
Abstract
Cognate tRNAs deliver specific amino acids to translating ribosomes according to the standard genetic code, and three codons with no cognate tRNAs serve as stop codons. Some protists have reassigned all stop codons as sense codons, neglecting this fundamental principle1,2,3,4. Here we analyse the in-frame stop codons in 7,259 predicted protein-coding genes of a previously undescribed trypanosomatid, Blastocrithidia nonstop. We reveal that in this species in-frame stop codons are underrepresented in genes expressed at high levels and that UAA serves as the only termination codon. Whereas new tRNAsGlu fully cognate to UAG and UAA evolved to reassign these stop codons, the UGA reassignment followed a different path through shortening the anticodon stem of tRNATrpCCA from five to four base pairs (bp). The canonical 5-bp tRNATrp recognizes UGG as dictated by the genetic code, whereas its shortened 4-bp variant incorporates tryptophan also into in-frame UGA. Mimicking this evolutionary twist by engineering both variants from B. nonstop, Trypanosoma brucei and Saccharomyces cerevisiae and expressing them in the last two species, we recorded a significantly higher readthrough for all 4-bp variants. Furthermore, a gene encoding B. nonstop release factor 1 acquired a mutation that specifically restricts UGA recognition, robustly potentiating the UGA reassignment. Virtually the same strategy has been adopted by the ciliate Condylostoma magnum. Hence, we describe a previously unknown, universal mechanism that has been exploited in unrelated eukaryotes with reassigned stop codons.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
All data generated during this study are included in this published article (and its Supplementary Information files). The MS data have been deposited to the ProteomeXchange Consortium through the PRIDE54 partner repository with the dataset identifier PXD033324. The high-throughput sequencing datasets were deposited in the National Center for Biotechnology Information under the number PRJNA790628 and in Figshare55. Additional data and analyses are available in Figshare56. Source data are provided with this paper.
Code availability
Custom computer code that was written for this project is publicly available on Zenodo57.
Change history
31 May 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41586-023-06260-9
18 January 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-07065-0
References
Záhonová, K., Kostygov, A. Y., Ševčíková, T., Yurchenko, V. & Eliáš, M. An unprecedented non-canonical nuclear genetic code with all three termination codons reassigned as sense codons. Curr. Biol. 26, 2364–2369 (2016).
Bachvaroff, T. R. A precedented nuclear genetic code with all three termination codons reassigned as sense codons in the syndinean Amoebophrya sp. ex Karlodinium veneficum. PLoS ONE 14, e0212912 (2019).
Swart, E. C., Serra, V., Petroni, G. & Nowacki, M. Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166, 691–702 (2016).
Heaphy, S. M., Mariotti, M., Gladyshev, V. N., Atkins, J. F. & Baranov, P. V. Novel ciliate genetic code variants including the reassignment of all three stop codons to sense codons in Condylostoma magnum. Mol. Biol. Evol. 33, 2885–2889 (2016).
Sella, G. & Ardell, D. H. The coevolution of genes and genetic codes: Crick’s frozen accident revisited. J. Mol. Evol. 63, 297–313 (2006).
Koonin, E. V. & Novozhilov, A. S. Origin and evolution of the genetic code: the universal enigma. IUBMB Life 61, 99–111 (2009).
Lobanov, A. V. et al. Position-dependent termination and widespread obligatory frameshifting in Euplotes translation. Nat. Struct. Mol. Biol. 24, 61–68 (2017).
Shulgina, Y. & Eddy, S. R. A computational screen for alternative genetic codes in over 250,000 genomes. eLife 10, e71402 (2021).
Keeling, P. J. Evolution of the genetic code. Curr. Biol. 26, R851–R853 (2016).
Baranov, P. V., Atkins, J. F. & Yordanova, M. M. Augmented genetic decoding: global, local and temporal alterations of decoding processes and codon meaning. Nat. Rev. Genet. 16, 517–529 (2015).
Keeling, P. J. & Leander, B. S. Characterisation of a non-canonical genetic code in the oxymonad Streblomastix strix. J. Mol. Biol. 326, 1337–1349 (2003).
Karpov, S. A. et al. Obligately phagotrophic aphelids turned out to branch with the earliest-diverging fungi. Protist 164, 195–205 (2013).
Lozupone, C. A., Knight, R. D. & Landweber, L. F. The molecular basis of nuclear genetic code change in ciliates. Curr. Biol. 11, 65–74 (2001).
