 1 h then 30 °C, 48 h; (iii) HBF4 (48% aq, 5 equivalents), t-BuONO (5 equivalents), CH2Cl2/IPA (1:1), 0 °C, 1 h, then KB(C6F5)4, then 30 °C, 48 h. b, Crystal and molecular structure of fused pentacyclic oxonium salt 10 from single-crystal X-ray diffraction studies. The tetrafluoroborate counterion is omitted for clarity. Numbers in parentheses indicate standard uncertainties. c, B3LYP-D3(BJ)/6.31+G(d,p) computed inversion barriers for a series of oxonium ions based upon a dibenzofuran-xanthene core (i) and a dibenzofuran-benzooxepine core (ii). h, Height of conceptualized triangular oblique pyramid with oxygen at its apex and the adjacent carbons of the attached O-aryl rings forming the base; Calc., calculated.
To enable us to rationalize the magnitude of the inversion barrier in 10 and the reactivity of more substituted compounds such as 11 we applied B3LYP-D3(BJ)/6-31+G(d,p) quantum chemical calculations (see page 33 of the Supplementary Information). We initially calculated barriers to the inversion of the oxonium ions based around the benzofuran−xanthene scaffold (Fig. 2c(i)). This gave excellent agreement with the experimental values for the overall geometry and barrier to pyramidal inversion for a model 2-methyl oxonium ion (R2 = Me, R3 = H: calculated at 59.0 kJ mol−1); other levels of theory and basis sets gave comparable quantitative results. These calculations also indicated that increasing the size of the substituent R2 would result in an increased inversion barrier (73.6 kJ mol−1 for R2 = iPr, R3 = H) and a marginal increase in pyramidalization about oxygen. Switching the (methyl) substitution to the xanthene portion gave predicted enantiomerization barriers similar to that previously calculated (R2 = H, R3 = Me, 54.4 kJ mol−1), but below the desired level for configurational stability. To investigate the impact of the addition of substituents on both rings, we examined a doubly alkyl-substituted oxonium (R2 = R3 = Me). This led to an increase in the predicted barrier to enantiomerization to 148.1 kJ mol−1 but also led to some changes in geometry that may explain the observed instability of molecules such as 11 and 12 at ambient temperature. The calculated sum of angles around the oxonium oxygen is 345.1° (R2 = R3 = Me), similar to the parent dibenzofuran oxonium ion, but we observe a notable increase in the external C–O–C angle to 129° and a reduction in apex height (h = 0.33 Å) consistent with a reduction in pyramidalization around oxygen. We also observe further lengthening of the longest C–O bond to 1.51 Å. This is a result of steric repulsion between the alkyl substituents, which leads to a twist in the xanthene ring and deformation from the boat-like conformation populated in 10. Calculating a series of hyperhomodesmotic bond separation reaction enthalpies44 enabled us to estimate the increase in steric strain in going from a compound such as 10 (R1 = H) to one bearing an extra alkyl group (R1 = Me) as 16.7 kJ mol−1 (see page 34 of the Supplementary Information for details). For 11, the calculated enthalpy difference versus 10 is significantly higher (45.6 kJ mol−1), probably indicating both an electronic and a steric contribution. We can conclude that the development of oxonium ions with large enough inversion barriers to attain configurational stability through the addition of multiple substituents on this ring system without compromising stability will probably be a challenge.
The design of configurationally stable molecules requires that we maximize the difference between ground states (GSs) and transition states (TSs). With a six-membered ring, our attempts to destabilize the TS (through installation of a larger R3 substituent) also results unavoidably in GS destabilization. Dihydrodibenzooxepines have a non-planar structure and we envisaged that the extra sp3 carbon in the seven-membered ring (versus xanthene) may enable the avoidance of steric destabilization in the GS. In contrast, we reasoned that as the molecule moves towards planarity in the TS, the extra sp3 carbon would actively lead to TS destabilization and a higher barrier to inversion.
