
Abstract
Filoviruses, including Ebola virus, pose an increasing threat to the public health. Although two therapeutic monoclonal antibodies have been approved to treat the Ebola virus disease1,2, there are no approved broadly reactive drugs to control diverse filovirus infection. Filovirus has a large polymerase (L) protein and the cofactor viral protein 35 (VP35), which constitute the basic functional unit responsible for virus genome RNA synthesis3. Owing to its conservation, the L–VP35 polymerase complex is a promising target for broadly reactive antiviral drugs. Here we determined the structure of Ebola virus L protein in complex with tetrameric VP35 using cryo-electron microscopy (state 1). Structural analysis revealed that Ebola virus L possesses a filovirus-specific insertion element that is essential for RNA synthesis, and that VP35 interacts extensively with the N-terminal region of L by three protomers of the VP35 tetramer. Notably, we captured the complex structure in a second conformation with the unambiguous priming loop and supporting helix away from polymerase active site (state 2). Moreover, we demonstrated that the century-old drug suramin could inhibit the activity of the Ebola virus polymerase in an enzymatic assay. The structure of the L–VP35–suramin complex reveals that suramin can bind at the highly conserved NTP entry channel to prevent substrates from entering the active site. These findings reveal the mechanism of Ebola virus replication and may guide the development of more powerful anti-filovirus drugs.
Similar content being viewed by others

Main
Infections with filoviruses such as Ebola virus (EBOV) and Marburg virus can cause severe clinical symptoms, including haemorrhagic fever and multiorgan failure4. Before December 2013, 35 outbreaks of filovirus disease had been recorded in remote African regions with infrequent spillover from animals to humans5. Since December 2013, atypically extensive ebolavirus disease outbreaks, including an unprecedented outbreak from 2013 to 2016 in West African countries, an outbreak from 2018 to the present in the Democratic Republic of the Congo, and a more recent brief outbreak in Guinea, have a profound impact on public health systems. At least 14,000 fatalities from ebolavirus disease were reported between December 2013 and August 2020 (ref. 6). The resurgence of EBOV in 2021 in Guinea suggests a persistent infection with reduced replication or a period of latency in humans7. Although filoviruses were initially thought to occur exclusively in Africa, recent studies have revealed that they are more widely distributed, including in Asia and Europe8,9. Moreover, previously unknown filovirus species have been reported10, and the genetic diversity of filoviruses and other potential zoonotic viruses may be greater than previously recognized11. This presents a great challenge for the development of virus-targeting prophylactic and therapeutic countermeasures.
Filoviruses, including ebolavirus, are non-segmented negative-sense RNA viruses (nsNSV) with seven genes, and belong to the order Mononegavirales12. The viral RNA genome is encapsidated by the nucleoprotein (NP) and is further associated with the polymerase complex consisting of the large (L) protein, the cofactor viral protein 35 (VP35) and the transcription activator VP30, forming the ribonucleoprotein (RNP) complex12. In virions, the RNP complex interacts with the nucleocapsid-associated VP24, and is surrounded by the matrix protein VP40, which drives the morphogenesis and budding of virus particles13. Finally, the matrix layer is covered by the host cell-derived envelope in which the viral glycoprotein GP is embedded12. Filoviruses follow the typical life cycle of the cytoplasmically replicating nsNSVs, including multiple key processes such as cell entry, genome replication and transcription, morphogenesis and budding4,14.
In recent years, antiviral therapeutic approaches have targeted the different processes of ebolavirus life cycle. The most developed area of antiviral development is focused on the entry process, including monoclonal antibodies (ZMapp, mAb114 and REGN-EB3) and small molecule inhibitors1,2,15,16,17. In a randomized clinical trial, the overall mortality rates among patients who received REGN-EB3 and mAb114 were 33.5% and 35.1%, respectively, much lower than that for patients treated with ZMapp (49.7%)18. Moreover, the viral RNA synthesis machinery has been a promising target for the design of broadly reactive drugs against other viral diseases19,20. Nucleoside analogue drugs (remdesivir, favipiravir and BCX4430) and small interfering RNA (siRNA)-based degradation of viral mRNAs (TKM-Ebola) targeting the viral RNA synthesis machinery have been designed and tested to combat ebolavirus infection21,22,23,24. However, these small molecule inhibitors have displayed poor clinical outcomes. The mortality of remdesivir-administered patients was 53.1% in a randomized clinical trial, much higher than that of antibody-treated groups18. In addition, the nucleoside analogue drug favipiravir showed significant effectiveness in protecting mice from lethal EBOV challenge, but provided low protection in a clinical trial25,26. The mortality of patients receiving TKM-Ebola was 79%, and the drug did not improve survival compared with historic mortality27. Although REGN-EB3 and mAb114 have been approved by the US Food and Drug Administration, these antibodies only provide protection against Zaire ebolavirus. Therefore, there is an urgent demand for broadly reactive drugs to control infection with diverse filoviruses. However, the current knowledge gap in the understanding of the filovirus RNA synthesis machinery is hampering the development of such drugs.
The filovirus RNP complex is responsible for viral RNA synthesis, and the L–VP35 complex is the competent core unit for efficient RNA-dependent RNA polymerization from the viral RNP template3. The structures of L polymerase in complex with the cofactor phosphoprotein (P) from several mononegaviruses, including vesicular stomatitis virus (VSV), rabies virus, human metapneumovirus (HMPV), human respiratory syncytial virus (HRSV) and human parainfluenza virus (HPIV) have revealed two binding modes: P interacting with the N-terminal region of L polymerase or with the C-terminal region of L polymerase28,29,30,31,32. P serves as an indispensable cofactor for RNA synthesis, and it can tether L to the RNP complex and act as a chaperone to prevent the non-specific aggregation of nascent N protein with host RNA, preserving monomeric RNA-free N protein for RNP assembly among Mononegavirales33. The respiratory syncytial virus P protein also interacts with M2-134 and the cellular phosphatase PP135 to coordinate viral transcription. In addition to its P-like chaperone function, EBOV VP35 binds to double stranded RNA (dsRNA) and is crucial for host immune evasion36. However, little is known about the structural basis of the filovirus L–VP35 complex, which precludes the molecular understanding of filovirus RNA synthesis.
Overall structure of EBOV polymerase
We used the baculovirus expression system to co-express the EBOV L polymerase and VP35 proteins and obtained soluble complex protein that was suitable for structural and functional studies. We analysed the purified protein complex using size-exclusion chromatography, and SDS-PAGE and western blotting profiles showed that the L protein was easily degraded into two bands, with molecular weights of about 180 kDa and 150 kDa (Extended Data Fig. 1). A primer-extension assay using 11-mer RNA template and 4-mer RNA primer confirmed that the L–VP35 complex possessed RNA-dependent RNA polymerization activity. The RNA products were inhomogeneous with a small percentage of full-length product, and the majority were abortive products probably caused by early termination (Extended Data Fig. 1).