Sanchez-Silva, R., Villalobo, E., Morin, L. & Torres, A. A new noncanonical nuclear genetic code: translation of UAA into glutamate. Curr. Biol. 13, 442–447 (2003).
Osawa, S. & Jukes, T. H. Codon reassignment (codon capture) in evolution. J. Mol. Evol. 28, 271–278 (1989).
Schultz, D. W. & Yarus, M. Transfer RNA mutation and the malleability of the genetic code. J. Mol. Biol. 235, 1377–1380 (1994).
Sengupta, S. & Higgs, P. G. A unified model of codon reassignment in alternative genetic codes. Genetics 170, 831–840 (2005).
Lukeš, J. et al. Trypanosomatids are much more than just trypanosomes: clues from the expanded family tree. Trends Parasitol. 34, 466–480 (2018).
Maslov, D. A. et al. Recent advances in trypanosomatid research: genome organization, expression, metabolism, taxonomy and evolution. Parasitology 146, 1–27 (2019).
He, F. & Jacobson, A. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu. Rev. Genet. 49, 339–366 (2015).
Baejen, C. et al. Transcriptome maps of mRNP biogenesis factors define pre-mRNA recognition. Mol. Cell 55, 745–757 (2014).
Kini, H. K., Silverman, I. M., Ji, X., Gregory, B. D. & Liebhaber, S. A. Cytoplasmic poly(A) binding protein-1 binds to genomically encoded sequences within mammalian mRNAs. RNA 22, 61–74 (2016).
Sladic, R. T., Lagnado, C. A., Bagley, C. J. & Goodall, G. J. Human PABP binds AU-rich RNA via RNA-binding domains 3 and 4. Eur. J. Biochem. 271, 450–457 (2004).
Alfonzo, J. D., Blanc, V., Estevez, A. M., Rubio, M. A. & Simpson, L. C to U editing of the anticodon of imported mitochondrial tRNA(Trp) allows decoding of the UGA stop codon in Leishmania tarentolae. EMBO J. 18, 7056–7062 (1999).
Wohlgamuth-Benedum, J. M. et al. Thiolation controls cytoplasmic tRNA stability and acts as a negative determinant for tRNA editing in mitochondria. J. Biol. Chem. 284, 23947–23953 (2009).
Paris, Z. et al. A mitochondrial cytidine deaminase is responsible for C to U editing of tRNA(Trp) to decode the UGA codon in Trypanosoma brucei. RNA Biol. 18, 278–286 (2021).
Hirsh, D. Tryptophan transfer RNA as the UGA suppressor. J. Mol. Biol. 58, 439–458 (1971).
Nenarokova, A. & Paris, Z. tRNAseq analysis of Blastocrithidia nonstop. figshare https://doi.org/10.6084/m9.figshare.17934200.v2 (2022).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004).
Van Haute, L., Powell, C. A. & Minczuk, M. Dealing with an unconventional genetic code in mitochondria: the biogenesis and pathogenic defects of the 5-formylcytosine modification in mitochondrial tRNA(Met). Biomolecules 7, 24 (2017).
Agris, P. F. et al. Celebrating wobble decoding: half a century and still much is new. RNA Biol 15, 537–553 (2018).
Beznosková, P., Gunisová, S. & Valášek, L. S. Rules of UGA-N decoding by near-cognate tRNAs and analysis of readthrough on short uORFs in yeast. RNA 22, 456–466 (2016).
Beznosková, P., Pavlíková, Z., Zeman, J., Echeverria Aitken, C. & Valášek, L. S. Yeast applied readthrough inducing system (YARIS): an in vivo assay for the comprehensive study of translational readthrough. Nucleic Acids Res. 47, 6339–6350 (2019).
Pineyro, D., Torres, A. G. & de Pouplana, L. R. In Fungal RNA Biology (eds Sesma, A. & von der Haar, T.) 233–267 (Springer, 2014).
Matheisl, S., Berninghausen, O., Becker, T. & Beckmann, R. Structure of a human translation termination complex. Nucleic Acids Res. 43, 8615–8626 (2015).
Brown, A., Shao, S., Murray, J., Hegde, R. S. & Ramakrishnan, V. Structural basis for stop codon recognition in eukaryotes. Nature 524, 493–496 (2015).
Blanchet, S. et al. New insights into stop codon recognition by eRF1. Nucleic Acids Res. 43, 3298–3308 (2015).
