
Abstract
Lysosomal dysfunction has been increasingly linked to disease and normal ageing1,2. Lysosomal membrane permeabilization (LMP), a hallmark of lysosome-related diseases, can be triggered by diverse cellular stressors3. Given the damaging contents of lysosomes, LMP must be rapidly resolved, although the underlying mechanisms are poorly understood. Here, using an unbiased proteomic approach, we show that LMP stimulates a phosphoinositide-initiated membrane tethering and lipid transport (PITT) pathway for rapid lysosomal repair. Upon LMP, phosphatidylinositol-4 kinase type 2α (PI4K2A) accumulates rapidly on damaged lysosomes, generating high levels of the lipid messenger phosphatidylinositol-4-phosphate. Lysosomal phosphatidylinositol-4-phosphate in turn recruits multiple oxysterol-binding protein (OSBP)-related protein (ORP) family members, including ORP9, ORP10, ORP11 and OSBP, to orchestrate extensive new membrane contact sites between damaged lysosomes and the endoplasmic reticulum. The ORPs subsequently catalyse robust endoplasmic reticulum-to-lysosome transfer of phosphatidylserine and cholesterol to support rapid lysosomal repair. Finally, the lipid transfer protein ATG2 is also recruited to damaged lysosomes where its activity is potently stimulated by phosphatidylserine. Independent of macroautophagy, ATG2 mediates rapid membrane repair through direct lysosomal lipid transfer. Together, our findings identify that the PITT pathway maintains lysosomal membrane integrity, with important implications for numerous age-related diseases characterized by impaired lysosomal function.
Similar content being viewed by others

Main
Lysosomes with severe LMP have been reported to be selectively degraded by macroautophagy, known as lysophagy4,5, whereas milder LMP can be fixed through faster, direct membrane repair by the endosomal sorting complex required for transport6,7,8 (ESCRT). However, depletion of ESCRT subunits causes only partial defects in rapid lysosomal repair7, suggesting the existence of additional repair mechanisms. To search for such pathways, we designed an unbiased proteomic approach to identify proteins that are recruited rapidly to damaged lysosomes. Targeting TurboID, a promiscuous biotin ligase9,10, to the lysosomal surface (Extended Data Fig. 1a,b) enabled rapid biotinylation of lysosomal surface proteins that can be identified by streptavidin (Extended Data Fig. 1c). l-Leucyl-l-leucine methyl ester (LLOME) is the best characterized lysosomotropic reagent that polymerizes inside lysosomes to induce fast but reversible lysosomal membrane damage5,11. We purified all biotinylated, membrane-associated proteins from 293T cells expressing lysosome-targeted TurboID (Lyso-TurboID) with or without LLOME treatment for mass spectrometry analysis (Fig. 1a and Extended Data Fig. 1d,e). The autophagy adaptor protein p62 (also known as SQSTM1), previously reported to accumulate on damaged lysosomes5, was highly enriched in the purified samples from LLOME-treated cells (Fig. 1a), confirming the utility of our approach.

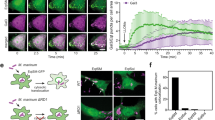
a, Schematic illustration of the Lyso-TurboID screen. P20-SA↓, streptavidin (SA) pulldown from P20, the pellet fraction after centrifugation at 20,000 g. b, Scatter plot of the top proteins from more than 1,800 mass spectrometry hits. Three categories of proteins are labelled in colour. ORP9/11 are known PtdIns4P effectors. c, Left, OSBP-PH–GFP is recruited to damaged lysosomes in U2OS cells; the outlined region is magnified on the right. Right, a schematic illustration of the response. d, Pearson’s correlation coefficient (R) for OSBP-PH–GFP and IST1. Data are mean ± s.e.m.; n = 15 (−LLOME), n = 27 (+LLOME) cells from 3 trials. e, Endogenous PI4K2A accumulates rapidly on damaged lysosomes in U2OS cells; the outlined region is magnified on the right. f, Pearson’s correlation coefficient for PI4K2A and IST1. Data are mean ± s.e.m.; n = 27 cells from 3 trials for each condition. g, Quantification of OSBP-PH–GFP puncta in PI4K2A-KO U2OS cells expressing the indicated PI4K2A mutants. Approximately 50–100 random cells were counted per condition. Data are mean ± s.e.m.; n = 3 per condition. See fluorescence images in Extended Data Fig. 4c. WT, wild type. h, Lyso-pHluorin quantification in U2OS cells with indicated genetic modifications expressing the indicated PI4K2A mutants. More than 100 cells were counted for each condition. Data are mean ± s.e.m.; n = 3 per condition. Dotted line indicates 50% of repair. See fluorescence images in Extended Data Fig. 4d,e. i, Quantification of galectin-3 intensity above threshold in U2OS cells with indicated genetic modifications expressing the indicated PI4K2A mutants. Between 50 and 100 random cells were quantified for each condition. Data are mean ± s.e.m.; n = 3 per condition. See fluorescence images in Extended Data Fig. 4f. j, Percentage of cells exhibiting tau spreading after tau fibril exposure. Data are mean ± s.e.m.; n = 4 per condition. See fluorescence images in Extended Data Fig. 4h. All experiments were based on stable cell lines unless otherwise indicated. Statistical significance was determined by unpaired, two-tailed t-tests. Scale bars, 10 μm.
PtdIns4P accumulates on damaged lysosomes
Besides p62 and multiple ESCRT subunits previously reported as enriched on damaged lysosomes5,6,7,8, mass spectrometry identified three top hits likely within the same phosphoinositide signalling pathway (Fig. 1b, Supplementary Table 1). These included PI4K2A, an enzyme that generates the lipid messenger phosphatidylinositol-4-phosphate12 (PtdIns4P), as well as ORP9 (also known as OSBPL9) and ORP11 (also known as OSBPL11), both of which are PtdIns4P-binding proteins13,14,15. We confirmed by immunoblotting that these proteins were enriched on damaged lysosomes (Extended Data Fig. 1f). To further test whether LMP stimulates lysosomal PtdIns4P signalling, we established a stable cell line expressing the pleckstrin homology (PH) domain of OSBP fused to GFP (OSBP-PH–GFP), a well-established probe for PtdIns4P16. Consistent with previous observations16, under basal conditions, OSBP-PH–GFP puncta were found exclusively in the Golgi region (Fig. 1c, top). By contrast, LLOME treatment induced many new OSBP-PH–GFP puncta that colocalized with increased sodium tolerance 1 homologue (IST1), one of the ESCRT subunits used to mark damaged lysosomes (Fig. 1c, bottom and Extended Data Fig. 1g), and with multiple lysosomal markers including LAMP1, LAMP2 and CD63 (Extended Data Figs. 1h and 2a,b and Supplementary Video 1). This phenomenon was recapitulated across multiple cell lines, observed using different PtdIns4P probes, was specific for PtdIns4P but not for other phosphoinositide species, and was not dependent on the pool of PtdIns4P on the trans-Golgi network (TGN) (Extended Data Fig. 2c–i). Thus, specific accumulation of PtdIns4P is triggered on damaged lysosomes.
PI4K2A generates PtdIns4P on damaged lysosomes
PtdIns4P can be generated by direct phosphorylation of phosphatidylinositol by PI4K or dephosphorylation of PtdIns(4,5)P2. Since PtdIns(4,5)P2 was barely detectable on damaged lysosomes (Extended Data Fig. 2e), PI4Ks were probably responsible for lysosomal PtdIns4P production. Using verified PI4K antibodies, we observed selective enrichment of PI4K2A on damaged lysosomes by immunoblotting (Extended Data Fig. 3a,b). This was supported by immunofluorescence showing robust lysosomal accumulation of PI4K2A but not PI4K2B upon LLOME treatment (Fig. 1e,f and Extended Data Fig. 3c–f), with no change in total PI4K2A protein levels (Extended Data Fig. 3g). Of note, the recruitment of PI4K2A and the ESCRT complex were independent of each other (Extended Data Fig. 3h).
Other LMP-inducing strategies including expression of the SARS-CoV-2 protein ORF3A17 or knockout of CLN3, a gene frequently mutated in the lysosomal storage condition known as Batten’s disease18, also stimulated robust lysosomal recruitment of PI4K2A (Extended Data Fig. 3i–m and Supplementary Table 2). Lysosomes are well-known Ca2+ stores; LMP-induced lysosomal Ca2+ release probably contributes to PI4K2A recruitment, as agonists of TRPML1—the main lysosomal Ca2+ channel19—were sufficient to trigger PI4K2A accumulation on lysosomes (Extended Data Fig. 3n). Thus, PI4K2A is specifically recruited to damaged lysosomes.
To investigate whether PI4K2A is responsible for PtdIns4P production on damaged lysosomes, we generated PI4K2A-knockout (PI4K2A-KO) U2OS cells using CRISPR technology20. Without affecting the basal Golgi targeting of OSBP-PH–GFP, PI4K2A-KO completely abolished LMP-induced lysosomal recruitment of the PtdIns4P probe (Extended Data Fig. 4a), which was fully rescued by re-expressing PI4K2A in knockout cells (Extended Data Fig. 4a,b). Pretreating cells with brefeldin A, which disassembles the Golgi complex21, confirmed the robust de novo generation of lysosomal PtdIns4P only in cells with wild-type PI4K2A (Fig. 1g and Extended Data Fig. 4c). Two kinase-deficient point mutations of PI4K2A, K152A and D308A, have been previously characterized2223. Lysosomal OSBP-PH–GFP recruitment in PI4K2A-KO cells was partially rescued by re-expression of PI4K2A(K152A), but not by either PI4K2A(D308A) or the double mutant K152A/D308A (KDAA) (Fig. 1g and Extended Data Fig. 4c). Together, these results demonstrate that PI4K2A is the enzyme that generates PtdIns4P on damaged lysosomes.
PtdIns4P mediates rapid lysosomal repair
We established a lysosomal repair assay based on lyso-pHluorin, a lysosomal pH sensor that is activated in damaged lysosomes owing to pH neutralization24 (Extended Data Fig. 4d, left). Lyso-pHluorin puncta were not detected in either wild-type or PI4K2A-KO cells in resting conditions (Extended Data Fig. 4d, top), and a brief LLOME treatment induced extensive puncta in almost all cells, suggesting equal lysosomal damage (Extended Data Fig. 4d, middle). While lyso-pHluorin puncta disappeared quickly in wild-type cells with an average half-life of 1.1 h, puncta clearance in two independent PI4K2A-KO cell lines was significantly slower, with a calculated half-life of 5.4 to 11 h (Fig. 1h and Extended Data Fig. 4d,e).