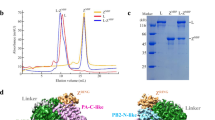
Using cryo-electron microscopy (cryo-EM), we determined structures of the L–VP35 complex and the VP35 oligomerization domain to resolutions of 3.0 Å and 3.4 Å, respectively (we refer to this as the state 1 conformation; Supplementary Figs. 1 and 2 and Supplementary Table 1). The final 3D reconstruction maps enabled us to build atomic models for residues 8–1383 of L and for the VP35 tetramer with different lengths (Fig. 1 and Supplementary Fig. 3). Although the L protein sequence identity is only about 25.5%, the overall ‘spoon-like’ structure of EBOV L–VP35 is similar to that of the HPIV5 L–P complex (Extended Data Fig. 2). The visible region of L polymerase contains an N-terminal domain (NTD), an RNA-dependent RNA polymerase (RdRp) domain and a GDP polyribonucleotidyltransferase (PRNTase) domain (Fig. 1). The RdRp domain of EBOV L folds into the canonical right-handed fingers–palm–thumb architecture observed in many RNA virus polymerase structures, containing six catalytic motifs (A–F) (Fig. 2a,b). The superposition of EBOV L and VSV L structures shows a root mean square deviation (r.m.s.d.) of 4.2 Å based on superimposition of Cα residues, illustrating the structural conservation of L polymerases during the evolution of Mononegavirales (Fig. 2c) and suggesting that RNA follows similar paths in nsNSV polymerases (Extended Data Fig. 3). However, we observed a filovirus-specific structural element consisting of a loop, which was absent in other mononegaviruses (Fig. 2d), owing to an insertion of ~30 residues in the NTD of L polymerase, roughly spanning residues 190 to 225 (Fig. 2e). Deletion of the insertion element abolishes the transcription activity of EBOV RNP (Extended Data Fig. 4), suggesting that this loop is essential for RNA synthesis. As in VSV L, the PRNTase domain of EBOV L has a large interface with the RdRp domain and is responsible for the 5′ capping of nascent viral mRNAs29,37. The structural organization of PRNTase domain highly resembles those of L proteins from other mononegaviruses and is supposed to have conserved sequence motifs (A′–E′). We used AlphaFold2 to predict the structure of the L protein and modelled the full-length EBOV L structure with bound VP35 tetramer (Extended Data Fig. 5). The putative model could be overlaid onto our structure, and showed the presence of a long loop (residues 1652–1761) between the connector domain and the methyltransferase domain, which may partially explain why the purified EBOV L protein tends to degrade.

a, Schematic diagram of the domain architecture of EBOV L and VP35. L protein can be divided into five regions. Structurally modelled domains are shown in colour: NTD, orange; fingers subdomain, blue; palm subdomain, red; thumb subdomain, green; PRNTase, cyan. The linkers, connector domain (CD), methyltransferase domain (MTase) and C-terminal domain (CTD) of L are not modelled in the structure, and are shown in white with dashed outline. The solved regions of the VP35 protomers vary in length and shown in different colours. The NTD of VP35 was not observed in all protomers owing to its flexibility. b,c, The cryo-EM density map (b) and atomic model (c) of the EBOV L–VP35 complex with coloured domains as depicted in a. The terminal residue numbers of the VP35 protomers are indicated.

a, The RdRp domain is shown as cartoon, with the fingers subdomain in blue, the palm subdomain in red, the thumb subdomain in green, and the remaining NTD in orange. The active site is highlighted with a red asterisk. The partial supporting helix is observed in the fingers subdomain. b, The same view as in a, with catalytic motifs highlighted. c, The superposition of the RdRp domains of the EBOV (coloured by subdomains as in a) and VSV (grey) (Protein Data Bank (PDB) ID: 5A22) L proteins highlights the difference in the NTD. d, Close-up view of the specific insertion region of the EBOV L RdRp domain. e, Sequence alignment of the above specific insertion region spanning residues 190 to 225. The sequence is unique to filoviruses compared with other nsNSVs. MARV, Marburg virus; LLOV, Lloviu cuevavirus; RABV, rabies virus; RSV, respiratory syncytial virus.
Previous structures of L polymerases from VSV and pneumoviruses (HRSV and HMPV) have revealed two different conformations of the catalytic chamber: the initiation state observed in VSV L polymerase, in which the priming loop from the PRNTase domain and the supporting helix from the RdRp domain largely occlude the central RNA-binding cavity37; and a possible elongation state observed in HRSV and HMPV L polymerases, in which the priming loop retracts completely into a cavity in the PRNTase domain and the supporting helix is not visible, leaving ample space for the dsRNA intermediate duplex28,30. In our structure of the free L–VP35 complex, we found that the priming loop (also referred to as motif B′ in the PRNTase domain), motif D′ of the PRNTase domain and the partial supporting helix are disordered, suggesting that these structural elements are flexible (Fig. 3a,b).

a–c, Overlay of EBOV L–VP35 complex structures in state 1 and state 2. a, The two structures could be superimposed with slight differences. For the L–VP35 complex in state 1, the motif B′ (also referred to as the priming loop) and D′ of the PRNTase domain, and the partial supporting helix of RdRp could not be modelled owing to their flexibilities; however, these regions are more stable and the main chain can be traced in state 2. b, Close-up view of the comparison of motifs B′ and D′, and the ends of unbuilt residues of motif B′ (magenta) and motif D′ (blue) are indicated by arrows related to the region outlined in a as indicated. c, Close-up view comparing the supporting helix and the ends of unmodelled residues of the supporting helix indicated by arrows, as circled in a. d, In state 2, the supporting helix flips outwards and moves away from the active site of RdRp, and three key residues of motif C in the RdRp domain form hydrogen bonds with the supporting helix to stabilize this conformation.
To further explore the elongation conformation of the L–VP35 complex, we incubated the free L–VP35 complex with template and primer RNAs in an enzyme reaction buffer and determined the complex structure to a resolution of 3.4 Å (we refer to this as the state 2 conformation; Supplementary Fig. 4). Although we do not observe clear density for the RNA in this L–VP35 complex structure, we were able to trace the complete priming loop (residues 1196–1216) and the supporting helix (residues from Ser610 to Thr623 are invisible in state 1 map) (Fig. 3a–d). The priming loop retracts completely into a cavity of the PRNTase domain, as seen in HRSV and HMPV L–P complex structures28,30 (Extended Data Fig. 6). The priming loop further stabilizes motif D′ (also referred to as the His–Arg (HR) motif), which consists of two catalytic residues—H1269 and R1270—critical for cap formation (Fig. 3a,b and Extended Data Fig. 6). This makes it possible for the flipping priming loop to approach the active site of the PRNTase domain, forming a compact conformation favourable for the capping function. The location of the supporting helix in the VSV L structure would clash with template-product dsRNA, indicating a necessary conformational change of this element during RNA elongation (Extended Data Fig. 7). In our EBOV L–VP35 structure, the supporting helix leaves from the central RNA-binding cavity and forms several hydrogen bonds with motif C to stabilize this conformation (Fig. 3d and Extended Data Fig. 7).