Eliseev, B., Kryuchkova, P., Alkalaeva, E. & Frolova, L. A single amino acid change of translation termination factor eRF1 switches between bipotent and omnipotent stop-codon specificity. Nucleic Acids Res. 39, 599–608 (2011).
Xue, H., Shen, W., Giege, R. & Wong, J. T. Identity elements of tRNA(Trp). Identification and evolutionary conservation. J. Biol. Chem. 268, 9316–9322 (1993).
Ulmasov, B., Topin, A., Chen, Z., He, S. H. & Folk, W. R. Identity elements and aminoacylation of plant tRNATrp. Nucleic Acids Res. 26, 5139–5141 (1998).
Sekine, S. et al. Major identity determinants in the “augmented D helix” of tRNA(Glu) from Escherichia coli. J. Mol. Biol. 256, 685–700 (1996).
Robertson, W. E. et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 372, 1057–1062 (2021).
Grybchuk, D. et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl Acad. Sci. USA 115, E506–E515 (2018).
Chin, J. W. Expanding and reprogramming the genetic code. Nature 550, 53–60 (2017).
Wang, J. et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. Nature 604, 343–348 (2022).
Janssen, B. D., Diner, E. J. & Hayes, C. S. Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Methods Mol. Biol. 905, 291–309 (2012).
Grentzmann, G., Ingram, J. A., Kelly, P. J., Gesteland, R. F. & Atkins, J. F. A dual-luciferase reporter system for studying recoding signals. RNA 4, 479–486 (1998).
Muhlrad, D. & Parker, R. Recognition of yeast mRNAs as “nonsense containing” leads to both inhibition of mRNA translation and mRNA degradation: implications for the control of mRNA decapping. Mol. Biol. Cell 10, 3971–3978 (1999).
Loughran, G., Howard, M. T., Firth, A. E. & Atkins, J. F. Avoidance of reporter assay distortions from fused dual reporters. RNA 23, 1285–1289 (2017).
Ross, R., Cao, X., Yu, N. & Limbach, P. A. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods 107, 73–78 (2016).
Beznosková, P. et al. Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS Genet. 9, e1003962 (2013).
Kouba, T. et al. Small ribosomal protein RPS0 stimulates translation initiation by mediating 40S-binding of eIF3 via its direct contact with the eIF3a/TIF32 subunit. PLoS ONE 7, e40464 (2012).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Nenarokova, A., Záhonová, K. & Nenarokov, S. The high-throughput sequencing datasets. figshare https://doi.org/10.6084/m9.figshare.21401541 (2022).
Nenarokova, A., Záhonová, K. & Nenarokov, S. Additional data and analyses. figshare https://figshare.com/projects/tRNA_anticodon_stem_length_variations_are_critical_for_stop_codon_reassignment/129167 (2022).
Nenarokov, S. & Nenarokova, A. Seraff/blasto: annotator & utilities for Blastocrithidia project (v1.0.2). Zenodo https://doi.org/10.5281/zenodo.7116082 (2022).
Potěšil, D. MS analysis of B. nonstop proteins. figshare https://doi.org/10.6084/m9.figshare.20105417.v2 (2022).
Acknowledgements
We thank M. Tesařová (Institute of Parasitology) for help with electron microscopy, O. Namy (Université Paris-Sud) for providing the eRF1 mutant strains, and D. Potěšil and Z. Zdráhal (CEITEC) for help with MS analyses. This work was supported by the Czech Science Foundation grants 18-15962S and 22-14356S (to J.L. and V.Y.), 20-11585S (to Z. Paris) and 20-00579S (to L.S.V.), the Charles University Grant Agency project GAUK 1192819 (to Z. Pavlíková), the Czech Ministry of Education ERD Funds 16_0000759 (to V.Y., Z. Paris and J.L.), the Gordon and Betty Moore Foundation GBMF no. 9354 (to J.L.) and the Praemium Academiae grant provided by the Czech Academy of Sciences (to L.S.V.). Supporting services were supplied by the project LM2018140, ERD Funds 18_046/0015974 and Czech BioImaging LM2018129.