Whereas lyso-pHluorin senses lysosomal pH, fluorescently-tagged galectin-3 has been used to detect large, unrepaired lysosomal membrane pores5,7. Stably expressed eGFP–galectin-3 in either wild-type or PI4K2A-KO cells showed no puncta under basal conditions (Extended Data Fig. 4f). Continuous LLOME treatment induced markedly more galectin-3 puncta in PI4K2A-KO cells than in wild-type cells, which was reversed by re-expressing wild-type but not kinase-dead PI4K2A (Fig. 1i and Extended Data Fig. 4f). Together, these data indicate that the kinase activity of PI4K2A is essential for rapid lysosomal repair. Of note, wild-type but not kinase-dead PI4K2B, when artificially anchored on the lysosomal surface, could also rescue the repair defects in PI4K2A-KO cells (Extended Data Fig. 4g).
In exploring the physiological significance of this repair pathway, we noted that loss of PI4K2A caused marked exacerbation of tau fibril spreading in a cell-based assay (Fig. 1j and Extended Data Fig. 4h), consistent with endolysosomal damage as a key step in tau fibril spreading25. Moreover, although CLN3-deleted skin fibroblasts normally accumulate lipofuscin, this lysosomal accumulation was further augmented upon loss of PI4K2A (Extended Data Fig. 4i).
PtdIns4P recruits ORPs to damaged lysosomes
The functions of phosphoinositide signalling pathways are often defined by lipid effectors26. Our mass spectrometry experiments using Lyso-TurboID identified four ORP family members—ORP9, ORP10, ORP11 and OSBP—as potential PtdIns4P effectors in this pathway (Supplementary Table 1). Using verified antibodies (Extended Data Fig. 5a), we found ORP9, ORP10 and ORP11 to be strongly enriched on damaged lysosomes, whereas OSBP was only weakly recruited to lysosomes (Fig. 2a and Extended Data Fig. 5b). Immunofluorescence revealed basal Golgi localization of ORP9 and ORP11, with robust lysosomal accumulation detected 10 min after LLOME treatment (Fig. 2b, Supplementary Videos 2 and 3 and Extended Data Fig. 5c–e). By contrast, ORP3 and ORP5 were not recruited to damaged lysosomes (Extended Data Fig. 5f,g). We confirmed that the LLOME-induced lysosomal recruitment of endogenous ORP9 and ORP11 and stably overexpressed eGFP–ORP9, eGFP–ORP10 and eGFP–ORP11 were all fully dependent on PI4K2A and its kinase activity (Fig. 2c and Extended Data Fig. 5h–m). Mutation of the PtdIns4P-binding residues in their N-terminal PH domains13,14 strongly suppressed the localization of ORP9, ORP10 and ORP11 to damaged lysosomes (Extended Data Fig. 5n–p). Thus, ORP9, ORP10 and ORP11 are recruited to damaged lysosomes in a PI4K2A- and PtdIns4P-dependent manner.

a, ORP family members are enriched on damaged lysosomes. Note that ORP10 is absent from S20 (supernatant after centrifugation at 20,000 g) (arrow head). Asterisk indicates a nonspecific band. b, Endogenous ORP9 is recruited to LAMP1-positive lysosomes upon brief LLOME treatment in U2OS cells. c, Punctate intensities of endogenous ORP9 and ORP11 before and after LLOME treatment. More than 30 random cells were quantified for each condition. Data are mean ± s.e.m.; n = 4 trials. See fluorescence images in Extended Data Fig. 5m. d, LLOME induces extensive wrapping of endogenous LAMP1 by eGFP–VAPA in U2OS cells. Right, schematic illustration of the process. e, Percentage of U2OS cells with more than five lysosomes extensively wrapped by eGFP–VAPA. More than 100 cells were counted for each condition. Data are mean ± s.e.m.; n = 3 per condition. See fluorescence images in Extended Data Fig. 6a. f, Representative microscopy and quantification of ER–lysosome wrapping in various ORP-knockout cells. More than 100 cells were counted for each condition. Data are mean ± s.e.m.; n = 3. See more details in Extended Data Fig. 6c–f. g, ORP-QKO cells stably expressing eGFP–VAPA and mCherry–ORP proteins were stimulated with LLOME for 10 min and the percentages of cells showing VAPA clusters on ORP proteins were quantified. Data are mean ± s.e.m.; n = 3 per condition. See more details in Extended Data Fig. 6j–m. ORP9/11 (FYAA/WT), co-expression of mCherry–ORP9(FYAA) and mCherry–ORP11(WT). h, Representative images and quantification of eGFP–galectin-3 puncta in wild-type and ORP-QKO cells. More than 100 cells were quantified per condition. Data are mean ± s.e.m.; n = 3 per condition. i, Quantification of eGFP–galectin-3 puncta in ORP-QKO cells stably expressing eGFP–galectin-3 and mCherry–ORP proteins. Approximately 50–100 cells were quantified for each condition. Data are mean ± s.e.m.; n = 5 per condition. Images are presented in Extended Data Fig. 6o. j, Schematic illustration of PtdIns4P-stimulated, ORP-mediated membrane tethering between the ER and damaged lysosomes. ORP9 carries a stronger FFAT motif than ORP10 and ORP11 and thus provides stronger ER tethering. Unpaired, two-tailed t-tests. NS, not significant. Scale bars, 10 μm.
ORPs establish ER–lysosome MCSs
The ORP family members often localize to membrane contact sites (MCSs) between the endoplasmic reticulum (ER) and other organelles27. Among the three lysosomal-recruited ORP family proteins, ORP9 carries a strong FFAT motif that binds to the ER transmembrane proteins, vesicle-associated membrane protein-associated proteins A and B13,14 (VAPA and VAPB) (Extended Data Fig. 5n,o). We observed extensive LMP-stimulated VAPA recruitment to lysosomes (Fig. 2d), which was completely dependent on PI4K2A and its kinase activity (Fig. 2e and Extended Data Fig. 6a) as well as the FFAT-binding residues in VAPA (Extended Data Fig. 6b). Extensive redundancy was detected in the recruitment of ORP proteins to damaged lysosomes (Extended Data Fig. 6c–e and Supplementary Table 3). Indeed, the deletion of all four recruited ORP family proteins (ORP9, ORP10, ORP11 and OSBP quadruple knockout (ORP-QKO)) (Supplementary Tables 1 and 4) was required to abolish VAPA clustering around damaged lysosomes (Fig. 2f and Extended Data Fig. 6f). ORP-QKO did not diminish upstream PtdIns4P signalling (Extended Data Fig. 6g,h) and in fact caused stronger and more prolonged lysosomal PtdIns4P production (Extended Data Fig. 6h,i). In agreement with the previously reported heterodimerization of ORP9 and ORP11 (ref. 15), re-expression of two ORPs (ORP9 and ORP10 or ORP9 and ORP11) was sufficient to fully rescue VAPA recruitment to damaged lysosomes in ORP-QKO cells, whereas individual expression of ORP9, ORP10 or ORP11 was insufficient (Fig. 2g and Extended Data Fig. 6j–l). Of note, although ORP9 could not homodimerize, it readily heterodimerized with either ORP10 or ORP11 (Extended Data Fig. 6m). By contrast, OSBP formed homodimers (Extended Data Fig. 6n) and was robustly recruited to damaged lysosomes in the absence of the other ORPs (Extended Data Fig. 6e,k). Similar to PI4K2A-KO cells (Fig. 1h,i), ORP-QKO cells also exhibited marked defects in rapid lysosomal repair (Fig. 2h), which were rescued by reconstitution with any two of ORP9, ORP10 and ORP11, with ORP9 and ORP11 reconstitution being the most robust (Fig. 2i and Extended Data Fig. 6o). Together, these data support the hypothesis that lysosomal PtdIns4P signalling recruits multiple ORP proteins, generating extensive ER–lysosomal MCSs, which are required for rapid lysosomal repair (Fig. 2j).
ORP9, 10 and 11 transport PS to damaged lysosomes
The ORP family are lipid transfer proteins that exchange PtdIns4P for sterols or phosphatidylserine (PS) at MCSs27,28,29. Among the twelve human ORP proteins, ORP5, ORP8, ORP9, ORP10 and ORP11 are evolutionarily closest to the yeast PS transporter Osh6, and previous reports have shown that ORP5, ORP8 and ORP10 specifically transfer PS but not sterols30,31,32. The main ORP proteins recruited to damaged lysosomes, ORP9, ORP10 and ORP11 (Supplementary Table 1), all carry highly conserved potential PS-binding sites (Extended Data Fig. 7a). Consistent with this structural homology, assessment of in vitro lipid transport using a fluorescence resonance energy transfer (FRET)-based assay demonstrated that ORP9 and ORP11 specifically transferred PS but not cholesterol (Fig. 3a,b and Extended Data Fig. 7b–d).