Interactions between L and VP35
The EBOV VP35 oligomerization domain can assemble into trimer or tetramer38 structures (Extended Data Fig. 8). Our L–VP35 complex structure shows that tetrameric EBOV VP35 binds to the L polymerase (Fig. 1 and Extended Data Fig. 9a). Large variations in conformation were observed in each of the four VP35 monomers (VP35a, VP35b, VP35c and VP35d) (Extended Data Fig. 9b). Clear densities enabled the tracing of VP35a residues 82–149, which does not interact with L, as well as VP35b 80–146, VP35c 81–179 and VP35d 81–340, which do interact with L (Fig. 1, Extended Data Fig. 9b and Supplementary Table 3). The oligomerization region (residues 83–145) of VP35 forms a coiled-coil structure of four helices to stabilize the tetramer, which is further stabilized by antiparallel β-sheets formed by VP35c residues 145–148 and VP35d residues 174–177, and hydrogen bonds formed between VP35c Arg151 and VP35d Gln168 and Pro169, and between VP35c Glu160 and VP35d Arg151 (Extended Data Fig. 9c). The VP35–L interaction is defined mainly by hydrogen bonds, electrostatic interactions and van der Waals contacts, contributed minimally by VP35b and largely by VP35c and VP35d (Fig. 4, Extended Data Fig. 10 and Supplementary Table 3). Residues Thr127 and Ser130 of VP35b form two hydrogen bonds with the Asn434 from the fingers subdomain of L, and VP35b Arg133 forms a hydrogen bond with Asp432 of L (Fig. 4b and Supplementary Table 3). van der Waals contacts are also formed between Thr127 and Ser130 of VP35b and Asn434 of L (Supplementary Table 3). About 30 additional residues are observed after the oligomerization region in VP35c compared with VP35a and VP35b, which form a β-strand that interacts with an antiparallel β-strand in VP35d and a α-helices connected by the loops (Extended Data Fig. 9b,c). The major interactions between VP35c and L contain five hydrogen bonds and a number of van der Waals contacts. Among them, residues Met147 and Thr149 of VP35c interact with the main chain of Leu399 and Lys397 from the fingers subdomain through two hydrogen bonds, respectively (Fig. 4c and Supplementary Table 3). Moreover, the side chain of Thr153 of VP35c forms hydrogen bond with Glu643 of L to further stabilize the interaction (Fig. 4c and Supplementary Table 3). For VP35d, all the regions (residues 146–340) after the oligomerization domain were traced, consisting of an α-helix, a β-strand, two consecutive α-helices and the rigid C-terminal RNA-binding (or interferon-inhibitory) domain (RBD/IID) which can inhibit induction of type I α- or β-interferon by sequestering dsRNA byproducts of viral replication and by interacting with the members of innate receptor pathway36. VP35d contacts residues from the fingers subdomain, palm subdomain and NTD of L polymerase (Fig. 4, Extended Data Fig. 10 and Supplementary Table 3). The region between the oligomerization domain and the C-terminal RBD/IID of VP35d forms a long loop structure that lies on top of the hole through which NTPs enter the active site chamber (Fig. 4d and Extended Data Figs. 3 and 10). This long loop is further stabilized by three hydrogen bonds formed by Leu209, Glu211 and Gly215 of VP35d interacting with Lys778 from the palm subdomain and Arg315 and Gln322 from the NTD, respectively. The C-terminal RBD/IID of VP35d contacts the NTD via two α-helices and leaves its positively charged RNA-binding cavity exposed to the solvent (Figs. 1 and 4a).

a, The overall structure of the L–VP35 complex in cartoon view. The L protein is coloured by subdomains as indicated. The VP35 protomers are shown in different colours. The interaction interfaces between EBOV L and VP35 can be distributed into four main regions, labelled I, II, III and IV. b–e, Close-up views of atomic interactions between L and VP35 in regions I (b), II (c), III (d) and IV (e). Key residues are shown as sticks. Hydrogen bonds are represented by yellow dashed lines.
Among the reported L–P complex structures, the EBOV L–VP35 structure most resembles the HPIV5 L–P complex. However, the resolution (4.3 Å) of the HPIV5 L–P structure is too low to accurately determine binding sites between L and P protein. Therefore, we analyse the conservation of these L protein recognition sites on the basis of the EBOV L–VP35 and HRSV L–P complex structures. Overlaying both structures, we find that the binding sites between EBOV L and VP35 are similar to those of HRSV L and P, involving the fingers, palm and NTD of L (Extended Data Fig. 10b). However, the critical residues contributing to the main interactions vary among the different nsNSV polymerase complexes (Extended Data Fig. 10c).
Inhibition of EBOV polymerase by suramin
Suramin is a potent inhibitor of Chikungunya virus and EBOV cell entry, but its mechanism of action remains largely unknown39. Suramin has also been reported to target the viral polymerases of norovirus and SARS-CoV-240,41. Here we show that suramin is also a potent inhibitor of the EBOV L–VP35 polymerase complex. Addition of 32 μM suramin completely abolished the polymerization activity of the EBOV L–VP35 complex in an in vitro enzymatic assay, with a half-maximal inhibition concentration (IC50) of about 11 μM (Fig. 5a and Extended Data Fig. 11b). Furthermore, in a cell-based assay using a stable EBOV replicon cell line, suramin potently inhibited EBOV replication, with a half-maximal effective concentration (EC50) of about 0.4 μM (Fig. 5b). The concentration of suramin required to reduce cell viability by 50% (CC50) is over 200 μM (Extended Data Fig. 11c), indicating its relatively low cytotoxicity and high selectivity index (SI > 500).

a, Suramin inhibits the replication activity of the EBOV L–VP35 complex in an enzymatic assay, with an IC50 value of 11.16 µM. b, Suramin inhibits EBOV RNP activity in EBOv-GLuc-Hyg replicon cells with an EC50 value of 0.4 µM. Data in a,b are mean ± s.d. of three or four independent experiments. c, Overall structure of the L–VP35–suramin complex. The L–VP35 complex is shown in cartoon representation with the same colours as in Fig. 4. Suramin is shown as a stick model in purple with a dashed outline. Top left, close-up view of the suramin-binding site in surface representation, showing that suramin is located in the NTP entry channel. d–f, Atomic interactions between suramin and EBOV L. Key residues that are responsible for suramin binding are shown as sticks and coloured according to subdomain. Hydrogen bonds and salt bridges are shown as yellow and green dashed lines, respectively.
To uncover the structural basis for suramin inhibition of EBOV RNA polymerase, we determined the cryo-EM structure of the EBOV L–VP35–suramin complex at 3.3 Å resolution (Supplementary Fig. 5 and Supplementary Table 2). The overall structure of the L–VP35–suramin complex resembles that of the apo L–VP35 complex, but residues 1,000–1,400 could not be modelled owing to the weak density in this region, indicating that suramin binding may make the PRNTase domain more flexible. On the basis of the clear density, we built an atomic model of the head half and ring D′ of suramin, spanning the putative NTP entry channel in the RdRp domain (Fig. 5c and Supplementary Fig. 5).