Author information
Authors and Affiliations
Contributions
A.N., E.H., Z. Paris, L.S.V. and J.L. conceived the study and designed the research. A.K., Z. Pavlíková, A.N., J.V., A.R., P.M., K.Z., S.N., I.M.D., E.H., R.L.R., V.Y., P.B. and Z. Paris carried out the experiments and analysed the data. A.N., Z. Pavlíková, Z. Paris, L.S.V. and J.L. wrote the manuscript. L.S.V. served as the lead contact, completed and submitted the work, and handled the revision and production tasks.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Pavel Baranov, Olivier Namy and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Morphology of Blastocrithidia nonstop sp. nov.
Light microscopy (1–9), scanning (10–12) and transmission electron microscopy (13–20). The DAPI-stained (1–3) and Giemsa-stained (4–9) cultured cells (1–6) and parasites from the host insect hindgut (7–9). Promastigote flagellates (P) dominate in the culture (1, 4, 10–11), while the epimastigotes (E) are more frequent in the insect gut (7–9), although they are also present in the culture (4). The cyst-like amastigotes (C) are present in both the culture and host-derived smears (9). The nucleus and the kinetoplast DNA are labelled with arrow and arrowhead, respectively. The scanning electron micrographs show a highly variable length of the flagellum in promastigotes (10) and the cyst-like amastigotes (11 and 12). The longitudinal section thru the cell reveals a deep flagellar pocket (FP; 13 and 18), oval nucleus (N), elongated mitochondria (Mt), oval glycosomes (Gl), and numerous dense acidocalcisomes (arrowhead; 18). The cross- (15) and longitudinally-sectioned (16) external flagellum is equipped with a prominent paraflagellar rod (PR; see also 13 and 18), which is absent in the part of the flagellum within the flagellar pocket (14; see also 13 and 18). Note a thin and wide kinetoplast DNA disk (K; 13, 17, and 18), virus-like particles within the cytoplasm (V; 19), the regularly spaced subpellicular microtubules (SM; 13), and the cyst-like amastigotes (20). Scale bars: 10 µm (1, 4, 7–9, and 10), 5 µm (2, 3, 5, 6, and 11), 1 µm (12, 13, 18, and 20), 200 nm (14–17), and 100 nm (19); repeated 3-times with similar results.
Extended Data Fig. 2 The 18S rRNA phylogenetic tree and consequences of AT mutational shift in Blastocrithidia ancestor.
(a) The 18S rRNA-based maximum likelihood phylogenetic tree of the family Trypanosomatidae. The branches are collapsed at the generic level to highlight the inter-generic relationships. The tree was rooted with Paratrypanosoma confusum; asterisks mark branches with maximal statistical support (bootstrap values for maximum likelihood >90; Bayesian posterior probabilities >0.95); double-crossed branches are 50% of the original length; the scale bar denotes the number of substitutions per site. The subfamily Leishmaniinae is subdivided into the genera Crithidia, Leishmania sensu lato, and Leptomonas. The genus Blastocrithidia is highlighted in red, the principal insect host and/or vectors (fleas, flies, and true bugs) are depicted by pictograms, and three dixenous genera infecting humans or plants are in bold. The presence of the key components (UPF1 and UPF2) of the nonsense-mediated decay pathway, which eliminates mRNAs with premature stops, is mapped onto the tree. The pathway was lost by the predecessor of the trypanosomatid lineage after the separation of the genus Trypanosoma. (b) Scheme illustrating possible consequences of AT mutational shift in Blastocrithidia ancestor. Frequent GC-to-AT substitutions in Blastocrithidia ancestor could have led to the following consequences that we observe in B. nonstop genome: overall AT-rich genome; extreme AT-richness of intergenic regions with frequent TAA codons; appearance of in-frame stop codons. Possible UAG-to-UAA and UGA-to-UAA substitutions are evolutionary neutral, but allowed TGG-to-TGA, GAG-to-TAG, and GAA-to-TAA substitutions.
Extended Data Fig. 3 Proteomic analysis and principles of the structural annotation of B. nonstop.
(a) Proteomic proof of the stop codon reassignment in B. nonstop. Four protein-coding genes were selected as they contain all three in-frame stop codons covered by identified peptides from mass spectrometry (MS) analysis. Nucleotide sequences with stop codons in red and their conceptual translations are shown. Below that, identified peptides are highlighted in light-pink. Above the sequences, MS spectra of identified peptide(s) are shown. All identified peptides are available on figshare58. (b) Principles of the structural annotation of B. nonstop. The genes in B. nonstop were predicted based on the combination of evidence inferred from transcriptome coverage, trans-splicing sites, and the alignments of the reference trypanosomatid proteins. For the detailed protocols, see Methods.