a, Schematic of the in vitro lipid transport assay for ORP9 and ORP11. Note that nitrobenzoxadiazole (NBD) is attached to one fatty acid chain but not the head group of PS. PE, phosphatidylethanolamine; Rh, rhodamine; DGS–NTA(Ni), a nickel-chelating lipid that binds His-tagged proteins. b, Lipid transfer assay demonstrating the increase in NBD fluorescence stimulated by ORP9 and ORP11 in the presence of NBD–PS but not NBD–cholesterol (Chol) or TopFluor (TF)-cholesterol. Data are mean ± s.e.m.; n = 3 per condition. c, U2OS cells stably expressing the PS probe GFP–Lact-C2 were treated with LLOME for 20 min and washed with detergent before immunostaining of endogenous IST1 and LAMP1. Scale bars, 10 μm. d, Colocalization of GFP–Lact-C2 and LAMP1 in the indicated U2OS cell lines. Data are mean ± s.e.m. of Pearson’s correlation coefficient; n = 30 cells from 3 trials per condition. See fluorescence images in Extended Data Fig. 7f. e, Schematic illustration of different ORP mutants used in f,h. f, U2OS cells stably expressing GFP–Lact-C2 and indicated mCherry–ORP9 and ORP11 proteins (mutants are notated as ORP9 mutant/ORP11 mutant) were treated as in c, and the Pearson’s correlation coefficient for GFP–Lact-C2 and LAMP1 were quantified. Data are mean ± s.e.m.; n = 30 cells from 3 trials for each condition. See fluorescence images in Extended Data Fig. 7k. g, Damaged lysosomes purified from wild-type cells, but not those from ORP-QKO cells, are enriched in PS. Total lysosomal lipids were spotted onto PVDF membranes and PS was detected using purified eGFP–Lact-C2. Data are mean ± s.e.m.; n = 3 per condition. See more details in Extended Data Fig. 7m,n. h, eGFP–galectin-3 assays demonstrating restored rapid lysosomal repair in ORP-QKO cells reconstituted with wild-type ORP9 and ORP11, but not indicated mutants of ORP9 and ORP11. Data are mean ± s.e.m.; 50–100 cells, n = 3 per condition. See fluorescence images in Extended Data Fig. 8a. i, Schematic of chimeric proteins generated to replace ORPs. The substrate specificity of each transporter is indicated. Chol, cholesterol. j, eGFP–galectin-3 assays showing restored rapid lysosomal repair in ORP-QKO cells expressing OSBP–ORP chimeric proteins. Data are mean ± s.e.m.; 50–100 cells quantified, n = 3 per condition. See fluorescence images in Extended Data Fig. 8d. Unpaired, two-tailed t-tests.
In cells, the PS-specific probe GFP–lactadherin C2 domain33 (GFP–Lact-C2) showed robust lysosomal accumulation upon LMP (Fig. 3c,d and Extended Data Fig. 7e), which was dependent on PI4K2A and its kinase activity, and required the ORP proteins (Fig. 3d and Extended Data Fig. 7f). Consistent with the ORP proteins being PS transporters, the recruitment of ORP9 preceded the accumulation of lysosomal PS (Extended Data Fig. 7g,h). In ORP-QKO cells, lysosomal PS accumulation was successfully rescued by re-expression of any two of ORP9, ORP10 and ORP11, with the strongest effects again seen with expression of ORP9 and ORP11 (Extended Data Fig. 7i,j). ORP mutants deficient in lysosomal targeting (RE) or PtdIns4P–PS counter transport (AAA or HHAA) were completely incapable of generating PS accumulation on damaged lysosomes (Fig. 3e,f and Extended Data Figs. 5n–p and 7k), although some were still able to rescue ER–lysosome tethering (Extended Data Fig. 7l). ORP-dependent lysosomal PS transport was further supported by demonstrating increased PS levels in lysosomes purified from LLOME-treated wild-type but not ORP-QKO cells (Fig. 3g and Extended Data Fig. 7m,n). These data are consistent with ORP9, ORP10 and ORP11 orchestrating PtdIns4P-driven PS transport from the ER to damaged lysosomes, analogous to the previously reported ER-to-plasma membrane PS transfer catalysed by ORP5 and ORP8 (ref. 31).
PS mediates rapid lysosomal repair
Whereas reconstitution with any two of the three wild type ORP proteins robustly rescued lysosome repair in ORP-QKO cells (Fig. 2i), various OPR mutants defective in PS transport were incapable of restoring the lysosomal repair capacity (Fig. 3h and Extended Data Fig. 8a). Exploiting the capacity of OSBP to homodimerize (Extended Data Fig. 6n), we generated OSBP–ORP chimeras that bypassed the ORP-heterodimerization requirement for lysosomal recruitment (Fig. 3i and Extended Data Fig. 8c), which enabled each individual chimeric PS transporter to rescue lysosomal repair in ORP-QKO cells (Fig. 3j and Extended Data Fig. 8d). We further demonstrated that the mammalian ORP proteins could be replaced by yeast Osh6 (OSBP–Osh6) (Fig. 3i), which fully rescued rapid lysosomal repair through its PS transport activity (Fig. 3j and Extended Data Fig. 8c,d). Thus, lysosomal transport of PS mediates rapid lysosomal repair.
While the major membrane tethers ORP9/10/11 mediate PS transport for lysosomal repair, we identified OSBP as a parallel, complementary membrane tether (Extended Data Fig. 6e) that transported cholesterol to damaged lysosomes (Extended Data Fig. 8e, f), consistent with its known function29,34. Interestingly, reconstituting lysosomal cholesterol transport in ORP-QKO cells was also sufficient to restore rapid lysosomal repair (Extended Data Fig. 8g–h), in line with cholesterol’s known role in increasing membrane stability35. Thus, OSBP provides an auxiliary cholesterol transport pathway for rapid lysosomal repair.
ATG2 is a PS effector in lysosomal repair
ATG2A, a recently reported lipid transporter potentially activated by PS36,37,38,39, was rapidly recruited to damaged lysosomes (Fig. 4a,b and Supplementary Video 4). Unlike PI4K2A, lysosomal Ca2+ release was insufficient to recruit ATG2A (Supplementary Video 5). Cells lacking both ATG2A and ATG2B had marked defects in rapid lysosomal repair, which was fully rescued by ATG2A re-expression (Fig. 4c and Extended Data Fig. 9a,b). The lysosomal repair defects in ATG2A and ATG2B double-knockout (ATG2A/B-DKO) cells took longer to develop (2–4 h) than in PI4K2A-KO or ORP-QKO cells (0.5–1 h), probably owing to OSBP-mediated compensatory repair activity (Extended Data Fig. 9c–e). Thus, ATG2A and ATG2B act in parallel with OSBP and cholesterol but not PS, suggesting that ATG2A and ATG2B might be downstream effectors of PS in rapid lysosomal repair. Supporting this notion, the in vitro lipid transport activity of ATG2A was potently stimulated by the addition of PS in the acceptor membrane (Fig. 4d,e).

a, Live-cell imaging demonstrating rapid recruitment of eGFP–ATG2A to damaged lysosomes in U2OS cells stably expressing eGFP–ATG2A and LAMP1–mCherry. Scale bar, 10 μm. b, Quantification of eGFP–ATG2A puncta in individual U2OS cells upon LLOME treatment. c, eGFP–galectin-3 assay showing defects of rapid lysosomal repair in four independent clones of ATG2A/B-DKO cells. Data are mean ± s.e.m.; 50–100 cells, n = 3 for each condition. See more details in Extended Data Fig. 9a,b. d, Experimental design for the reconstitution of PS-stimulated lipid transport by ATG2A using a FRET-based assay. e, PS in acceptor liposomes potently stimulates ATG2A-dependent NBD fluorescence. Data are mean ± s.e.m.; n = 3 per condition. f, Illustration of lipid transport mutants of ATG2A. Flexible loops outside of the main structure are not shown. g, In vitro lipid transport assays testing the activity of various ATG2A mutants. Data are mean ± s.e.m.; n = 3 per condition. h, eGFP–galectin-3 assay demonstrating that ATG2A lipid transport mutants cannot rescue rapid lysosomal repair in ATG2A/B-DKO cells. Data are mean ± s.e.m.; 50–100 cells, n = 3 per condition. See fluorescence images in Extended Data Fig. 9f. i, In vitro ATG2A lipid transport assays in the presence of 1 μM maltose-binding protein (MBP) or MBP-tagged ATG2A-CT (MBP–CT). Data are mean ± s.e.m.; n = 3 per condition. j, Lysosomal repair defects in wild-type cells stably expressing LAMP1–CT but not in cells expressing LAMP1–mCherry or mCherry–CT. Data are mean ± s.e.m.; 50–100 cells were quantified, n = 3 per condition. See fluorescence images in Extended Data Fig. 9h. k, A cartwheel drawing of amino acids 1757–1789 from ATG2A-CT. Conserved residues mutated in the 5E mutant are indicated. l, The ATG2A(5E) mutant is not activated by PS-positive acceptor liposomes (A3) in lipid transport assays. Data are mean ± s.e.m.; n = 4. A2, charge-matched control acceptor liposomes. m, The ATG2A(5E) mutant cannot rescue rapid lysosomal repair in ATG2A/B-DKO cells. Data are mean ± s.e.m.; 50–100 cells were quantified, n = 3 per condition. See fluorescence images in Extended Data Fig. 9n. Unpaired, two-tailed t-tests.
ATG2 repairs lysosomes by lipid transport
ATG2 is proposed to transfer lipids between membranes through its hydrophobic tunnel36,37,38,39. Similar to previously reported mutants of another tunnel-like protein—VPS13 (ref. 40)—we designed three independent mutants (Mut1–3) in which the central hydrophobic tunnel of ATG2A is blocked (Fig. 4f), based on its predicted AlphaFold structure41,42. All three mutants were completely deficient in lipid transport in vitro (Fig. 4g) and were unable to rescue rapid lysosomal repair (Fig. 4h and Extended Data Fig. 9f) or macroautophagy (Extended Data Fig. 9g) when expressed in ATG2A/B-DKO cells. In addition, ATG2A without its carboxyl terminus (ΔCT) lost its in vitro lipid transport activity (Fig. 4g) and was unable to reconstitute lysosomal repair activity in ATG2A/B-DKO cells (Fig. 4h and Extended Data Fig. 9f). Therefore, the lipid transport activity of ATG2A is required for rapid lysosomal repair.
We next focused on the CT domain of ATG2, which is predicted to form amphipathic helices (Fig. 4f), a structure that is well known for membrane embedding43,44. Since ATG2A probably binds to the acceptor membrane (for example, damaged lysosomes) through its CT domain, we reasoned that the CT fragment could potentially compete with full-length ATG2A and ATG2B for membrane binding. Supporting this notion, a purified CT fragment (ATG2A amino acids 1723–1938) acted in a dominant-negative manner to inhibit in vitro lipid transport by full-length ATG2A (Fig. 4i). Similarly, overexpression of lysosomal-anchored ATG2A-CT (LAMP1–CT), but not soluble mCherry-tagged ATG2A-CT (mCh-CT), caused marked lysosomal repair defects in wild-type cells, which phenocopied ATG2A/B-DKO cells (Fig. 4j and Extended Data Fig. 9h) without however, affecting macroautophagy (Extended Data Fig. 9i). Indeed, rapid lysosomal repair, macroautophagy and lysosomal damage-induced LC3 lipidation appear to be three distinct pathways (Extended Data Fig. 9h,i,o–u and Supplementary Table 6). These data suggest that ATG2 uses its amphipathic CT to interact with lysosomal membranes for lipid delivery and that the role of ATG2A and ATG2B in lysosomal repair and macroautophagy are separable, although both processes require the lipid transport activity of ATG2.