The chemical structure of suramin (C51H40N6O23S6) is a symmetric polysulfonated naphthylurea, which includes four benzene rings, two naphthalenes and six sulfonic acid groups with a urea linker at the centre (Extended Data Fig. 11a). In the electron microscopy density map, we could clearly see the head half of the suramin molecule occluding the NTP entry channel (Fig. 5c), and the key interactions include hydrogen bonds, electrostatic and hydrophobic interactions with conserved NTD and RdRp residues. The sulfonate at position 3 forms a salt bridge with the side chain of Lys392 and a hydrogen bond with the side chain of His392 from the fingers subdomain (Fig. 5d). The sulfonate at position 5 forms a salt bridge with the side chain of Lys293 from the NTD and a hydrogen bond with the main chain of Val559 from the fingers subdomain (Fig. 5e), whereas the sulfonate at position 1 has little interaction with the polymerase. A hydrophobic residue (Phe793) in palm subdomain undergoes substantial conformational change, and together with Phe319 and Met323 provides a stable hydrophobic cavity to accommodate benzene ring D of suramin, further tethering the suramin in the NTP entry channel (Fig. 5f). For the tail half of suramin, the density is only visible at a very low threshold, but we can model the full suramin molecule on the basis of the density and stereochemistry of suramin; the tail half of suramin occupies the space where the nascent RNA would go, where the surrounding basic residues may form electrostatic interactions with the negatively charged sulfonate groups of suramin (Extended Data Fig. 11d–g).
Discussion
The high-resolution structures of the EBOV polymerase complex presented here provide first structural characterization of EBOV L protein in complex with VP35. VP35 is functionally analogous to P proteins of other mononegaviruses, which not only acts as a cofactor of L protein but also facilitates nucleocapsid formation by binding the monomeric NP to prevent premature and non-specific assembly42,43. In addition, the binding of VP35 to L protein can prevent the self-aggregation of L protein caused by hydrophobic interactions, as there are several hydrophobic patches in the VP35-binding regions (Supplementary Table 3). Moreover, VP35 and VP24 are required for the proper condensation of filovirus nucleocapsid, which acts as a template for genome transcription and replication44. Structurally, VP35 is composed of an NTD, an oligomerization domain and C-terminal domain connecting with a linker. The NTD is responsible for interacting with free NP and the LC8 subunit of cellular dynein, which is also involved in viral RNA synthesis42,43,45, however, this region is not visible in our cryo-EM map, indicating that it is flexible and may adopt distinct conformations to bind different partners. In addition to functioning as an interferon antagonist36, the C-terminal domain can also interact with the NP protein46, and the L–VP35 complex structure presented here reveals that the three free C-terminal domains may function as anchors to underpin the L protein moving along the nucleocapsid during genome transcription and replication.
Mononegavirus L protein contains all the domains required for RNA synthesis, capping and methylation. The switch from initiation to elongation involves significant conformational changes of key elements, including the supporting helix and priming loop. Previous studies have revealed the different conformations of the priming loop28,30,32,37, but could not determine the conformation of the supporting helix in the non-initiation state. The outward state of the supporting helix that we captured in this study contributes towards the understanding of the conformational dynamics during the catalytic cycle. A filovirus-specific insertion element was observed in the NTD of EBOV L, which is essential for its transcription activity, as revealed by the replicon assay. A previous study reported that the NTD of the VSV L protein is essential for viral genome transcription47. Thus, we deduce that the NTD of the EBOV L protein might be involved in genome transcription. Notably, compared with other nsNSVs, filoviruses, including EBOV, have a unique cofactor protein VP30, which is crucial for transcription initiation; we therefore suspect that the insertion element in NTD may evolve with VP30 to regulate the transcription process.
The reoccurrence of EBOV in 2018–2020 in the Democratic Republic of the Congo and resurgence of EBOV in 2021 in Guinea is evidence of the need for economical and effective drugs to treat the disease. Suramin is a multifunctional drug with inhibition activity against parasites, viruses and cancers48. The L–VP35–suramin structure indicates that suramin can bind to the RdRp domain, blocking the NTP entry channel and occupying the space that the nascent RNA chain would occupy, thus hindering polymerization activity (Fig. 5 and Extended Data Fig. 11). Previous studies have shown that the suramin-binding sites in norovirus polymerase overlap the proposed NTP entry channel40, and in SARS-CoV-2 RdRp, they occupy the chamber where the RNA template and primer go41 (Extended Data Fig. 12). In both of the above structures, only half of a suramin molecule was seen, owing to the other half moiety remaining unrestricted by protein. In this study, we could trace the whole suramin molecule, with clear density at the head moiety and relatively weak density at the tail part, which could be stabilized by the surrounding positively charged residues. Our result suggests that the NTP entry channel would be an attractive target for the development of antiviral drugs.
Although suramin has been used to treat African sleeping sickness for about 100 years, we noted that the relatively large molecular weight and multiple negative charges could be shortcomings for clinical use. In addition, suramin also binds to cellular DNA polymerase, primase, helicases and transcription factors48,49,50, which might lead to off-target effects. The details of the interaction between suramin and L protein described here could guide optimization of the molecule to increase its affinity and specificity, such as by enhancing the hydrophobic interaction between ring D of suramin and the hydrophobic residues of L protein, or reducing the negative charge by deleting the sulfonate at position 1, which provided few interactions. In sum, the findings for suramin can be used as a proof of principle for the development of a broad-spectrum inhibitor.
In summary, we determine the structure of EBOV L protein in complex with tetrameric VP35 and capture a previously missing structural snapshot of mononegavirus polymerase in the non-initiation state, with a supporting helix and priming loop getting away from the active site. Of note, we demonstrate that suramin could inhibit EBOV L–VP35 activity and elucidate the structural basis of suramin binding to RdRp, suggesting the feasibility of developing non-nucleoside antiviral drugs to treat filovirus infection.
Methods
Protein expression and purification
The coding sequences of EBOV L (GenBank: AHX24663.1) and VP35 (GenBank: AHX24647.1) were synthesized and codon-optimized for Bac-to-Bac expression system using pFastBac Dual transfer vector. The sequences of EBOV L and VP35 were fused with an N-terminal 2×Strep tag and a C-terminal His tag, respectively. The purification process is performed as previously reported51. Insect SF9 cells (11496015; Invitrogen) and High Five cells (B85502; Invitrogen) were used to prepare the recombinant baculoviruses and express EBOV L–VP35 complex protein, respectively. Both cell lines are routinely maintained in our lab and tested negative for mycoplasma contamination. The High Five cells were harvested by centrifugation (3,000 rpm, 4 °C, 10 min) at 48 h post-infection and lysed by sonication in buffer A containing 25 mM HEPES, pH 7.8, 1 M NaCl, 5% glycerol, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 0.01% Tween 20, 4 mM MgCl2 and 1 mM Phenylmethanesulfonyl fluoride (PMSF). Cell debris were discarded using super-centrifugation (30,000 rpm, 4 °C, 1 h) and 0.22 μm filter to collect supernatant. The protein solution was loaded into a HisTrap column (GE Healthcare, 5 ml) with equilibrium of buffer B containing 25 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 4 mM MgCl2, and the bound protein was eluted using 300 mM imidazole supplemented in buffer B. The eluted fractions were pooled and subjected to further purification using StrepTrap column (GE Healthcare, 5 ml). The EBOV L–VP35 complex protein was eluted using 2.5 mM d-desthiobiotin dissolved in buffer B. The protein was concentrated with a 100 kDa-cutoff Millipore Ultra centrifugal filter, and then loaded onto a size-exclusion chromatography (GE Healthcare, Superdex 200) with buffer B. The purified L–VP35 complex protein was confirmed by western blot using mouse monoclonal antibody against Strep tag (Easybio BE2038, dilution 1:3,000). The final products were collected and concentrated to ~5 mg ml−1, which was calculated by Nanodrop at 280 nm, and finally stored at −80 °C until to use.