Extended Data Fig. 4 GO-enrichment analysis and Blastocrithidia transcripts with UAA genuine stop codons.
(a) GO-enrichment analysis of genes devoid of reassigned stop codons. The pie chart summarizes the predicted functions of 228 proteins of B. nonstop without reassigned stop codons. The complete list of corresponding proteins with annotations is available in Source Data Extended Data Fig. 4. (b) UAA is the only genuine stop codon in transcripts from Blastocrithidia sp. ex Lygus hesperus. Shown are open reading frames from Blastocrithidia sp. ex L. hesperus that were previously predicted with premature ends1. The alignment with B. nonstop revealed that all of them can be extended until the next UAA stop codon. Nucleotide identities are highlighted by white font on black background.
Extended Data Fig. 5 Distribution of codons in 3′ UTRs of B. nonstop transcripts in three coding frames.
The graph shows the summary counts for each of 64 codon triplets in 100 triplets (300 nt) after the stop codon in 1,569 B. nonstop transcripts calculated in three coding frames. Frame 1 corresponds to the gene coding frame, while frames 2 and 3 are shifted by 1 and 2 nucleotides, respectively.
Extended Data Fig. 6 Stop codon context analysis and schematic of the dual luciferase assay.
(a) GC-content around the termination codon position in the 3′ ends of protein-coding genes in trypanosomatids. The GC distribution in the B. nonstop genome dramatically differs inside of the coding regions versus the intergenic regions, in contrast to other trypanosomatids. The former are more GC-rich, however, immediately after the 3′end of genes the genome sequences become substantially more A+T-rich. (b) Sequence logo for first 100 nt after the stop codon. A is the most abundant nucleotide in the first ~39 nt past the stop codon with T taking over afterwards. Equiprobable abundance of As and Ts in the first 40 nucleotides after stop codon as a null hypothesis was rejected with the two-tailed p-value <0.0001; χ²1 = 1218.98. (c) Schematic representation of the dual luciferase assay. A cassette composed of an in-frame UGA codon positioned between the Firefly and Renilla luciferase genes was electroporated into T. brucei or S. cerevisiae expressing either 4- or 5-bp-long variants of tRNATrpCCA.
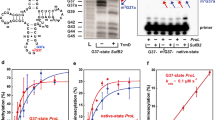
Extended Data Fig. 7 Detection and charging of tRNATrpCCA variants expressed in T. brucei.
(a) Increased gene dosage of all analyzed tRNATrp variants substantially increases their cellular levels in vivo. Total RNA was extracted from T. brucei strains expressing plasmid-borne high copy 4-bp vs. 5-bp-long tRNATrp (hc tW) variants from S. cerevisiae, B. nonstop, T. brucei, and C. magnum (indicated at the top of each panel). Ten µg of RNA was resolved by urea-PAGE, followed by the northern blot analysis using 32P-labeled oligonucleotides specific for tRNA variants; 5.8S rRNA was used as a loading control. Repeated 3 times with similar results; for gel source data, see Supplementary Fig. 1. (b) All expressed tRNA variants show an equal level of aminoacylation. Samples were prepared as in panel a, except that acidic conditions were applied to protect charged tRNAs. Aliquots of RNA samples were subjected to deacylation (labelled as “base +”). Ten µg of acylated or deacylated RNA samples were resolved on the acid-urea 12% polyacrylamide gel, transferred onto a nylon membrane and hybridized with 32P-labeled oligonucleotides specific for given tRNA variants; 5.8S rRNA was used as a loading control. The nature of hydrogen bonding between bases of the anticodon stem 5th base pair (26:42) is indicated by a vertical black line (base paring) or a red cross (no paring). Repeated 3 times with similar results; for gel source data, see Supplementary Fig. 1, for quantifications, see Source Data Extended Data Figs. 7 and 9.
Extended Data Fig. 8 Control stop codon readthrough analysis.