We then tested whether PS regulates the membrane binding of ATG2A-CT. In vitro liposome pulldown assays revealed substantial PS-stimulated membrane binding of ATG2A-CT (Extended Data Fig. 9j). Positively charged residues on the polar face of amphipathic helices in other proteins have been reported to sense PS-positive membranes45,46. A similar group of highly conserved basic residues was identified on the polar face of ATG2A-CT, on the basis of which we generated the 5E mutant (Fig. 4k and Extended Data Fig. 9k,l). Purified ATG2A-CT(5E) retained normal basal membrane binding (Extended Data Fig. 9j), suggesting correct amphipathic folding, but did not demonstrate PS-stimulated membrane binding (Extended Data Fig. 9j). Notably, the 5E mutant of full-length ATG2A (ATG2A(5E)) acted similarly to ATG2A lacking the CT domain (ATG2A(ΔCT)). ATG2A(5E) was not activated by PS in lipid transport assays in vitro (Fig. 4l), exhibited weaker retention on damaged lysosomes (Extended Data Fig. 9m), and did not rescue lysosomal repair in ATG2A/B-DKO cells (Fig. 4m and Extended Data Fig. 9n). Thus, rapid lysosomal repair requires PS-stimulated lysosomal lipid transport by ATG2 (Supplementary Table 5).
Discussion
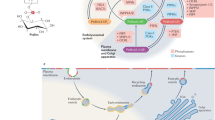
Our study reveals a phosphoinositide-initiated membrane tethering and lipid transport (PITT) pathway that is essential for rapid lysosomal repair (Extended Data Fig. 10). PI4K2A generates PtdIns4P, the initiating signalling lipid that decorates damaged lysosomes. In response to PtdIns4P, the recruitment of multiple ORPs establish new and extensive ER MCSs, leading to ER-to-lysosome transport of PS and cholesterol, as complementary and parallel mechanisms to support rapid lysosomal repair. These observations are broadly consistent with other recent reports implicating OSBP in PtdIns4P-dependent endolysosomal cholesterol transport34,47. Finally, lysosomal PS accumulation activates ATG2-mediated lipid delivery for direct membrane repair, a function for ATG2 that is independent of its canonical role in autophagosome formation (Supplementary Table 6). The current work, as well as previous studies, are limited by the lack of evidence demonstrating high volume in vitro lipid transport by ATG2. Such in vitro activity might be achieved once the factors ensuring directional lipid transport by ATG2 are identified. Nevertheless, we demonstrated strict dependence of rapid lysosomal repair on ATG2 lipid transport activity, which suggests that cellular ATG2 activity is likely sufficiently robust to mediate membrane repair, although additional studies are required to solidify this conclusion.
Increasing evidence implicates a decline in lysosomal integrity and function in normal ageing and in age-related disease1,2,3. In cell-based models, we observed that deletion of PI4K2A caused exacerbation of a lysosomal storage disease (Extended Data Fig. 4i) and tau fibril spreading (Fig. 1j, Extended Data Fig. 4h). Coupled with the neurodegenerative phenotype previously reported in Pi4k2a-KO mice48, our observations suggest that the PITT pathway may have therapeutic implications for a wide range of age-dependent diseases characterized by impaired lysosomal function.
Methods
Cell culture, chemicals, and treatments
All cell lines were from ATCC. 293T, U2OS, PC3 and BJ cells were authenticated through short tandem repeat profiling and the profiling data are publicly available from ATCC. Cell lines in this study have different morphologies and growth rates and contamination were constantly monitored. All cell lines used in this study were free from mycoplasma contamination based on PCR detection and were regularly maintained with mycoplasma reagent. 293T, U2OS, COS7, and BJ cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM); PC3 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640. Both media were supplemented with 8% fetal bovine serum (FBS) and penicillin/streptomycin. All cells were maintained at 37 °C with 5% CO2. LLOME (Sigma, L7393) was dissolved in ethanol and stored in aliquots at −20 °C. Gly-Phe-β-naphthylamide (GPN, Cayman Chemical, 14634), Brefeldin A (Sigma, B5936), ML-SA1 (Cayman Chemical, 29958), MEK6–83 (Cayman Chemical, 21944), O-methyl-serine dodecylamide hydrochloride (MSDH, Avanti Polar Lipids, 850546) were dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C. Nano-silica (Invivogen, tlrl-sio-2) was suspended in water and sonicated right before each use. Tau fibrils (SPR-329) were from StressMarq. Chemicals, silica, or tau fibrils were directly added to cell culture media to induce lysosomal membrane damage with equal volume of vehicle (ethanol or DMSO) added into control wells. For cell treatment that requires medium change, fresh media were pre-warmed overnight in an empty dish in the same incubator, which minimizes disturbance to cells caused by medium change.
Antibodies
The following antibodies were from Santa Cruz Biotechnology: ORP9 (A-7, sc-398961, immunofluorescence 1:1,000); PI4K2A (sc-390026, immunofluorescence 1:500); LAMP2 (sc-18822, immunofluorescence 1:200); CD2AP (sc-25272, western blot 1:1,000); CHMP3 (sc-166361, immunofluorescence 1:1,000); Golgin-97 (sc-59820, immunofluorescence 1: 500); LAMP1 (sc-20011, immunofluorescence 1:200); SQSTM1/p62 (sc-28359, immunofluorescence 1: 1,000, western blot 1: 2,000); GAPDH (sc-365062, western blot 1:5,000); tubulin (sc-5286, western blot 1:3,000); GFP (sc-9996, western blot 1:3,000, immunoprecipitation 2 μg per reaction). The PI4KB antibody (611816, western blot 1:1,000, immunofluorescence 1:200) and GM130 antibody (610822, immunofluorescence 1:1,000) were from BD Biosciences. Anti-rabbit IgG (H+L) CF350 (SAB4600412, immunofluorescence 1:200), Flag (M2, immunofluorescence 1:1,000; F7425, western blot 1:3,000), Flag M2 agarose (immunoprecipitation 10 μl beads per reaction) were from Sigma. Rabbit anti-LAMP1 monoclonal antibody (9091, immunofluorescence 1:100) was from Cell Signaling. Mouse anti-Alix antibody (634502, immunofluorescence 1:1,000) was from Biolegend. The following antibodies produced in rabbits were from Proteintech Group: STAM (12434-1-AP, western blot 1:1,000); IST1 (19842-1-AP, immunofluorescence 1:1,000, western blot 1:2,000); PI4KA (12411-1-AP, western blot 1:1,000); STAMBP (11346-1-AP, western blot 1:1,000); ORP9 (11879-1-AP, western blot 1:1,000); OSBP (11096-1-AP, western blot 1:1,000, immunofluorescence 1:1,000); PI4K2A (15318-1-AP, western blot 1:1,000); ATG2A (23226-1-AP, western blot 1:500); ATG2B (25155-1-AP, western blot 1:500), galectin-3 (14979-1-AP, immunofluorescence 1:200). The following antibodies were from Bethyl Laboratories: ORP11 (A304-580A, western blot 1:2,000, immunofluorescence 1:1,000) and ORP10 (A304-885A, western blot 1:1,000). Alexa-488/594- and Pacific Blue-conjugated secondary antibodies were obtained from ThermoFisher Scientific.
DNA cloning
For stable expression of proteins in this study, the relevant DNA sequences were cloned into pCDH-CMV-MCS between restriction sites BamHI and NotI. All cDNA sequences were of human origin unless otherwise specified. The cDNAs for ORP3, ORP5, ORP9, PI4K2A, ATG2A, Osh6 and Kes1 were from the DNASU plasmid repository at Arizona State University (HsCD00829348, HsCD00439845, HsCD00820675, HsCD00618068, HsCD00863023, ScCD00012609 and ScCD00009548, respectively). The ORP10 cDNA was a gift from V. Olkkonen. The ORP11 cDNA was purchased from Sino Biological (HG22085-U). The cDNA for PI4K2B was originally from Addgene (pDONR223-PI4K2B, 23507) which contained a 15-bp insert in the middle that introduced a premature stop codon, leading to the production of an inactive fragment majorly localized into the nucleus. This insert was removed when cloning PI4K2B into pCDH-CMV-MCS to express the full-length wild-type protein. For most fusion proteins, the peptide linker GSGSGS was used. To generate CRISPR guide RNA-resistant mutations or other point mutations, two fragments of the cDNA of interest were amplified with their joining ends containing the target mutations and then fused together into pCDH-CMV-MCS through infusion reactions. To generate the Lyso-TurboID construct (pCDH-LAMP1-mGFP-TurboID), the LAMP1–mGFP sequence was amplified from Addgene Plasmid 34831 and fused with TurboID sequence into pCDH-CMV-MCS. To constitutively target PI4K2B to lysosomes, the open reading frame (ORF) of PI4K2B was fused with the LAMP1–mCherry sequence into pCDH to achieve stable expression of LAMP1– mCherry–PI4K2B. Amino acids 1–382 of OSBP without its C-terminal cholesterol transport domain were used as an ER–lysosome tether when designing artificial fusion proteins to rescue ER-to-lysosome lipid transport in ORP-QKO cells. In mCherry–OSBP–ORP9, the sequences encoding mCherry, OSBP residues 1–382 and ORP9 residues 337–746 were fused together with GSGSGS linkers in between. Similarly, OSBP residues 1–382 was fused with ORP10 residues 348–764, ORP11 residues 322–747, and the full-length sequences of Kes1 or Osh6 to generate the relevant fusion proteins.
Stable cell line generation
Except for RNA interference and transient expression of GFP–P4M and SARS-CoV-2-ORF3A, all other experiments were based on stable cell lines with genetic knockout and/or stable ectopic expression of proteins. pCDH vectors carrying the ORF sequences of specific genes were used to generate lentiviruses to infect recipient cells, after which the cells were treated with puromycin to select against uninfected cells. In most situations, no selection was necessary, as the infection rates were close to 100%. A titration of viruses was usually performed to find out the lowest viral dosage that can lead to expression of the target protein in more than 90% of cells. To generate knockout cell lines, multiple CRISPR–Cas9 guide sequences for each gene were tested to identify the guide that most effectively diminished the target protein in the CRISPR pool. LentiCRISPR.v2 (Addgene, 52961) carrying the selected guide sequence was used for lentivirus packaging. The infection rate of U2OS cells by CRISPR viruses was remarkably high that no cell death was observed after puromycin treatment of infected pools when all uninfected control cells died. See Supplementary Table 4 for the CRISPR guide sequences.