RdRp enzymatic activity assay and its inhibition by suramin
To determine whether the purified L–VP35 protein is biologically active, we performed the primer-extension assay52,53. A mixture of 1.5 μM L–VP35 complex, 1 μM template RNA (5′-UUUGUUCGCGU-3′) and 200 μM primer RNA (5′-ACGC-3′) was added into a reaction buffer containing 20 mM Tris-HCl, pH 8.0, 20 mM NaCl, 4 mM MgCl2, 1 mM TCEP, 0.01% Tween 20, 1 mM CTP/UTP/ATP and 0.12 μCi μl−1 [α-32P]GTP at 30 °C for 1 h. The reaction system was quenched with addition of formamide and boiled at 100 °C for 10 min. The RNA products were separated by 25% polyacrylamide gels containing 7 M urea in 0.5× TBE buffer. Images were taken by exposing the gels on a storage phosphor screen and read with a Typhoon scanner (GE Healthcare). For assays with inhibition by suramin, the setup was similar to the above, except that the suramin was added at final concentrations of 1, 2, 4, 8, 32 and 64 μM. The intensity of each band was quantified with ImageJ software.
Mini-replicon assay
Plasmids expressing EBOV RNP (pCAGGS-L or pCAGGS-L(196–225)GS (in which residues 196–225 were replaced with GS), pCAGGS-VP35, pCAGGS-VP30 and pCAGGS-NP) and T7 polymerase (pCAGGS-T7), and a reporter plasmid encoded GFP flanked by T7 promoter, 5′- and 3′-terminal untranslated region sequences, were co-transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen)54. EBOV RNP activity was measured using the expression level of GFP calculated by CellVoyager CQ1 (Yokogawa) after 72 h. The expressions of VP35, NP and VP30 were detected using polyclonal antibodies (dilution 1:1,000) which were derived by immunizing rabbits (3–4 months old) with purified VP35, NP and VP30 proteins, respectively. Tubulin was used as a loading control and detected using mouse anti-tubulin (Easybio, BE0031, dilution 1:3,000). All rabbit experiments were performed according to the procedures approved by the Institute of Microbiology, Chinese Academy of Sciences and complied with all relevant ethical laws. Due to lack of antibody against L protein, the mRNA transcription levels of L (both wild type and L(196–225)GS mutant) were measured by quantitative real-time PCR (RT–qPCR). Total RNA was extracted using Cell Total RNA Isolation Kit (Foregene) and the concentration of RNA was measured by Nanodrop at 260 nm. About 5 μg total RNA was treated by Hifair III 1st Strand cDNA Synthesis SuperMix for quantitative PCR (qPCR) (purchased from YEASEN) to generate cDNA. RT–qPCR was performed using Hieff UNICON qPCR SYBR Green Master Mix (purchased from YEASEN) on the Bio-Rad CFX96 real-time PCR system. RT–qPCR primers for L mRNA were as follows: forward primer, 5′-GGACGAATCACAAAACTAGTCAATG-3′; reverse primer, 5′-CGGAAATAAACTCAGAAGCCCTG-3′. L mRNA copy numbers were calculated based on a standard curve generated with purified PCR products.
Cell-based assay for antiviral activity of suramin
Cell-based assay for inhibitory activity of suramin against EBOV polymerase was performed on the stable EBOV-GLuc-Hyg replicon cell line55. The cells were cultured overnight in Dulbecco’s modified Eagle medium (DMEM, Gibco) containing 100 μg ml−1 hygromycin and 10% fetal bovine serum (FBS, Gibco) at 37 °C, 5% CO2. The medium was replaced with FBS-free substrate before the addition of drugs. The cells were incubated with different concentrations of suramin and further cultured in 96-well plate. After 72 h incubation, a 30 μl volume of supernatant from each well was pipetted after centrifugation and then added to a new 96-well white plate and mixed with same volume of Gluc assay solution (Gaussia Luciferase Assay Kit, GeneCopoeia) immediately. The values of luminescence were measured by Glmax Reader (Promega).
Cytotoxicity assay
The cytotoxicity of suramin against EBOV-Gluc-Hyg replicon cells were measured by CCK8 reagent (MCE). Briefly, the drug-treated cells were washed twice with PBS and mixed with reaction solution containing CCK8. After incubation at 37 °C for 1.5 h, the A450 values were read by microplate reader (Thermo Fisher Scientific).
Statistical analysis
IC50, EC50 and CC50 values were represented as mean ± s.d. from at least three independent experiments. All values were determined by the nonlinear regression analysis using GraphPad Prism software v9.0.0 (https://www.graphpad.com/).
Cryo-EM sample preparation and data collection
To prepare the cryo-EM sample of L–VP35, 3 μl protein solution (0.6 mg ml−1) was loaded into a cleaned Nitai grid (R1.2/1.3), which was blotted for 3 s with a humidity of 100% at 4 °C, and then plunged into liquid ethane using an FEI Vitrobot Mark IV. The well-prepared cryogenic specimens were transferred onto an FEI Titan Krios transmission electron microscope for data collection. Cryo-EM micrographs were automatically collected using Serial-EM software using beam-image shift imaging scheme. Images were recorded with a K3-subunit detector using the super-resolution counting mode at a calibrated magnification of 22,500, corresponding to a pixel size of 1.07 Å. The exposure was performed with a dose rate of 20 e−1 pixel−1 s−1 and an accumulative dose of 60 e−1 Å2 for each micrograph, which fractionated into 32 frames. The defocus range of this dataset was roughly −1.5 to −2.6 μm.
For L–VP35 complex in the non-initiation state, the protein sample was diluted with enzymatic reaction buffer and then mixed with RNA at a molar ratio of protein:RNA of 1:2. After incubation for 1 h on ice, the complex was applied to a glow-discharged graphene coated grid (R1.2/1.3). The process of data collection was almost as same as above with a modified magnification of 29,000, yielding to a pixel size of 0.83 Å. Each image was exposed with a dose rate at 20 e−1 pixel−1 s−1 and total dose of 60 e−1 Å−2 and fractionated into a movie stack of 32 frames. The defocus range of this dataset was roughly −1.2 to −3.0 μm.
For the L–VP35–suramin complex, the purified protein was diluted to 0.4 mg ml−1 using salt-free buffer to reduce the concentration of NaCl to 300 mM and incubated with suramin at 4 °C for 1 h. An aliquot of 3 μl L–VP35–suramin complex was loaded onto glow-discharged Nitai grid (R1.2/1.3) following the same protocol for L–VP35. The micrographs were collected on a 300 kV Titan Krios transmission electron microscope equipped with a post-column GIF Quantum energy filter (Gantan) with a silt width of 20 eV. The images were recorded with K2 direct detection camara using the super-resolution counting mode with a calibrated pixel size of 1.04 Å on micrographs. Each stack was exposed with a dose rate at 10 e−1 pixel−1 s−1 and total dose of 60 e−1 Å−2 and fractionated into a movie stack of 32 frames. The defocus range of this dataset was roughly −0.9 to −2.4 μm.