(a-c) The “anticodon stem length” effect is specific only for UGA. (a) T. brucei cells were transformed with empty vector (EV) or tRNATrpCCA (tW) with 4-bp or 5-bp AS versions from B. nonstop and processed for UAG or UAA readthrough measurements as described in Methods. Readthrough values were normalized to the control cell line (containing dual-luciferase cassette without in-frame stops). The nature of hydrogen bonding between bases of the anticodon stem 5th base pair (26:42) is indicated by a vertical black line (base paring) or a red cross (no paring). Each box in the box-and-whisker plot is represented by n = 9 readthrough values (3 individual experiments each including n = 3 biological replicates). The mean value is marked (+); whiskers range from minimal to maximal values. Statistical significance was determined by the unpaired, two-tailed Welch’s t test; **** p < 0.0001; *** p < 0.001; * p < 0.05; ns = non-significant. (b-c) The yeast strain H541 was transformed with a corresponding dual luciferase readthrough reporter construct YEp-R/T-UAGC-L (panel b) or YEp-R/T-UAAC-L (panel c) together with EV or a given high copy tRNATrp variant or a control, readthrough-inducing yeast tRNATyr (tY). The resulting transformants were grown in synthetic media, processed for stop codon readthrough measurements as described in Methods, and analyzed and plotted as in panel a; **** p < 0.0001; *** p = 0.0002; * p < 0.03; n = 15 values (3 individual experiments each including 5 biological replicates; see also raw data of Firefly and Renilla measurements in Source Data Figs. 3, 4 and Extended Data Fig. 8). (d) The “anticodon stem length” effect is independent of amino acid starvation stress. The yeast strain ZH252 was transformed with a corresponding dual luciferase readthrough reporter construct YEp-R/T-UGAC-L together with an EV or a given high copy tRNATrp variant. The resulting transformants were grown in synthetic media to an O.D. of ~1 and either treated with 3-AT (3-amino-1,2,4-triazole; final concentration 10 mmol/l) for 6 h or not (nt). Subsequently, both cultures were processed for stop codon readthrough measurements as described in Methods, and analyzed and plotted as in panel a; n = 10 values (2 individual experiments each including 5 biological replicates; see also raw data of Firefly and Renilla measurements in Source Data Figs. 3, 4 and Extended Data Fig. 8). (e) The nature of the UGA stop codon tetranucleotide does not impact on the difference between readthrough levels displayed by 4-bp-long vs. 5-bp-long anticodon stem tRNATrp variants from various eukaryotes. The yeast strain H541 was transformed with a corresponding dual luciferase readthrough reporter construct (from left to right: YEp-R/T-UGAC-L; YEp-R/T-UGAA-L; YEp-R/T-UGAG-L; YEp-R/T-UGAU-L or YEp-R/T-CAAC-L) together with EV or a given high copy tRNATrp variant. The resulting transformants were grown in synthetic media and processed for stop codon readthrough measurements as described in Methods, and analyzed and plotted as in panel a; n = 9 values (3 individual experiments each including 3 biological replicates; see also raw data of Firefly and Renilla measurements in Source Data Figs. 3, 4 and Extended Data Fig. 8). (f) Shortening of the anticodon stem of S. cerevisiae Cys and Arg tRNAs from 5 bp to 4 bp has no effect on UGA-TMV readthrough levels in yeast. The yeast strain H541 was transformed with a corresponding dual luciferase readthrough reporter construct (YEp-R/T-UGA-TMV-L or YEp-R/T-CAAC-L) and subsequently with EV or a given high copy of either Cys or Arg tRNA variant. The resulting transformants were grown in synthetic media, processed for stop codon readthrough measurements as described in Methods, and analyzed and plotted as in panel a; n = 12 values (3 individual experiments, including 3, 4 and 5 biological replicates, respectively; see also raw data of Firefly and Renilla measurements in Source Data Figs. 3, 4 and Extended Data Fig. 8). (g) The sup45S67G substitution of yeast eRF1 strictly restricts UGA decoding. The yeast strains bearing wild type eRF1 (sup45Δ + SUP45) or the S67G eRF1 substitution (sup45Δ + sup45S67G) were transformed with a corresponding dual luciferase readthrough reporter construct (YEp-R/T-UGAC-L; YEp-R/T-UAAC-L or YEp-R/T-UAG-L). The resulting transformants were grown in synthetic media, processed for stop codon readthrough measurements as described in Methods, and analyzed and plotted as in panel a; n = 11 values (3 individual experiments, including 3, 4 and 4 biological replicates, respectively; see also raw data of Firefly and Renilla measurements in Source Data Figs. 3, 4 and Extended Data Fig. 8). Statistical significance was determined by the unpaired, two-tailed Welch’s t test; **** p < 0.0001; *** p < 0.001.
Extended Data Fig. 9 Expression levels and charging efficiency of all tRNAs tested in S. cerevisiae in this study.