We computed GS and TS structures for oxepin-containing oxonium ions (Fig. 2c(ii)). There is a substantial predicted increase in the barrier to inversion upon changing from the six to the seven-membered ring oxonium, from 59.0 to 117.0 kJ mol−1 (for R2 = Me, R3 = H) and this is augmented to 127.2 kJ mol−1 and 154.4 kJ mol−1 upon introduction of larger iso-propyl and tert-butyl groups respectively. In the calculated dihydrodibenzooxepine GS structures, the sum of angles about oxygen is generally lower than in the xanthene series and decreases as the size of the substituent on the benzofuran ring increases; this is consistent with a greater apex height in the seven-ring series and greater pyramidalization about oxygen.
With this information in hand, we focussed on the synthesis of seven-ring-containing compounds, by using a strategically similar approach to that adopted earlier: dibenzooxepine 13 (made in four steps) was cross-coupled with aryl boronic ester 14 to afford a biaryl-containing intermediate in 67% yield. Hydrogenation gave 15, which was diazotised under standard conditions and gently warmed to 30 °C to enable O-arylation and to afford the oxonium tetrafluoroborate rac-16 in 75% yield (Fig. 3a). Counterion metathesis with sodium hexafluorophosphate enabled the synthesis of oxonium ion rac-17 in 56% yield. Oxonium tetrafluoroborate rac-16 and hexafluorophosphate rac-17 are room-temperature stable solids that can be stored indefinitely without special precautions. The structure of rac-16 was confirmed by single-crystal X-ray diffraction studies (space group P−1), which illustrate the expected puckered conformation of the oxepin ring and a pyramidal arrangement of atoms around the oxygen (the sum of the angles around oxygen is 335.9°, Fig. 3b). The apex height h = 0.428(1) Å, reflects the increased pyramidalization about oxygen versus 10. The external C–O–C angle is 117.78(7)° and the internal oxepin C–O–C angle is 113.03(7)°; these constitute relatively minor angular deviations from those observed in the parent heterocycles and are consistent with a ring system that does not experience substantial angle strain. Computed hyperhomodesmotic reaction enthalpies suggest that oxonium ion 16 is less strained than 10 by 13.4 kJ mol−1. Examination of the unit cell does not show any short C–H or C–C distances less than normal van der Waals contact distances, which is indicative that the conformation of the oxonium cation is not influenced by crystal packing. The longest C–O bond length is 1.4918(12) Å; this is longer than the corresponding measurement in benzofuran-derived triaryloxonium ions (1.459(6) Å) and the average C–O bond length in triphenyloxonium tetraphenylborate (at 1.47 Å).

a, Reagents and conditions: (i) Pd(dppf)Cl2·CH2Cl2 (5 mol%), Na2CO3 (4 equivalents), PhMe/H2O (1:1, v/v), 95 °C, 24 h (67% yield); (ii) H2 (g, 1 atmosphere), Pd/C (2.5 mol%), EtOAc, RT, 16 h (quantitative); (iii) HBF4 (48% aq, 5 equvialents), t-BuONO (5 equivalents), CH2Cl2/IPA (1:1), 0 °C, 1 h then 30 °C, 48 h (75% yield); (iv) NaPF6 (aq), CH2Cl2 (56% yield); (v) Na[B((R)−BINOL)2] (1.0 equivalent), MeCN, RT then CH2Cl2, wash with water; (vi) dissolve in CHCl3 and modulate concentration. b. Crystal and molecular structure of fused pentacyclic oxonium salt rac-16 from single-crystal X-ray diffraction studies. The tetrafluoroborate counterion is omitted for clarity. Numbers in parentheses indicate standard uncertainties. h, height of conceptualized triangular oblique pyramid with oxygen at its apex and the adjacent carbons of the attached O-aryl rings forming the base.