Image processing
The movie frames were aligned using MotionCor256 and the contrast transfer function (CTF) values of each micrograph were determined using CTFFind457. Fifty micrographs were selected for automatic particles picking using Laplacian-of-Gaussian bolb detection, and were subjected to two-dimensional (2D) classification to generate templates for autopicking against entire dataset. All subsequent classification and reconstruction procedures were performed using Relion-3.058. For L–VP35 complex, a total of ~6,000,000 particles were selected from 4,432 micrographs, and were reduced to ~3,300,000 particles after three rounds of 2D classification. Due to lack of homologous structure, we generated the ab initio model (6 classes) using Relion-3.0 and selected the best class low-passed to 60 Å as the initial reference model for 3D classification. After two iterative rounds of 3D classification, a clean dataset of 1,083,293 particles from two classes with clear features of secondary-structure elements were subjected to 3D refinement, which yielded a reconstruction map at 3.5 Å. To further improve the map resolution, we performed dose weighting using MotionCor2 discarding the first two and last 14 frames in each stack to generate a reduced dataset with total dose of 30 e−1 Å−2. In addition, CTF refinement was performed to correct the local CTF values of each particle. After a final round of 3D refinement, we obtained a better map with a resolution at 3.0 Å. During above image processing, we noticed that extra densities belonging to VP35 oligomerization domain were cut off due to small box size. We extracted these particles with larger box size and re-centred them into the centre of whole particle not focused on L protein. Additional round of 3D classification was performed to discard bad particles among eight classes, and two of them showed obvious structural features. To avoid the density of L protein affecting the alignment, we performed particle subtraction that was subjected to the final round of 3D classification and 3D refinement with local angular search, and generated a reconstruction map at 3.4 Å for VP35 oligomerization domain (Supplementary Fig. 2). The processing of dataset for EBOV L–VP35 in the non-initiation state was quite straightforward. A total of 4,625 micrographs were collected, and ~5,000,000 particles were picked from the micrographs. After three rounds of 2D classification and two rounds of 3D classification, a clean dataset of 475,325 particles were subjected to 3D refinement, which yielded a final map at 3.4 Å resolution. Although we found a mass of density at C-terminal domain of VP35d, we were unable to reconstruct this region using local refinement or signal subtraction (Supplementary Fig. 4). The data processing for L–VP35–suramin complex was similar to the previous procedure. A total of 3,200,000 particles were auto-picked from 2,845 images, and after several rounds of 2D classification and three rounds of 3D classification, the remaining 193,982 particles were subjected to 3D refinement which yielded the density map at 3.3 Å resolution (Supplementary Fig. 5). The local resolution maps were evaluated by ResMap59.
Model building and refinement
The low sequence identity between EBOV L and other polymerases prevented us using any reported structure as a starting model. The quality of map is good enough for us manually modelling ab initio. The density for VP35 C-termini was well fitted with the reported crystal structure (PDB ID: 3L26). However, the oligomerization domain could not be fitted satisfactorily and was built manually. The initial coordinates were refined against the corresponding maps using PHENIX60 with secondary-structure restraints and Ramachandran restraints applied. And then, we performed manual model building to improve local fit using COOT61. The stereochemical quality of each model was assessed using MolProbity62. Structural figures were prepared with Pymol (https://pymol.org/) and CHIMERAX63.
AlphaFold2 prediction of EBOV L
The whole structure of EBOV L protein was predicted by AlphaFold264 with default settings. We compared the top five ranked outputs with our solved structure and selected the most similar one to prepare the figures.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The cryo-EM density maps and atomic coordinates have been deposited to the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB), respectively. The accession numbers are listed as follows: EBOV L–VP35 complex in state 1 (7YER, EMD-33775), EBOV L–VP35 complex in state 2 (7YES, EMD-33776), EBOV L–VP35-suramin complex (7YET, EMD-33777). All other data are available from the authors upon reasonable request.
References
Markham, A. REGN-EB3: first approval. Drugs 81, 175–178 (2021).
Gaudinski, M. R. et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study. Lancet 393, 889–898 (2019).
Muhlberger, E. Filovirus replication and transcription. Future Virol. 2, 205–215 (2007).
Jacob, S. T. et al. Ebola virus disease. Nat. Rev. Dis. Primers 6, 13 (2020).
Languon, S. & Quaye, O. Filovirus disease outbreaks: a chronological overview. Virology 10, 1178122X19849927 (2019).
Languon, S. & Quaye, O. Impacts of the Filoviridae family. Curr. Opin. Pharmacol. 60, 268–274 (2021).
Keita, A. K. et al. Resurgence of Ebola virus in 2021 in Guinea suggests a new paradigm for outbreaks. Nature 597, 539–543 (2021).
Dovih, P. et al. Filovirus-reactive antibodies in humans and bats in Northeast India imply zoonotic spillover. PLoS Negl. Trop. Dis. 13, e0007733 (2019).
Negredo, A. et al. Discovery of an ebolavirus-like filovirus in europe. PLoS Pathog. 7, e1002304 (2011).
Yang, X. L. et al. Characterization of a filovirus (Mengla virus) from Rousettus bats in China. Nat. Microbiol. 4, 390–395 (2019).
Shi, Y. New Virus, New Challenge. Innovation https://doi.org/10.1016/j.xinn.2020.04.005 (2020).
Feldmann, H., Sprecher, A. & Geisbert, T. W. Ebola. N. Engl. J. Med. 382, 1832–1842 (2020).
Stahelin, R. V. Membrane binding and bending in Ebola VP40 assembly and egress. Front. Microbiol. 5, 300 (2014).
Yu, D. S. et al. The lifecycle of the Ebola virus in host cells. Oncotarget 8, 55750–55759 (2017).
Davey, R. T. et al. A randomized, controlled trial of ZMapp for Ebola virus infection. N. Engl. J. Med. 375, 1448–1456 (2016).
Basu, A. et al. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 85, 3106–3119 (2011).
Li, H. Y. et al. Chemically modified human serum albumin potently blocks entry of Ebola pseudoviruses and viruslike particles. Antimicrob. Agents Chemother. 61, e02168–16 (2017).
Mulangu, S. et al. A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 381, 2293–2303 (2019).
Takashita, E. Influenza polymerase inhibitors: mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 11, a038687 (2021).
Peng, Q. et al. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovation 2, 100080 (2021).
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014).
Oestereich, L. et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antivir. Res. 105, 17–21 (2014).
Madelain, V. et al. Ebola virus dynamics in mice treated with favipiravir. Antivir. Res. 123, 70–77 (2015).
Warren, T. K. et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016).
Sissoko, D. et al. Experimental treatment with favipiravir for Ebola virus disease (the JIKI trial): a historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, e1001967 (2016).