(a) Increased gene dosage of all analyzed tRNATrp variants substantially increases their cellular levels in vivo. Total RNA samples were isolated from the yeast strain H541 expressing plasmid-borne high copy 4-bp vs. 5-bp-long tRNATrp (tW) variants from S. cerevisiae, B. nonstop, T. brucei, or C. magnum (indicated at the top of each panel). 0.5 or 1 µg of total RNA were resolved on the Criterion Precast gel, transferred onto a nylon membrane and hybridized with a particular DIG-labeled probe against tRNATrp. The 5.8S rRNA was used as a loading control. Repeated 3 times with similar results; for gel source data, see Supplementary Fig. 1. (b) Increased gene dosage of all analyzed tRNATrp variants (4 bp vs. 5 bp) shows an equal level of aminoacylation. Total RNA samples were prepared as in panel a, except that acidic conditions were applied to protect charged tRNAs. Aliquots of RNA samples were subjected to deacylation (labelled as “base +”). 0.5 µg of acylated or deacylated RNA samples were resolved on the acid-urea 12% polyacrylamide gel, transferred onto a nylon membrane and hybridized with a particular DIG-labeled probe against tRNATrp. The 5.8S rRNA was used as a loading control. The nature of hydrogen bonding between bases of the anticodon stem 5th base pair (26:42) is indicated by a vertical black line (base paring) or a red cross (no paring). Repeated 3 times with similar results; for gel source data, see Supplementary Fig. 1, for quantifications, see Source Data Extended Data Figs. 7 and 9.
Extended Data Fig. 10 Alignment of eRF1 from various species.
Species with non-canonical genetic codes are highlighted by a grey background. X in the sequences represents in-frame stop codons. Blue and yellow boxed alignment regions correspond to the conserved motifs required to recognize stop codons by eRF1. All available Blastocrithidia spp. exhibit critical substitution Ser70Gly (numbering according to the human sequence; Ser67 in S. cerevisiae or Gly74 in B. nonstop). All other motifs are unchanged.
Supplementary information
Supplementary Information
This file contains Supplementary Discussion, Taxonomic Summary, Methods, Tables 1–4 and References.
Supplementary Information 2
Supplementary spreadsheet containing the total nucleoside analysis of purified B. nonstop tRNATrpCCA by MS.
Supplementary Fig. 1
The uncropped gel source data for Figs. 2–4 and Extended Data Figs 7 and 9.
Supplementary Data 1
Fasta file containing the sequences of 70 tRNA genes of B. nonstop.
Supplementary Data 2
Fasta.tree file containing the full phylogenetic tree of tRNAGlu of B. nonstop and related trypanosomatids.
Supplementary Video 1
UGA reassignment in Blastocrithidia nonstop. When a premature, in-frame UGA stop codon enters the ribosomal A-site, eukaryotic release factor 1 (eRF1) of all organisms with a canonical genetic code recognizes it and terminates translation. Tryptophanyl tRNA (Trp-tRNATrp), which is nearcognate to UGA (with the third base wobble), is very inefficient at recognizing it. However, the newly isolated trypanosomatid B. nonstop with all three stop codons reassigned to sense codons has accumulated two mutations that enable very effective reading of UGA as tryptophan, allowing translation to continue. These include the shortening of the tRNATrp anticodon stem from 5 to 4 base pairs, which greatly enhances Trp-tRNATrp accommodation in the A-site occupied by UGA, and the Ser74Gly substitution in the stop codon recognition motif of eRF1, which severely restricts UGA recognition.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kachale, A., Pavlíková, Z., Nenarokova, A. et al. Short tRNA anticodon stem and mutant eRF1 allow stop codon reassignment. Nature 613, 751–758 (2023). https://doi.org/10.1038/s41586-022-05584-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05584-2
This article is cited by
-
In silico prediction of the metabolism of Blastocrithidia nonstop, a trypanosomatid with non-canonical genetic code
BMC Genomics (2024)
-
tRNA therapeutics for genetic diseases
Nature Reviews Drug Discovery (2024)
-
Extended stop codon context predicts nonsense codon readthrough efficiency in human cells
Nature Communications (2024)
-
Shining the spotlight on the neglected: new high-quality genome assemblies as a gateway to understanding the evolution of Trypanosomatidae
BMC Genomics (2023)
-
No stopping with a short-stem transfer RNA
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