Treatment of rac-16 with a stoichiometric amount of sodium bis-(R)-BINOL borate led to complete conversion to a 1:1 mixture of diastereoisomeric salts (18, Fig. 3 and 19 (not shown)). Exploitation of the differential solubility of these two salts in chloroform enabled precipitation of one to yield the enriched diastereoisomeric pair 18 (ratio 94:6–99:1). We were unable to grow crystals suitable for X-ray analysis of diastereoisomerically enriched salt 18, and hence we attempted to exchange the counterion in a highly diastereoisomerically enriched sample by stirring with an excess of tetrafluoroboric acid. From this mixture we were able to isolate crystals of the desired tetrafluoroborate salt 16; this crystallized in P212121, with an absolute configuration of P-(R)O (determined by refinement of the Flack parameter x against X-ray diffraction datasets collected from three separate crystals; CCDC 21911038-2191040)45, which thus also indicates the absolute configuration of the oxonium ion in 18 (see page 19 of the Supplementary Information). We probed the configurational stability of dichloromethane solutions of 18, and observed a small change in the diastereoisomeric ratio over 7 days at 25 °C. With this data and the equilibrium ratio of 50.5:49.5 (18, P-(R)O, (R); 19, M-(S)O,(R)), we can calculate the barriers to helical inversion to be 111.8 ± 0.1 kJ mol−1 (for the P-(R)O, (R) 18 to M-(S)O, (R) 19 conversion) and 111.7 ± 0.1 kJ mol−1 (for the M-(S)O, (R) 19 to P-(R)O, (R) 18 conversion); attempts to examine equilibration at higher temperatures (more than 50 °C) led to decomposition at a rate faster than equilibration. We were also able to resolve oxonium hexafluorophosphate rac-17 by chiral stationary phase high-performance liquid chromatography, and use this method to determine its enantiomerization barrier as 111.0 ± 0.1 kJ mol−1. This demonstrates that the influence of the counterion on the magnitude of the inversion barrier is probably negligible, consistent with the observed values for diastereoisomeric salts 18 and 19 (which are almost energetically degenerate).
The experimentally determined barriers to inversion in 17 and 18 (111.0–111.8 kJ mol−1) are close to that predicted by calculation (117.0 kJ mol−1). A calculated reaction energy profile for the reversible enantiomerization of this oxonium ion is shown in Fig. 4a (ref. 46); this indicates that the enantiomerization process from P-(R)O A consists of a high barrier helical inversion and a much lower barrier conformational relaxation associated with the seven-membered ring. The transition structure (TS) B for the helical inversion (Grel = 117 kJ mol−1) possesses inverted pyramidal geometry at the oxygen, albeit reduced from the GS A (h = 0.32 Å, sum of angles around oxygen 346.7° for B versus 335.9° for A). A structure in which the oxygen is trigonal planar lies earlier on the reaction coordinate from A to B; this also demonstrates that O-inversion is directly coupled with overall inversion of the helical chirality of the molecule. In TS B, both the internal oxepin and external C–O–C angles are close to 121° (Fig. 4b), which represents a notable change in structure around the oxygen atom versus the GS. Lengthening of the C–O bond in the oxepin ring versus the GS is also observed (1.51 versus 1.49 Å). TS B leads directly to intermediate C (Grel = 32 kJ mol−1) in which the oxepin ring populates a boat-like conformation; this undergoes a facile ring flip via TS D (Grel = 35 kJ mol−1) to the more stable boat-like conformation in GS structure M-(S)O A. To rationalize why the inversion barrier of the oxepin-containing oxonium ion is greater than 40 kJ mol−1 higher than its xanthene-containing analogue, we calculated how ring strain in the six- and seven-membered rings in oxonium ions changes on going from the GS to the TS (Fig. 4c). In the xanthene-containing system, the boat-like GS E changes to an almost perfectly planar TS F through movement of the two aromatic rings into the same plane. This leads to a decrease in strain in the six-membered heterocycle of 12.1 kJ mol−1. This