Smither, S. J. et al. Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res. 104, 153–155 (2014).
Dunning, J. et al. Experimental treatment of Ebola virus disease with TKM-130803: a single-arm phase 2 clinical trial. PLoS Med. 13, e1001997 (2016).
Pan, J. H. et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 577, 275–279 (2020).
Jenni, S. et al. Structure of the vesicular stomatitis virus L protein in complex with its phosphoprotein cofactor. Cell Rep. 30, 53–60 (2020).
Gilman, M. S. A. et al. Structure of the respiratory syncytial virus polymerase complex. Cell 179, 193–204 (2019).
Horwitz, J. A., Jenni, S., Harrison, S. C. & Whelan, S. P. J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl Acad. Sci. USA 117, 2099–2107 (2020).
Abdella, R., Aggarwal, M., Okura, T., Lamb, R. A. & He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl Acad. Sci. USA 117, 4931–4941 (2020).
Liang, B. Structures of the mononegavirales polymerases. J. Virol. 94, e00175–20 (2020).
Blondot, M. L. et al. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog. 8, e1002734 (2012).
Richard, C. A. et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 14, e1006920 (2018).
Leung, D. W. et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat. Struct. Mol. Biol. 17, 165–U165 (2010).
Liang, B. et al. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162, 314–327 (2015).
Zinzula, L. et al. Structures of Ebola and Reston virus VP35 oligomerization domains and comparative biophysical characterization in all ebolavirus species. Structure 27, 39–54 (2019).
Henss, L. et al. Suramin is a potent inhibitor of Chikungunya and Ebola virus cell entry. Virol J. 13, 149 (2016).
Mastrangelo, E. et al. Structure-based inhibition of norovirus RNA-dependent RNA polymerases. J. Mol. Biol. 419, 198–210 (2012).
Yin, W. et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 28, 319–325 (2021).
Kirchdoerfer, R. N., Abelson, D. M., Li, S., Wood, M. R. & Saphire, E. O. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep. 12, 140–149 (2015).
Leung, D. W. et al. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein–RNA interactions. Cell Rep. 11, 376–389 (2015).
Wan, W. et al. Structure and assembly of the Ebola virus nucleocapsid. Nature 551, 394–397 (2017).
Luthra, P., Jordan, D. S., Leung, D. W., Amarasinghe, G. K. & Basler, C. F. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J. Virol. 89, 5148–5153 (2015).
Prins, K. C. et al. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J. Virol. 84, 10581–10591 (2010).
Qiu, S. H., Ogino, M., Luo, M., Ogino, T. & Green, T. J. Structure and function of the N-terminal domain of the vesicular stomatitis virus RNA polymerase. J. Virol. 90, 715–724 (2016).
Wiedemar, N., Hauser, D. A. & Maser, P. 100 years of suramin. Antimicrob. Agents Chemother. 64, e01168–19 (2020).
Nardone, V. et al. Structural basis of inhibition of the pioneer transcription factor NF-Y by suramin. Cells 9, 2370 (2020).
Ono, K., Nakane, H. & Fukushima, M. Differential inhibition of various deoxyribonucleic and ribonucleic acid polymerases by suramin. Eur. J. Biochem. 172, 349–353 (1988).
Peng, R. C. et al. Structural insight into arenavirus replication machinery. Nature 579, 615–619 (2020).
Tchesnokov, E. P., Raeisimakiani, P., Ngure, M., Marchant, D. & Gotte, M. Recombinant RNA-dependent RNA polymerase complex of Ebola virus. Sci Rep. 8, 3970 (2018).
Tchesnokov, E. P., Feng, J. Y., Porter, D. P. & Gotte, M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 11, 326 (2019).
Du, X. et al. Combinatorial screening of a panel of FDA-approved drugs identifies several candidates with anti-Ebola activities. Biochem. Biophys. Res. Commun. 522, 862–868 (2020).
Tao, W. Y., Gan, T. Y., Guo, M. Z., Xu, Y. F. & Zhong, J. Novel stable Ebola virus minigenome replicon reveals remarkable stability of the viral genome. J. Virol. 91, e01316–17 (2017).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Acknowledgements
We thank all the staff members at the Center for Biological Imaging (CBI), Institute of Biophysics (IBP), Chinese Academy of Sciences (CAS) for assistance with data collection. We thank the staff of the State Key Laboratory of Membrane Biology, Institute of Zoology (IOZ), CAS for technical support of electron microscope operation. This study was supported by the National Key Research and Development Program of China (2021YFC2300700 to Y.S.), Strategic Priority Research Program of CAS (XDB29010000 to Y.S. and G.F.G.), and National Natural Science Foundation of China (NSFC) (81871658 and 32192452 to Y.S. and 32100119 to Q.P.). Y.S. is also partially supported by the Youth Innovation Promotion Association of CAS (Y201921).
Author information
Authors and Affiliations
Contributions
Y.S. and G.F.G. conceived the study. B.Y., J.C. and M.W. purified the protein samples and conducted biochemical and cellular experiments. B.Y. and Q.P. prepared the cryo-EM specimens and collected data. Q.P. conducted the image processing and reconstruction. J.Q. and Q.P. built the atomic models. Y.S., G.F.G., J.Q., Q.P. and B.Y. analysed the structure. Q.P., B.Y., J.Z., G.F.G. and Y.S. wrote the manuscript. All authors participated in the discussion and manuscript editing. B.Y. and Q.P. contributed equally to this work. Y.S. supervised all the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Ming Luo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 The purified EBOV L-VP35 complex has polymerization activity.
(a, b) Size-exclusion chromatography, SDS-PAGE and western blotting profiles of EBOV L-VP35 WT (a) and D742A mutant (b) proteins. Molecular weights (in kilodaltons, kDa) of ladder makers are shown in the left, and the L and VP35 bands are labeled on the right. (c, d) In vitro primer extension assay of L-VP35 complex. It demonstrates the purified complex protein possesses polymerase activity, and the product bands are inhomogeneous. The active site D742A mutant can abolish the production of RNA. Otherwise, a small percentage of full-length product (indicated by red arrow) can be clearly seen when the product bands are overexposed (d). The data shown above are representative results of at least three independent experiments using different protein preparations.
Extended Data Fig. 2 Structural comparison of EBOV L-VP35 and HPIV5 L-P complex.
Overall structure of EBOV L-VP35 (a) and HPIV5 L-P complex (PDB 6v85) (b). Overlay of the NTD (c), RdRp (d), PRNTase (e) domains and tetrameric P/VP35 protein (f) between EBOV L-VP35 and HPIV5 L-P complex shows a similar architecture, but with local differences.
Extended Data Fig. 3 The paths for RNA synthesis within EBOV L-VP35 structure.
(a) The EBOV L-VP35 structure is shown in surface representation to highlight the entry channel of NTP substrate which was indicated by a red cycle. (b) RNA elongation model of EBOV L-VP35 complex. Template RNA entry and exit channels are indicated as black arrows, and NTP entry and nascent RNA product exit tunnels are indicated by red and purple arrows. (c) Cutoff view of the L protein shown in electrostatic surface representation (blue, positive charge; red, negative charge). The paths are filled by the template and nascent RNA strands which are modeled based on the structure of rotavirus polymerase complex with in situ elongation conformation (PDB 6OGZ).