RNA interference
Double knockdown of ALIX and TSG101 was performed to block the recruitment of ESCRT subunits to damaged lysosomes, as previously described7. For transient knockdown of PI4K2A and PI4KA, two different siRNA sequences were pooled and transfected at the same time using Lipofectamine RNAiMAX, following the manufacturer’s instructions. See Supplementary Table 4 for the siRNA sequences.
Biotinylation and purification of lysosomal surface proteins
293T cells stably expressing Lyso-TurboID were cultured until 80% confluent (Extended Data Fig. 1d). Cells were treated with 500 μM LLOME or ethanol (viechle control) for 30 min to induce lysosomal membrane damages and membrane repair events, followed by the addition of 50 μM biotin. After 30 min of incubation allowing for biotinylation of lysosomal surface proteins, cells were washed twice with 10 ml pre-chilled PBS, scraped into 1 ml of cold PBS per dish, and transferred into 1.5 ml tubes. Cells were spun down at 1,000g for 3 min and resuspended in 500 μl hypotonic buffer (10 mM HEPES, pH 7.5, 5 mM MgCl2, 10 mM NaCl). Resuspended cells were passed through a 25 G needle 10 times and spun at 1,000g for 3 min to remove intact cells and nuclei in the pellet (P1). The supernatant S1 was further spun at 20,000g for 20 min to precipitate most membranes including lysosomes with biotinylated proteins (P20). P20 was resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Triton X-100, 2 mM NaF, 5 mM MgCl2, protease inhibitor cocktail) and briefly sonicated to completely dissolve the membrane. The samples were further spun at 15,000 g for 5 min to remove any undissolved aggregates. The supernatants were transferred to a new tube with 100 μl well-resuspended streptavidin magnetic beads and rotated at 4 °C for 2 h. The beads were washed twice sequentially with three different buffers: buffer 1 (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.8% SDS), buffer 2 (50 mM Tris-HCl, pH 8.0, 1 M NaCl, 0.5% Triton X-100), buffer 3 (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Triton X-100). Proteins bound on the beads were eluted by adding 50 μl 2× SDS sample buffer (0.1 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.01% bromphenol blue) and heated to 95 °C for 5 min.
Immunofluorescence
Cells were seeded on glass coverslips (Warner Instruments, 64–0712) in 24-well plates. PC3 cells took 2 to 3 days to firmly attach to glass surfaces and were treated 2 to 3 days after seeding. All the other cell lines attached to glass coverslips quickly and were treated 24 h after seeding. LLOME and GPN were directly added to each well to induce lysosomal membrane damage. When medium change was needed after lysosomal damage was induced, fresh media were pre-warmed overnight in an empty dish in the same incubator to minimize any potential disturbance to the cells induced by medium change. Cells were fixed with 4% paraformaldehyde (PFA) in PBS for 30 min at room temperature, permeabilized with 0.1% Triton X-100 for 5 min at room temperature, and blocked for 1 h at room temperature with 1× fluorescent blocking buffer (ThermoFisher Scientific, 37565) which was also used to dilute all primary and secondary antibodies in immunofluorescence staining. Primary antibody incubation was performed at room temperature for 2 h or at 4 °C overnight. Cells on glass coverslips were washed with PBS three times and incubated with fluorescently labelled secondary antibodies at room temperature for 30 to 60 min, followed by PBS wash for three times. When necessary, DAPI staining of the nuclei was carried out after secondary antibody staining. The coverslips were mounted on slides using VECTASHIELD HardSet Antifade Mounting Medium (Vector Laboratories, H-1400). Images were taken using a Leica SP8 LIGHTNING confocal system using the built-in software Leica Application Suite X 3.5.5.19976. Multiple controls were included to avoid nonspecific staining and cross talks between channels. An Okolabstage-top incubator was used for live-cell imaging on the same confocal system. All images in the same figure panel were taken under the same software setting and equally processed in Adobe Photoshop. Colocalization was quantified by Coloc2 in ImageJ with Person’s coefficient used to determine the level of colocalization.
Immunoprecipitation
Immunoprecipitation was done as previously described49. 293T cells stably expressing Flag- and eGFP-tagged ORPs proteins were grown to confluent in six-well plates. Cells were washed briefly with cold PBS and lysed in 1 ml immunoprecipitation buffer per well (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2 mM NaF, 2 mM Na3VO4, 0.5% Triton X-100, protease inhibitor cocktail). Lysed cells were scraped into 1.5 ml tubes and briefly sonicated before centrifugation at 15,000 g for 10 min. The supernatants were collected as whole cell lysates, 100 μl of which were saved as input controls. The remaining 900 ul lysates were split into 3× 300 μl aliquots for immunoprecipitation with anti-Flag, anti-GFP, and normal IgG, respectively. Mouse anti-Flag M2 agarose gel (Sigma) was used to immunoprecipitate Flag-tagged proteins, whereas mouse anti-GFP (sc-9996, Santa Cruz) or normal IgG were used with protein A/G beads. The immunoprecipitation mixtures were rotated at 4 °C for 2 h and then washed three times with immunoprecipitation buffer without protease inhibitors. The immunoprecipitated proteins on beads were eluted by heating in 2× SDS loading buffer for 5 min, followed by immunoblotting analysis of the ORP proteins.
Lipofuscin detection
Cells on coverslips were fixed with 4% PFA in PBS for 30 min at room temperature and washed with PBS before mounted on slides using VECTASHIELD HardSet Antifade Mounting Medium (Vector Laboratories, H-1400). Lipofuscin was excited at 448 nm and emissions from 453–750 were collected as confocal images.
Immunoblotting
Cells were washed twice with cold PBS and scraped into lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 2 mM NaF, 5 mM MgCl2, protease inhibitor cocktail). The samples were briefly sonicated to disrupt membrane aggregates and then spun at 15,000 g for 10 min to remove insoluble components. The supernatants were collected as whole cell lysates which is mixed with equal volume of 2× SDS loading buffer (0.1 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.01% bromphenol blue) and heated at 95 °C for 5 min. Equal amounts of total proteins were separated on a 4–20% precast polyacrylamide gel (Biorad, 4561096) and transferred onto 0.2 µm nitrocellulose membranes using the Trans-Blot Turbo system. The membrane was blocked with StartingBlock Blocking Buffer (ThermoFisher, 37542) for 30 min at room temperature and then incubated with primary antibody at 4 °C overnight. The membrane was then washed and incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. The membrane was washed and exposed with SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher, 34580) in a ChemiDoc MP Imaging System.
Lysosomal PtdIns4P production assay
The production of PtdIns4P on damaged lysosomes were tracked by a stably expressed PtdIns4P probe OSBP-PH–GFP17 or alternatively by transiently transfected GFP–P4M50 of which the stable cell line generation was not successful. Stable expression of OSBP-PH–GFP was achieved by a lentiviral approach. The DNA sequence of OSBP PH domain was cloned into pCDH, a lentiviral vector, with GFP fused on the C-terminal end. All cell types in which we have tried stable expression of OSBP-PH–GFP showed constitutive targeting of the probe to the Golgi complex with the other regions of the cell demonstrating only a diffuse signal. The PtdIns4P production on damaged lysosomes was assessed by tracking the lysosomal recruitment of OSBP-PH–GFP upon lysosomal damage by either 1 mM LLOME for 30 min or 100 μM GPN for 5 min. To rule out the interference of pre-existing Golgi-localized OSBP-PH-GFP puncta in assessing LMP-induced lysosomal puncta of the probe, cells were pre-treated with brefeldin A to disassemble the whole Golgi complex before LLOME-mediated lysosomal damage. Note that the lysosomal recruitment of PI4K2A and ORP9/10/11 happened much more quickly (within 10 min) than the recruitment of OSBP-PH–GFP (peaks at 30 min). This is likely because ORP9/10/11 have higher affinity to PtdIns4P than does OSBP-PH–GFP. Live-cell imaging and confocal analysis of fixed cells stained for endogenous markers of damaged lysosomes were performed. To ensure the lysosomal recruitment of OSBP-PH-GFP was due to lysosomal PtdIns4P production, we identified the enzyme PI4K2A as the source of PtdIns4P on damaged lysosomes. We confirmed that the lysosomal recruitment of OSBP-PH–GFP was abolished in cells lacking PI4K2A, even though their lysosomes could still be damaged.
Cell-based PS transport assay
Cells stably expressing the genetically encoded PS probe GFP–Lact-C2 (ref. 28) were treated with 1 mM LLOME for 20. This probe shows strong background signal from the cytosol and the plasma membrane, which was removed by a brief detergent wash before fixation. Specifically, cells with or without LLOME treatment were gently rinsed for 5–10 s with 0.1% Triton X-100, followed by PBS rinse for another 5–10 s. Cells were then immediately fixed in 4% PFA for 10 min. Fixed cells were blocked with 1× fluorescent blocking buffer (ThermoFisher Scientific, 37565) for 30 min at room temperature and stained with antibodies against the lysosome marker LAMP1 and/or the damaged lysosome marker IST1. The colocalization of GFP–Lact-C2 and LAMP1 was quantified by Person’s correlation coefficient.
Lysosomal repair assays
We aimed to analyse lysosomal repair in live cells with minimal interference from chemical staining or washing steps. Two approaches were established to monitor lysosomal repair in live cells, with one based on the stable expression of lyso-pHluorin and the other of eGFP–galectin-3.
Lyso-pHluorin is based on the lysosomal marker CD63 as the backbone with the GFP variant pHluorin that exhibits no fluorescence in the acidic lysosomal lumen but does emit when the lysosomal membrane is damaged with its lumenal pH neutralized20. Thus, the de-quenching and re-quenching of the lyso-pHluorin signal reflects lysosomal damage and repair after LLOME treatment. A U2OS lyso-pHluorin stable cell line was treated with 1 mM LLOME for 5 min and then washed and chased for up to 12 h. To minimize disturbance to cells caused by medium change especially after acute lysosomal damage, fresh media were pre-warmed overnight in an empty dish in the same incubator, which were used to gently rinse cells twice to wash out LLOME. Note, the washing step is critical to ensure efficient lysosomal repair, which can cause cell death and detachment if not properly handled. The lyso-pHluorin signals were checked every 1–2 h with live-cell imaging. Cells with more than 5 lyso-pHluorin puncta were defined as unrepaired. The number of repaired and unrepaired cells were manually counted. In each condition, 100–500 cells were counted. The whole process was independently repeated at least three times.