is owing to the relief of angle strain in the xanthene ring, which can be seen in the increase in internal C–O–C angle from 111.0° to 116.6° (GS to TS). In this system, the TS resembles the GS conformation of xanthene itself. In contrast, there is an increase in strain of 33.9 kJ mol−1 in the dihydrodibenzooxepine ring going from GS G to TS H. To attain the TS geometry demonstrated in H, the two aromatic rings twist to enable the inversion of the seven-membered ring. This raises the energy through a combination of torsional strain (between the two sp3-carbon atoms in the backbone (H–C–C–H torsion = 38.1°)), an increase in allylic strain between the backbone sp3 C–H bonds and the aromatic rings, internal angle strain and a minor deviation from planarity in the two aromatic rings. The seven-membered ring conformation in oxonium ion G is boat-like and resembles dihydrodibenzooxepine itself: deviation from this geometry in TS H raises the energy substantially. Delocalization of the oxygen lone pair changes along the reaction coordinate for inversion and this is also a prominent source of differentiation between dihydrodibenzooxepine and xanthene-derived oxonium ions. Natural bond orbital47 calculations highlight how oxygen lone-pair delocalization into the adjacent π systems stabilizes the inversion TS. For the xanthene oxonium ion, the increasing planarity about oxygen stabilizes the TS by 54.1 kJ mol−1 relative to the GS, whereas for the dihydrodibenzooxepine oxonium ion this is lower at 12.0 kJ mol−1, a difference of 33.1 kJ mol−1. Increased strain in the seven-membered ring oxonium backbone resists planarization in the inversion TS, reducing the extent of lone-pair delocalization, which consequently increases the activation energy barrier.

a, B3LYP-D3(BJ)/6-31+G(d,p) computed Gibbs energy profile for enantiomerization demonstrates that the process consists of a high barrier oxygen inversion and a low barrier conformational relaxation of the seven-membered ring. b, Geometrical measurements of TS B. ϕ = (C6–C7–C8–C9) dihedral angle. c, Computed change in ring strain (ΔEdist) in highlighted rings in xanthene and dihydrodibenzooxepine oxonium ions on going from GS to TS. The relative energies of truncated xanthene and dihydrodibenzooxepine substructures are computed at geometries taken from the parent oxonium ion. d, Comparison of computed barriers to inversion show that triaryloxonium ions have higher barriers to inversion than corresponding triaryl amines (B3LYP-D3(BJ)/6-31+G(d,p)). Grel = relative Gibbs free energy. ‡ = transition state.
To examine how the identity of the heteroatom influences the inversion barrier, we studied this class of oxonium ions and their nitrogen analogues computationally (Fig. 4d). The calculated barriers to inversion of triaryloxonium ions are systematically higher than their triarylamine counterparts. This can be rationalized based on the resistance to rehybridization of the heteroatom, which is greater in the oxonium ion (see page 38 of the Supplementary Information). For the nitrogen analogues there is greater lone-pair delocalization in the inversion TS structures. This is consistent with calculations on the dihydrodibenzooxepine oxonium (ΔG‡inv = 117.0 kJ mol−1) and the dihydrodibenzazepine (ΔG‡inv = 99.7 kJ mol−1) systems, indicating that barrier height depends on both the geometric restraint provided by the ring structure and the identity of the heteroatom.
Data availability
All data (experimental procedures, characterization data and cartesian coordinates for all DFT calculations) supporting the findings of this study are available within the article and its supplementary information. Crystallographic data for compounds 10, rac-16 and P-(R)O-16 (three datasets) have been deposited with the Cambridge Crystallographic data Centre under deposition numbers CCDC 2124020, CCDC 2124019 and CCDC 21911038–2191040. These data can be obtained free of charge from www.ccdc.cam.ac.uk/data_request/cif.
References
Lehn, J. M. Nitrogen inversion: experiment and theory. Fortschr. Chem. Forsch. 15, 311–377 (1970).