Extended Data Fig. 4 The critical role of the insertion element in transcription activity of EBOV RNP.
(a) Clear green fluorescence can be visualized with the wild type L protein. However, for the insertion-element-deletion L(196-225)GS construct (residues 196 to 225 consisting of the insertion loop structure were deleted and two ends were linked with GS residues), no green fluorescence was observed. As a negative control, the L gene was not transfected in the replicon system. (b) The expression levels of NP, VP35 and VP30 were measured using Western Blotting assay and the tubulin was used as loading control. The data shown are representative of three independent experiments. (c) The transcription levels of L mRNA were analyzed by RT-PCR. The data represent mean values (histograms) ± s.d. (error bars) from three independent experiments.
Extended Data Fig. 5 The predicted full-length structure of EBOV L by AlphaFold2.
(a) Superimposition of the predicted and solved structures of EBOV L protein, and they could be overlaid well. The predicted structure is colored in grey, while the solved structure is colored as depicted in Fig. 1. (b) The modeled full-length structure of EBOV L-VP35 complex. (c–e) Close-up view of the predicted structures of CD (purple), MTase (magenta) and CTD (black) domains.
Extended Data Fig. 6 Structural analysis of the EBOV L PRNTase domain.
(a) Overlay of the PRNTase domains of L proteins from the EBOV (colored by cyan) and RSV (grey). (b) The same view as in (a) but with catalytic motifs indicated by different colors (motif A’, red; motif B’, purple; motif C’, yellow; motif D’, blue; motif E’, orange). The Cα atoms of the conserved glycines in GxxT motif and motif A’ (Gly1129) are shown in sphere representation.
Extended Data Fig. 7 Conformational change of the priming loop and supporting helix.
The priming loop and supporting helix would clash with RNA duplex formed during the RNA elongation process (a), and would undergo conformational change to release adequate space for RNA elongation (b) with the priming loop retracting into PRNTase domain and the supporting helix moving outward. The RNA was modeled based on the structure of rotavirus polymerase complex with in situ elongation conformation (PDB 6OGZ), and the RdRp active site was indicated by a red star.
Extended Data Fig. 8 Structural comparison of filovirus VP35 ODs.
(a) The structure of tetrameric VP35 OD from EBOV L-VP35 complex. (b–d) The crystal structures of tetrameric REBOV VP35 OD (PDB 6GBQ), trimeric EBOV VP35 OD (PDB 6GBO) and trimeric MARV VP35 OD (PDB 5TOI). (e) The sequence alignment of VP35 OD regions from different filoviruses.
Extended Data Fig. 9 The entangled tetrameric VP35 wraps around L protein.
(a) EBOV L is shown in grey surface representation, and VP35 protomers are shown in cartoon representation with different colors. (b) Structural conformations of different VP35 protomers. (c) The major atomic interactions between VP35 protomers. The key interacting residues are shown in stick representation. Hydrogen bonds are presented in yellow dash lines, while the hydrogen bonds between the anti-parallel β strands are not shown (left side).
Extended Data Fig. 10 Binding interface between VP35 and EBOV L and its comparison with other nsNSV polymerase complexes.
(a) The binding footprints of VP35 protomers on EBOV L are indicated by black dash line. The interacting residues of L are colored and labelled according to the bound VP35 protomers, and the overlapping regions are shown in orange (VP35b/d) and pink (VP35c/d), respectively. (b) Comparison of the binding interfaces of EBOV L-VP35 and RSV L-P polymerase complexes. The L and P of RSV are colored in black and white, respectively. The L and VP35 of EBOV are colored as in Fig. 4. (c) Sequence alignment of critical interactive residues from L proteins among Mononegavirales. The L residues that formed hydrogen bonds with the residues from VP35 of EBOV and P of RSV are indicated by red and blue stars, respectively.
Extended Data Fig. 11 The inhibition mechanism of suramin against EBOV L-VP35.
(a) The chemical structure of suramin. Each benzene ring group is labeled by a unique symbol. (b) Inhibitory activity of suramin against EBOV L-VP35 complex was measured at enzymatic level. The RNA products were shown in urea-PAGE, and a series of concentration of suramin were added in the enzyme reaction system. The data shown are representative of three independent experiments using different protein preparations. (c) The 50% cytotoxicity concentration (CC50) of suramin was determined with the stable replicon cell. Each data point indicates the mean value of three independent experiments and the error bars represent standard deviation. (d–e) The structures of EBOV L-VP35 (d) and L-VP35-suramin (e) complex are shown in surface representation, and the NTP entry channel is indicated by a dashed circle. The suramin is stuck in the NTP entry channel to prevent NTP substrates reaching active site of RdRp. (f-g) The suramin could also hinder the activity of RdRp by occupying the spaces for product RNA strand. Cutoff view of the L-VP35-suramin complex overlapped with the modeled RNA (f). The tail part of suramin molecule would clash with nascent RNA product strand (g). The template (golden) and product (black) RNA strands are modeled based on the structure of rotavirus polymerase with in situ elongation conformation (PDB 6OGZ).
Extended Data Fig. 12 Structural comparison of EBOV L-VP35-suramin complex with SARS-CoV-2 RdRp-suramin and Murine Noroviruses (MNV) RdRp-suramin complex.
(a-b) Superimposition of the EBOV L-VP35-suramin with SARS-CoV-2 RdRp-suramin complex (PDB 7D4F) and MNV RdRp-suramin complex (PDB 3UR0) was performed based on the RdRp domain. A close-up view of suramin within the catalytic chamber in the SARS-CoV-2 RdRp-suramin complex (c), EBOV L-VP35-suramin complex (d) and MNV RdRp-suramin complex (e).
Supplementary information
Supplementary Information
This file contains Supplementary Figs. 1-6 and Supplementary Tables 1-3. Supplementary Figs. 1-5 are the cryo-EM analysis processes of structures determined in this paper and Supplementary Fig. 6 shows the uncropped gels, western blotting images and autoradiographs used for preparing Extended Data Fig. 1, 4 and 11. Supplementary Tables 1-2 contain cryo-EM data processing and refinement statistics for L-VP35 complex in two states and L-VP35-suramin complex and Supplementary Table 3 shows interactions between EBOV L protein and VP35.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yuan, B., Peng, Q., Cheng, J. et al. Structure of the Ebola virus polymerase complex. Nature 610, 394–401 (2022). https://doi.org/10.1038/s41586-022-05271-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05271-2
This article is cited by
-
Structural basis for dimerization of a paramyxovirus polymerase complex
Nature Communications (2024)
-
Structures of the promoter-bound respiratory syncytial virus polymerase
Nature (2024)
-
Puerarin attenuates valproate-induced features of ASD in male mice via regulating Slc7a11-dependent ferroptosis
Neuropsychopharmacology (2024)
-
Navigating the Complex Landscape of Ebola Infection Treatment: A Review of Emerging Pharmacological Approaches
Infectious Diseases and Therapy (2024)
-
Molecular mechanism of de novo replication by the Ebola virus polymerase
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