The other approach is based on stably expressed eGFP–galectin-3 which accumulates in the lysosomal lumen when the membrane pore is big enough to allow its entrance from the cytosol7,51. When stably expressed, eGFP–galectin-3 only shows diffuse cytosol signal similar to that of lyso-pHluorin. Upon lysosomal membrane damage, the number and sizes of eGFP–galectin-3 puncta correlate with the degree of membrane damage5,7. U2OS cells stably expressing eGFP–galectin-3 were treated continuously with 1 mM LLOME for 1 to 5 h and monitored for the formation of eGFP–galectin-3 puncta over time using live cells. The total punctate intensities above threshold were quantified in ImageJ. Note that images should be taken using live cells, as fixation causes puncta formation of eGFP–galectin-3.
Protein purification
ORP9, ORP11 and OSBP were purified by two-step purifications using 6×His tag and twin-strep-tag. All three proteins carry an N-terminal Flag-tag and a C-terminal twin-strep-tag. The FATT motif of ORP9/OSBP was replaced with a 6×His tag. A GGGS linker was added after the 6×His tag and a GSGSGS linker was added between the C-terminal end of each protein and the twin-strep-tag. As ORP11 does not carry a strong FATT motif, a 6×His tag followed with a GGGS linker was inserted into ORP11 between amino acids 321 and 322. The coding sequence for the three fusion proteins were cloned into pCDH for stable protein expression in HEK293T cells. The cells were washed with PBS, pelleted, and resuspended in lysis buffer containing 100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 1% Triton X-100, and protease inhibitor cocktail. Cells were sonicated and centrifuged at 15,000 g for 10 min. His-tagged proteins from the supernatant were purified using Ni-NTA agarose and eluted into elution buffer containing 100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 300 mM imidazole. The eluted protein was dialysed and further purified using Strep-Tactin column and eluted into 20 mM Tris-HCl, pH 8.0, 200 mM NaCl. The eluted protein was further dialysed with 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT, aliquoted, and stored at −80 °C.
Flag–MBP, Flag–MBP–CT, Flag–ATG2A and all related mutants were purified with anti-Flag M2 beads. Flag–MBP and Flag–MBP–CT were cloned into pCDH and then stably expressed in 293T cells by a lentiviral approach. The coding sequence for Flag–ATG2A with a 26-amino acid linker followed by a 6xHis tag at its C-terminal end was cloned into pCDNA3.0 and transiently transfected into 293T cells. Forty-eight hours after transfection, cells were washed with PBS and resuspended into lysis buffer containing 100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 1% Triton X-100, and protease inhibitor cocktail. After sonication and clearance centrifugation, the lysates were incubated with M2 beads for 1 h at 4 °C with rotations. The mixture was then transferred into an open column and sequentially, extensively washed with three buffers. Buffer 1: 100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol; buffer 2: 100 mM Tris-HCl, pH 8.0, 500 mM NaCl; buffer 3: 20 mM Tris-HCl, pH 8.0, 200 mM NaCl. Bound proteins were then eluted with 0.2 mg ml−1 3×Flag peptide in buffer 3. DTT was added to the eluted proteins at a final concentration of 1 mM. Proteins were aliquoted and stored at −80 °C.
Liposome preparation
The following lipids used for liposome preparation were from Avanti polar lipids: 18:1-12:0 NBD–PS (810195), 18:1 NBD–phosphatidylinositol (810145), NBD–cholesterol (810252), 18:1 lissamine–rhodamine–PE (810150), DOPC (850375), DOPE (850725), DOPS (840035), DGS–NTA (790404), brain PtdIns4P (840045), biotin–phosphatidylinositol (870282). Lipids dissolved in chloroform were mixed in glass tubes and dried to thin films under nitrogen flow and were desiccated overnight. Five hundred microlitres of rehydration buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT; or 25 mM HEPES, pH 7.5 150 mM NaCl, 1 mM DTT) was added to dried lipids to make 1 mM final total lipid concentration. The tubes was shaken vigorously for 30 min at room temperature, subject to 10 freeze–thaw cycles using liquid nitrogen and 37 °C water bath, and then extruded through a 100-nm filter 21 times for ORP9/11-mediated lipid transport assays. The donor liposomes for ATG2A-mediated lipid transport tests were sonicated, and acceptor liposomes were extruded through 50 nm filter 31 times. ATG2A lipid transport activity was only observed when sonicated donors were used, whereas the sizes of extruded ATG2A acceptor liposomes did not affect the overall lipid transport activity.
In vitro reconstitution of lipid transport
Lipid transport in vitro was measured in 40 μl reaction buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT) in a 384-well plate through a FRET-based assay29,36,37,38,39. The donor and acceptor liposomes (25 μM total lipids for each) were mixed and the NBD fluorescence was monitored (excitation at 460 nm, emission at 535 nm) upon the addition of 100 nM purified proteins.
For the ORP9/11-mediated lipid transport assays, the donor liposomes contained 66% DOPC, 25% DOPE, 5% DGS–NTA (Ni), 2% 18:1-12:0 NBD–PS (or 2% NBD–cholesterol or 2% TopFluor(TF)-cholesterol), and 2% Rh–phosphatidylethanolamine, the PtdIns4P acceptor liposomes contained 70% DOPC, 25% DOPE, 5% PtdIns4P, and the neutral acceptor liposomes contained 75% DOPC, 25% DOPE. Note that, to allow for efficient PS recognition by ORPs, 18:1-12:0 NBD–PS carries the NBD tag at the end of one fatty acid chain instead of the serine head group. We introduced a His tag in the middle of both ORP9/11 (replacing the FATT motif in ORP9) so that they can bind to the donor membrane containing lipids labelled with DGS–NTA (Ni), mimicking ORP interactions with VAPA/B on the ER through FATT motifs. The acceptor membrane carries 5% of PtdIns4P for ORP recruitment to mimic damaged lysosomes without fluorescent lipids. When testing the lipid transport activity of individual ORP9 or ORP11, 100 nM of each was used. When testing the activity of ORP9–ORP11 heterodimers, the two proteins were premixed at a molar ratio of 1:1 and 100 nM of total proteins (50 nM each) were added to the liposome mixtures to start the reaction.
For ATG2A assays, the donor liposomes contained 63% DOPC, 25% DOPE, 5% DGS–NTA (Ni), 2% 18:1 NBD–phosphatidylethanolamine, 2% Rh–phosphatidylethanolamine, and 3% DOPS, the PS-positive acceptor liposomes (A3) contained 50% DOPC, 25% DOPE, 5% PtdIns4P, and 20% DOPS, the PS-negative acceptor liposomes (A1) contained 70% DOPC, 25% DOPE, and 5% PtdIns4P, an additional set of charge-matched control acceptor liposomes (A2) contained 50% DOPC, 25% DOPE, and 25% PtdIns4P. ATG2A carried a 26 aa linker followed with His tag at its C-terminal end, as previously described37. OSBP, which does not transport PS or NBD–phosphatidylethanolamine, was used as a physiologically relevant membrane tether between the donor and acceptor liposomes. Note that OSBP appeared to exhibit nonspecific activity towards NBD–phosphatidylethanolamine which could be avoided after a pre-incubation of OSBP at room temperature for 1 h. Similar to ORP9/11, a His tag was added to the middle of OSBP (see protein purification for details) so that it can tether the donor and acceptor membrane through the His tag and its N-terminal PH domain. After mixing donor and acceptor liposomes with 100 nM OSBP, 100 nM ATG2A was added immediately before fluorescence reading. All ATG2A lipid transport mutants and the impact of MBP–CT on ATG2A activity were tested with A3 acceptor liposomes.
Lysosomal purification and lipid extraction
Lysosomes were purified through LAMP1-Twin-Strep and streptavidin beads as described52. U2OS cells stably expressing LAMP1-Twin-Strep grown to confluent in 15 cm dishes were washed three times with chilled PBS, scraped into 1 ml potassium buffer (KPBS, 136 mM KCl, 10 mM KH2PO4, pH 7.5). Cells were centrifuged for 5 min at 1,000 g, resuspend in 1 ml of potassium buffer, and then passed through a 25 G needle 10 times. Cell homogenates were centrifuged at 1,000 g for 2 min and 800 μl supernatant was transferred to a new tube with 100 μl of streptavidin magnetic beads. The mixture was incubated with rotation for 5 min, followed by bead collection and three washes with a magnetic stand. For each wash, the beads were resuspended in 1 ml potassium buffer and transferred into a new tube. After washing, half of the beads were directly heated in SDS loading buffer to extract lysosomal proteins as input controls. The other half of the beads were used for total lysosomal lipid extraction using the methyl-tert-butyl ether (MTBE) extraction protocol which allows fast and clean lipid recovery53. The washed beads were suspended in 200 ul water and transferred to a glass tube with a Teflon-lined cap followed by the addition of 1.5 ml methanol. The tube was vortexed and 5 ml of MTBE was added. The mixture was then incubated at room temperature on a shaker for 1 h. Phase separation was induced by the addition of 1.25 ml ultrapure water. After 10 min of incubation at room temperature, the tube was centrifuged at 1,000 g for 10 min. The upper organic phase was collected, and the lower phase was re-extracted with 2 ml of the MTBE:methanol:water (10:3:2.5, v/v/v) mixture. Combined organic phase solutions containing lysosomal lipids were dried under a nitrogen flow. Extracted lipids were dissolved in 200 μl of CHCl3:methanol:water (60:30:4.5, v/v/v) and 10 μl of each sample was spotted onto a PVDF membrane using a glass capillary. The membrane was completely dried for 1 h with air flow in a fume hood and then immediately used for PS detection by GFP–Lact-C2.
PS detection by lipid–protein overlay assay
PIP strips membrane or lipid-spotted PVDF membrane was incubated overnight at 4 °C with binding buffer (1× PBS, 3% BSA, 0.1% Tween 20) containing 1 μg ml−1 purified GFP–Lact-C2. The membrane was washed the next day with the binding buffer 3 times 5 min each, followed by imaging to capture GFP signal.