Baechler, R. D. & Mislow, K. Effect of structure on the rate of pyramidal inversion of acyclic phosphines. J. Am. Chem. Soc. 92, 3090–3093 (1970).
Andersen, K. K., Cinquini, M. & Papanikolaou, N. E. Synthesis and stereochemistry of triarylsulfonium salts. J. Org. Chem. 35, 706–710 (1970).
Rauk, A., Allen, L. C. & Mislow, K. Pyramidal inversion. Angew. Chem. Int. Ed. Engl. 9, 400–414 (1970).
Chan, H. S., Nguyen, Q. N. N., Paton, R. S. & Burton, J. W. Synthesis, characterization and reactivity of complex oxonium ions, proposed intermediates in natural product biosynthesis. J. Am. Chem. Soc. 141, 15951–15962 (2019).
Meerwein, H., Hinz, G., Hofmann, P., Kroning, E. & Pfeil, E. Über tertiäre oxoniumsalze I. J. Prakt. Chem. 147, 257–285 (1937).
Lambert, J. B. & Johnson, D. H. The stereochemistry and inversion of trivalent oxygen. J. Am. Chem. Soc. 90, 1349–1350 (1968).
Tröger, J. Ueber einige mittelst nascirenden formaldehydes entstehende basen. J. Prakt. Chem. 36, 225–245 (1887).
Prelog, V. & Wieland, P. Über die spaltung der Tröger’schen base in optische antipoden, ein beitrag zur stereochemie des dreiwertigen stickstoffs. Helv. Chim. Acta 27, 1127–1134 (1944).
Rigol, S., Beyer, L., Hennig, L., Sieler, J. & Giannis, A. Hunlich base: (re)discovery, synthesis, and structure elucidation after a century. Org. Lett. 15, 1418–1420 (2013).
Boratyński P.J., Zielińska-Błajet M. & Skarżewski J. Cinchona alkaloids-derivatives and Applications. Alkaloids Chem. Biol. 82, 29−145 (2019).
Huang, S. et al. Organocatalytic enantioselective construction of chiral azepine skeleton bearing multiple-stereogenic elements. Angew. Chem. Int. Ed. Engl. 60, 21486–21493 (2021).
Rowley, J. H., Yau, S. C., Kariuki, B. M., Kennedy, A. R. & Tomkinson, N. C. O. Readily accessible chiral at nitrogen cage structures. Org. Biomol. Chem. 11, 2198–2205 (2013).
Walsh, M. P., Phelps, J. M., Lennon, M. E., Yufit, D. S. & Kitching, M. O. Enantioselective synthesis of ammonium cations. Nature 597, 70–76 (2021).
Koeppl, G. W., Sagatys, D. S., Krishnamurthy, G. S. & Miller, S. I. Inversion barriers of pyramidal (XY3) and related planar (=XY) species. J. Am. Chem. Soc. 89, 3396–34051 (1967).
Penner, G. H., Polson, K. M., Daleman, S. & Reid, K. Deuterium NMR study of molecular dynamics and geometry in two classes of onium salts: (CH3)E+X− and C6H5M(CH3)3+I−. Can. J. Chem. 71, 417–426 (1993).
Mikata, Y., Fujimoto, T., Noko, I. & Kondo, S. Differentiation of oxygen atom chirality in copper(II) complexes with dipicolylamine-derived ligands.Eur. J. Inorg. Chem. 2012, 4310–4317 (2012).
Mikata, Y., Sugai, Y., Obata, M., Harada, M. & Yano, S. Novel oxygen chirality induced by asymmetric coordination of an ether oxygen atom to a metal center in a series of sugar-pendant dipicolylamine copper(II) complexes. Inorg. Chem. 45, 1543–1551 (2006).
Braddock, D. C. A hypothesis concerning the biosynthesis of the obtusallene family of marine natural products via electrophilic bromination. Org. Lett. 8, 6055–6058 (2006).