Liposome binding assay
Liposomes were made similarly to acceptors in the ATG2A lipid transport assay except the addition of 2% Biotin–phosphatidylethanolamine to all liposomes. Magnetic streptavidin beads (10 μl) were incubated with 10 μl 500 uM liposomes (total lipid concentration) in 500 μl binding buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT) on a rotation mixer for 30 min at room temperature. Following this, 1 μg of purified MBP–CT or its 5E mutant was added together with 100 μg BSA. The samples were rotated for 10 min at room temperature to allow for liposome binding and then washed twice with the binding buffer above plus 0.04% BSA. For each wash, the beads were resuspended and transferred to new Eppendorf tubes. Proteins bound to immobilized liposomes were extracted by directly heating the beads in SDS loading buffer for 5 min, followed by immunoblotting analysis.
Tau fibril spreading assay
Active human recombinant tau441 (2N4R) P301S mutant protein pre-formed fibrils (SPR-329) were purchased from StressMarq and used for the cell-based tau spreading assay. U2OS cells stably expressing tau K18-P301L/V337M-mRuby2 without pre-formed mRuby2 puncta were incubated with 250 nM sonicated tau fibrils and the formation of K18–mRuby2 aggregates in live cells were examined by fluorescence microscopy on a daily basis.
Software
Confocal images were taken using a Leica SP8 LIGHTNING confocal system with the built-in software Leica Application Suite X 3.5.5.19976. Images were processed and assembled in Adobe Photoshop 20.0.4. Schematic illustrations were made with Microsoft Office Power Point Professional Plus 2016 and Biorender. AlphaFold structure of ATG2A was visualized in the PyMOL Molecular Graphics System, Version 2.4.0 Schrödinger, LLC. Cartwheel drawing of amino acid sequence was performed with HeliQuest54. Unpaired, two-tailed t-tests Statistical analysis was performed in Microsoft Office Excel Professional Plus 2016. Graphs were generated in Excel and GraphPd Prism 9.0.0. For colocalization quantification, Pearson's correlation coefficient was quantified by Coloc 2 version 3.0.5 in ImageJ (Fiji 1.53f51).
Statistics and reproducibility
All experiments were independently reproduced at least three times, except the initial Lyso-TurboID screen which was performed once. Strict standards were applied to screen for robust and unbiased results. No statistical methods were used to predetermine sample size. The investigators were not blinded to allocation. Data were presented as mean ± s.e.m. Statistical significance was determined by unpaired, two-tailed t-tests.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE55 partner repository with the dataset identifier PXD028852 and 10.6019/PXD028852. Source data are provided with this paper.
References
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 4, 27 (2018).
Gómez-Sintes, R., Ledesma, M. D. & Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168 (2016).
Hung, Y.-H., Chen, L. M.-W., Yang, J.-Y. & Yang, W. Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 4, 2111 (2013).
Maejima, I. et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347 (2013).
Radulovic, M. et al. ESCRT‐mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 37, e99753 (2018).
Skowyra, M. L., Schlesinger, P. H., Naismith, T. V. & Hanson, P. I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078 (2018).
López-Jiménez, A. T. et al. The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PLoS Pathog. 14, e1007501 (2018).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Thiele, D. L. & Lipsky, P. E. Mechanism of l-leucyl-l-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc. Natl Acad. Sci. USA 87, 83–87 (1990).
Balla, A. & Balla, T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16, 351–361 (2006).
Liu, X. & Ridgway, N. D. Characterization of the sterol and phosphatidylinositol 4-phosphate binding properties of Golgi-associated OSBP-related protein 9 (ORP9). PLoS ONE 9, e108368 (2014).
Ngo, M. & Ridgway, N. D. Oxysterol binding protein–related protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol. Biol. Cell 20, 1388–1399 (2009).
Zhou, Y. et al. OSBP-related protein 11 (ORP11) dimerizes with ORP9 and localizes at the Golgi–late endosome interface. Exp. Cell. Res. 316, 3304–3316 (2010).
Levine, T. P. & Munro, S. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and-independent components. Curr. Biol. 12, 695–704 (2002).
Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 56, 427–442.e5 (2021).
Mirza, M. et al. The CLN3 gene and protein: What we know. Mol. Genet. Genomic Med. 7, e859 (2019).
Cheng, X., Shen, D., Samie, M. & Xu, H. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 584, 2013–2021 (2010).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783 (2014).
Fujiwara, T., Oda, K., Yokota, S., Takatsuki, A. & Ikehara, Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 263, 18545–18552 (1988).
Baumlova, A. et al. The crystal structure of the phosphatidylinositol 4‐kinase II α. EMBO Rep. 15, 1085–1092 (2014).
Zhou, Q. et al. Molecular insights into the membrane-associated phosphatidylinositol 4-kinase IIα. Nat. Commun. 5, 3552 (2014).
Rost, B. R. et al. Optogenetic acidification of synaptic vesicles and lysosomes. Nat. Neurosci. 18, 1845 (2015).
Gibbons, G. S., Lee, V. M. & Trojanowski, J. Q. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 76, 101–108 (2019).
Hammond, G. R. & Balla, T. Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758 (2015).
Antonny, B., Bigay, J. & Mesmin, B. The oxysterol-binding protein cycle: burning off PI(4)P to transport cholesterol. Annu. Rev. Biochem. 87, 809–837 (2018).
von Filseck, J. M. et al. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 349, 432–436 (2015).
Mesmin, B. et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843 (2013).
Maeda, K. et al. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature 501, 257–261 (2013).
Chung, J. et al. PI4P/phosphatidylserine countertransport at ORP5-and ORP8-mediated ER–plasma membrane contacts. Science 349, 428–432 (2015).
Kawasaki, A. et al. PI4P/PS countertransport by ORP10 at ER–endosome membrane contact sites regulates endosome fission. J. Cell Biol. 221, e202103141 (2021).
Yeung, T. et al. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 (2008).
Lim, C.-Y. et al. ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–Pick type C. Nat. Cell Biol. 21, 1206–1218 (2019).
Subczynski, W. K., Pasenkiewicz-Gierula, M., Widomska, J., Mainali, L. & Raguz, M. High cholesterol/low cholesterol: effects in biological membranes: a review. Cell Biochem. Biophys. 75, 369–385 (2017).
Osawa, T., Ishii, Y. & Noda, N. N. Human ATG2B possesses a lipid transfer activity which is accelerated by negatively charged lipids and WIPI4. Genes Cells 25, 65–70 (2020).
Valverde, D. P. et al. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798 (2019).
Osawa, T. et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 26, 281–288 (2019).
Maeda, S., Otomo, C. & Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 8, e45777 (2019).
Li, P., Lees, J. A., Lusk, C. P. & Reinisch, K. M. Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J. Cell Biol. 219, e202001161 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Giménez-Andrés, M., Čopič, A. & Antonny, B. The many faces of amphipathic helices. Biomolecules 8, 45 (2018).
Drin, G. & Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 584, 1840–1847 (2010).
Pranke, I. M. et al. α-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J. Cell Biol. 194, 89–103 (2011).
Opaliński, Ł., Kiel, J. A., Williams, C., Veenhuis, M. & Van Der Klei, I. J. Membrane curvature during peroxisome fission requires Pex11. EMBO J. 30, 5–16 (2011).
Dong, R. et al. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423 (2016).
Simons, J. P. et al. Loss of phosphatidylinositol 4-kinase 2α activity causes late onset degeneration of spinal cord axons. Proc. Natl Acad. Sci. USA 106, 11535–11539 (2009).
Tan, X., Thapa, N., Sun, Y. & Anderson, R. A. A kinase-independent role for EGF receptor in autophagy initiation. Cell 160, 145–160 (2015).
Hammond, G. R., Machner, M. P. & Balla, T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 205, 113–126 (2014).
Paz, I. et al. Galectin‐3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 12, 530–544 (2010).
Xiong, J. et al. Rapid affinity purification of intracellular organelles using a twin strep tag. J. Cell Sci. 132, jcs235390 (2019).
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A. & Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146 (2008).
Gautier, R., Douguet, D., Antonny, B. & Drin, G. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 (2008).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Acknowledgements
We thank members of the Aging Institute at the University of Pittsburgh for helpful discussions and reagents; K. Zhang, G. Shang and M. Hanna for discussions on protein purification and liposome preparation; Z. Wang, H. G. Wang and P. L. Opresko for sharing reagents; MS Bioworks for mass spectrometry services; G. D. Fairn for providing the mCherry-D4H plasmid; V. Olkkonen for the ORP10 plasmid; A. Y. Ting for the TurboID DNA; T. Levine for the OSBP-PH plasmid; G. Du for the LAMP1-GFP-Twin-Strep plasmid; M. Kampmann the eGFP–galectin-3 and Tau.K18(P301L/V337M)-mRuby2 plasmid; C. Rosenmund for the lyso-pHluorin plasmid; T. Balla for the GFP-P4M plasmid; R. Parton for the 2xFYVE_hrs plasmid; S. Grinstein for the Lact-C2 plasmid; C. Tomasetto for the VAPA WT and KD/MD plasmids; and W. Hahn and D. Root for the PI4K2B cDNA. This work was supported by Competitive Medical Research Fund (CMRF) of the University of Pittsburgh Medical Center (UPMC) Health System (J.X.T.) and National Institutes of Health (NIH) grants 1K01AG075142 (J.X.T.), 1R01HL142663 (T.F.), 1R01HL142589 (T.F.), U54AG075931 (T.F.) and P30AG024827 (T.F.). Figs. 1a, 2d, 2j, 3a, 3e and 4d and Extended Data Figs. 5n, 7n and 10 were generated using Biorender.
Author information
Authors and Affiliations
Contributions
J.X.T. designed and performed all experiments, collected and analysed data, designed figures and wrote the manuscript; T.F. supervised the work, provided advice on experiments and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.X.T. declares no competing interests. T.F. is a co-founder and stockholder in Generian Pharmaceuticals.