Murai, A. in Comprehensive Natural Products Chemistry 1st edn (eds Barton, D. H. R., Meth-Cohn & O. Nakanishi, K.) 303−324 (Elsevier, 1999).
Kikuchi, H., Suzuki, T., Kurosawa, E. & Suzuki, M. The structure of notoryne, a halogenated C15 nonterpenoid with a novel carbon skeleton from the red alga Laurenda nipponica yamada. Bull. Chem. Soc. Jpn. 64, 1763–1775 (1991).
Bonney, K. J. & Braddock, D. C. A unifying stereochemical analysis for the formation of halogenated C15-acetogenin medium-ring ethers from laurencia species via intramolecular bromonium ion assisted epoxide ring-opening and experimental corroboration with a model epoxide. J. Org. Chem. 77, 9574–9584 (2012).
Kim, B. et al. Biomimetic asymmetric total synthesis of (−)-laurefucin via an organoselenium-mediated intramolecular hydroxyetherification. J. Am. Chem. Soc. 130, 16807–16811 (2008).
Zhang, Y. A., Yaw, N. & Snyder, S. A. General synthetic approach for the laurencia family of natural products empowered by a potentially biomimetic ring expansion. J. Am. Chem. Soc. 141, 7776–7788 (2019).
Stoyanov, E. S. et al. The R3O+···H+ hydrogen bond: toward a tetracoordinate oxadionium(2+) ion. J. Am. Chem. Soc. 134, 707–714 (2012).
Meerwein, H., Bettenberg, E., Gold, H., Pfeil, E. & Willfang, G. Über tertiäre oxoniumsalze II. J. Prakt. Chem. 154, 83–156 (1940).
Klages, F. & Jung, H. A. Synthese und eigenschaften eines bicyclischen oxoniumsalzes. Chem. Ber. 98, 3757–3762 (1965).
Gargulio, R. J. & Tarbell, S. Cyclic oxonium ions and related structures. Proc. Natl Acad. Sci. USA. 62, 52–55 (1969).
Olah, G. A., Doggweiler, H., Felberg, J. D. & Frohlich, S. Onium ions. 33. (Trimethylsilyl)- and [(trimethylsilyl)methyl]oxonium and -halonium ions. J. Org. Chem. 50, 4847–4851 (1985).
Etzhorn, M.et al. 1‐Oxoniaadamantane. Eur. J. Org. Chem. 2008, 4555–4558 (2008).
Mascal, M., Hafezi, N., Meher, N. K. & Fettinger, J. C. Oxatriquinane and oxatriquinacene: extraordinary oxonium ions. J. Am. Chem. Soc. 130, 13532–13533 (2008).
Mascal, M., Hafezi, N. & Toney, M. D. 1,4,7-Trimethyloxatriquinane: SN2 reaction at tertiary carbon. J. Am. Chem. Soc. 132, 10662–10664 (2010).
Gunbas, G. et al. Extreme oxatriquinanes and a record C-O bond length. Nat. Chem. 4, 1018–1023 (2012).
Suzuki, H. & Muratake, H. Functionalized oxatriquinanes and their structural equilibrium in protic solvent. Chem. Pharm. Bull. 62, 921–926 (2014).
Nesmeyanov, A. N., Makarova, L. G. & Tolstaya, T. P. Heterolytic decomposition of onium compounds (diphenyl halogenonium and triphenyloxonium salts). Tetrahedron 1, 145–157 (1957).
Watkins, M. I., Ip, W. M., Olah, G. A. & Bau, R. Onium ions. 23. Structure of oxonium ions: an X-ray crystallographic study of triethyloxonium hexafluorophosphate and triphenyloxonium tetraphenylborate. J. Am. Chem. Soc. 104, 2365–2372 (1982).
Tolstoya, T. P. et al. Nucleophilic substitution in O-phenyldibenzofuranium and 10-phenylxanthonium cations. Russ. Chem. Bull. 46, 789–797 (1997).
Hellwinkel, D. & Seifert, H. Ringschlußreaktionen von 2′-heterosubstituierten biphenyl-2-diazonium-salzen zu (spiro)cyclischen tetraarylammonium-salzen und tribenz[b.d.f]azepinen. Chem. Ber. 105, 880–906 (1972).
Oki, M. in Topics in Stereochemistry Vol. 14 (eds Allinger, N. L., Eliel, E. L. & Wilen, S. H.) 1–81 (Wiley, 1983).
Lu, M. et al. Peraryl-x-onium ions of nitrogen and oxygen. Org. Chem. Front. 6, 2640–2646 (2019).
Bosson, J., Gouin, J. & Lacour, J. Cationic triangulenes and helicenes: synthesis, chemical stability, optical properties and extended applications of these unusual dyes. Chem. Soc. Rev. 43, 2824–2840 (2014).
Wu, Y.-T., Hayama, T., Baldridge, K. K., Linden, A. & Siegel, J. S. Synthesis of fluoranthenes and indenocorannulenes: elucidation of chiral stereoisomers on the basis of static molecular bowls. J. Am. Chem. Soc. 128, 6870–6884 (2006).
Zhukhlistova, N. E., Tishchenko, G. N., Tsarev, D. A. & Tolstaya, T. P. Crystal and molecular structure of O-phenyldibenzofuranium tetrafluoroborate. Kristallografiya 42, 863–869 (1997).
Wheeler, S. E., Houk, K. N., Schleyer, P. V. R. & Allan, W. D. A hierarchy of homodesmotic reactions for thermochemistry. J. Am. Chem. Soc. 131, 2547–2560 (2009).
Flack, H. D. On enantiomorph-polarity estimation. Acta Crystallogr. A39, 876–881 (1983).
Reist, M., Testa, B., Carrupt, P. A., Jung, M. & Schurig, V. Racemization, enantiomerization, diastereomerization and epimerization: their meaning and pharmacological significance. Chirality 7, 396–400 (1995).
Glendening, E. D. et al. NBO v. 7.0. Theoretical Chemistry Institute, University of Wisconsin, Madison https://nbo6.chem.wisc.edu/biblio_css.htm (2018).
Acknowledgements
The EPSRC has provided financial support for studentships (to O.S. and M.V.P.) via the Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/ L015838/1) and to M.J.H. via a DTP studentship (EP/T517811/1; project no. 2446204). R.P. acknowledges support from the NSF (CHE-1955876) and computational resources from the RMACC Summit supercomputer supported by the National Science Foundation (ACI-1532235 and ACI-1532236), the University of Colorado Boulder and Colorado State University and the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation TG-CHE180056. We are grateful to A. Thompson (University of Oxford) for assistance with X-ray crystallography and P. Solon for assistance with the high-performance liquid chromatography measurements. A CC-BY licence is applied to the author-accepted manuscript arising from this submission, in accordance with open-access conditions of the EPSRC.
Author information
Authors and Affiliations
Contributions
O.S., M.V.P., M.J.H., R.P., J.W.B. and M.D.S. conceived and designed the study. O.S. and M.J.H. performed the synthetic experiments and analysed data for all compounds. O.S. performed the X-ray crystallography. M.V.P. and R.P. performed the computational study. O.S., M.V.P., M.J.H., R.P., J.W.B. and M.D.S. cowrote the paper. Correspondence regarding computation should be addressed to R.P.; all other correspondence should be addressed to M.D.S. and J.W.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Jonathan Clayden, Ganna Gryn'ova and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, O., Popescu, M.V., Hindson, M.J. et al. Control of stereogenic oxygen in a helically chiral oxonium ion. Nature 615, 430–435 (2023). https://doi.org/10.1038/s41586-023-05719-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-05719-z
This article is cited by
-
Harnessing triaryloxonium ions for aryne generation
Nature Synthesis (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