Peer review
Peer review information
Nature thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Identification of proteins enriched on damaged lysosomes using Lyso-TurboID cells.
a, Schematic illustration of Lyso-TurboID cells. r1 is the estimated distance between TurboID and the lysosomal membrane; r2 is the estimated radius within which a protein can be biotinylated by TurboID. b, Lyso-TurboID stably expressed in 293T cells colocalizes with the late endosome/lysosome marker CD63. c, Biotin addition enhances biotinylation of proteins that colocalize with Lyso-TurboID. Biotinylated proteins were stained by streptavidin. Note, lysosomal membrane damage by LLOME is known to cause lysosomal enlargement. 293T Lyso-TurboID (green) cells were treated with (500 μM) LLOME for 30 min, followed by another 30 min of biotin treatment (50 μM) without removal of LLOME. Cells were then fixed and permeabilized for streptavidin staining (red). d, Schematic illustration for the purification of proteins recruited to normal and damaged lysosomes. e, Lyso-TurboID is enriched in P20 (pellet after 20,000xg centrifugation) membrane fraction. Samples collected in (d) was analyzed by anti-GFP immunoblotting. Note that some lysosomes shifted from P20 to the heavier P1 fraction in LLOME-treated cells. This is likely because damaged lysosomes are extensively tethered to the ER (See Fig. 2) and thus a fraction of lysosomes might pellet with larger ER fragments in P1. f, Immunoblot analysis confirming the enrichment of mass spectrometry-identified proteins on damaged lysosomes. Protein samples were purified as in (a) from 293T Lyso-TurboID cells. S20, the supernatant above P20, contains the whole cytosol and some light membranes. g, The endosomal sorting complex required for transport (ESCRT) subunits IST1, ALIX, and CHMP3 are all rapidly recruited to damaged lysosomes in multiple cell lines. Cells were treated with 1 mM LLOME for 10 min and stained for the indicated endogenous ESCRT subunits and lysosomal marker LAMP1 after fixation and permeabilization. h, The PtdIns4P probe OSBP-PH-GFP is recruited to damaged lysosomes in U2OS cells as shown by the three channel colocalization between OSBP-PH, LAMP1, and IST1. Cells were treated with 1 mM LLOME for 30 min and then fixed for the staining of endogenous LAMP1 and IST1. DAPI stains the nuclei. Bar, 10 μm. Uncropped western blot images are provided in Supplementary Fig. 1.
Extended Data Fig. 2 Selective PtdIns4P production on damaged lysosomes in different cell lines.
a, b, OSBP-PH-GFP is recruited to damaged lysosomes in U2OS cells as shown by its colocalization with the lysosomal markers LAMP2 (a) and CD63 (b) upon lysosome damage. U2OS cells stably expressing OSBP-PH-GFP were treated with 1 mM LLOME for 30 min and then fixed for immunostaining of the endogenous indicated lysosomal markers. c, OSBP-PH-GFP is recruited to damaged lysosomes in human PC3 prostate adenocarcinoma cells. Cells stably expressing OSBP-PH-GFP were treated with 1 mM LLOME for 30 min and then fixed for immunostaining of endogenous IST1, an ESCRT-III subunit and marker for damaged lysosomes. Pearson’s correlation coefficient of OSBP-PH-GFP and IST1 were quantified; mean ± sem; n = 11 cells from 3 trials for each condition. d, An alternative PtdIns4P probe GFP-P4M is recruited to damaged lysosomes in COS7 cells. Cells were transiently transfected with GFP-P4M. After 24 h, cells were treated with 1 mM LLOME for 30 min and then fixed for immunostaining of endogenous IST1 and LAMP1. Pearson’s correlation coefficient of GFP-P4M and IST1 were quantified; mean ± sem; n = 9 (− LLOME) and 24 (+ LLOME) cells over 3 trials. e, f, Probes for PI(4,5)P2 (e) or PI3P (f) are not recruited to damaged lysosomes. U2OS cells stably expressing the indicated probe were treated with 1 mM LLOME for indicated time periods and then fixed for immunostaining of LAMP1 or IST1. g, Schematic illustration of the dispensability for an intact Golgi in the recruitment of OSBP-PH-GFP to damaged lysosomes. h, Disassembly of the Golgi complex by Brefeldin A does not affect the recruitment of the PtdIns4P probe OSBP-PH-GFP to damaged lysosomes. Cells were pretreated or not with 2 μM Brefeldin A for 10 min followed by the addition of 1 mM LLOME for 30 min. Cells were then fixed for the staining of endogenous LAMP1. i, Lysosomal damage by LLOME does not affect the overall morphologies of the cis- (GM130) and trans-Golgi (Golgin-97) complex. U2OS cells were treated as indicated and stained for endogenous markers of the Golgi complex. DAPI stains the nuclei. Bar, 10 μm. Source data for graphs in this Figure are provided.
Extended Data Fig. 3 Lysosomal damage by diverse factors triggers specific lysosomal recruitment of PI4K2A.
a, Antibody verification for PI4KA, PI4KB, and PI4K2A using the indicated knockdown or knockout cells. Asterisk indicates a nonspecific band. b, Immunoblotting shows the specific enrichment of PI4K2A but not PI4KA or PI4KB on damaged lysosomes. Streptavidin blot was used as a loading control. Note, consistent with subcellular redistribution, PI4K2A levels in the S20 fraction were reduced upon lysosome damage. c, Endogenous PI4K2A forms puncta outside of the Golgi complex upon lysosomal damage by LLOME. GM130 is a marker for the cis-Golgi. Cells were treated with 1 mM LLOME for 10 min, followed by co-staining of endogenous GM130 and PI4K2A. d, Endogenous PI4K2A is rapidly recruited to damaged lysosomes. Cells were treated with 1 mM LLOME for 10 min, followed by co-staining of endogenous LAMP1 and PI4K2A. Quantification of Pearson’s correlation coefficient of PI4K2A and LAMP1 is shown on the right. Mean ± sem; n = 27 cells over three trials for each condition. e, LLOME treatment of COS7 cells induces the formation of endogenous PI4K2A puncta that colocalize with IST1, a marker for damaged lysosomes. f, PI4K2B is not recruited to damaged lysosomes as it does not colocalize with IST1. U2OS cells stably expressing Flag-PI4K2B were stimulated with LLOME for 10 min and stained for endogenous IST1. g, The protein level of PI4K2A does not change within one hour of LLOME treatment. Asterisk indicates a nonspecific band, as demonstrated in panel (a) where knockout of PI4K2A only causes the loss of the lower band. h, The recruitment of endogenous PI4K2A and the ESCRT subunits to damaged lysosomes are independent of each other. Cells were transfected with indicated siRNA; 72 h later, cells were treated with 1 mM LLOME for 10 min and then fixed for co-staining of PI4K2A and IST1. See Supplementary Results for additional discussions about different lysosomal repair pathways. i–k, PI4K2A is recruited to lysosomes damaged by the lysosomotropic detergent O-methyl-serine dodecylamine hydrochloride, MSDH (i), the dipeptide glycyl-l-phenylalanine 2-naphthylamide GPN (j), or the transient expression of SARS-COV2-ORF3A (k). See Supplementary Table 2 for details about the mechanisms of lysosomal damage mediated by these factors. Quantification of Pearson’s correlation coefficient of PI4K2A and LAMP1 is shown for each LMP inducer. Mean ± sem; n = 23 (DMSH), 22 (GPN), 13 (SARS-COV2-ORF3A) cells over three trials for each condition. l, PI4K2A is recruited to phagolysosomes damaged by nano-silica (SiO2). Galectin3 is used as a marker for damaged phagolysosomes. The percentage of phagolysosomes positive for either PI4K2A or Galectin3 or both are quantified on the right. A total of 324 phagolysosomes from 35 silica-treated cells were quantified over three experiments. ND, not detected. Note that the vast majority of Galectin3-positive (indication of severe membrane damage) phagolysosomes are positive for PI4K2A and that a small fraction of PI4K2A-positive large vesicles are negative for Galectin3, likely due to small membrane damages not severe enough to allow the entry of Galectin3. m, CRISPR-based CLN3 knockout causes robust expansion of LAMP1-positive lysosomes that recruit PI4K2A. CLN3 knockout cells were CRISPR pools, with two different sgRNAs showing similar LAMP1 accumulation and PI4K2A recruitment. Quantification of Pearson’s correlation coefficient of PI4K2A and LAMP1 is shown on the right. Mean ± sem; n = 30 cells over three trials for each condition. n, ML-SA1 and MK6–83, two structurally distinct chemical agonists of the main lysosomal Ca2+ channel TRPML1, are each sufficient to trigger lysosomal PI4K2A recruitment. Cells treated with 25 μM of either ML-SA1 or MK6–83 for 40 min were fixed for the staining of endogenous PI4K2A and LAMP1. LLOME treatment for 10 min was used as a positive control. The colocalization of PI4K2A and LAMP1 in ML-SA1 treated cells was quantified. Mean ± sem; n = 26 cells over three trials for each condition. See also Supplementary Results. DAPI stains the nuclei. Bar, 10 μm. Uncropped western blot images are provided in Supplementary Fig. 1. Source data for graphs in this Figure are provided.
Extended Data Fig. 4 PI4K2A-meidated lysosomal PtdIns4P signaling is essential for rapid lysosomal repair.
a, PI4K2A is required for PtdIns4P production on damaged lysosomes. Wild type (WT) or two independent PI4K2A-KO cell lines stably expressing the PtdIns4P probe OSBP-PH-GFP were treated with or without LLOME for 30 min before being fixed for immunostaining of endogenous IST1, a marker for damaged lysosomes. The images show the loss of LMP-induced lysosomal recruitment of OSBP-PH-GFP in PI4K2A-KO cells, which is fully rescued by re-expression of WT PI4K2A. Note, some green puncta still remain in PI4K2A-KO cells, corresponding to the Golgi pool of PtdIns4P. These puncta are absent after Brefeldin A treatment. See panel (c) and Fig. 1g. Bar, 10 μm. b, Western blot verification of PI4K2A levels in wild type or genetically modified U2OS cells. Asterisk indicates a nonspecific band. c, PI4K2A recruitment is responsible for PtdIns4P production on damaged lysosomes. OSBP-PH-GFP-expressing U2OS cells with the indicated genetic modifications were pretreated for 10 min with Brefeldin A to disassemble the Golgi, removing background GFP puncta on the Golgi complex. Cells were then treated with or without 1 mM LLOME for 30 min before immunostaining of endogenous IST1. See quantification in Fig. 1g. Bar, 10 μm. d, The Lyso-pHluorin assay reveals the requirement of PI4K2A and its kinase activity in rapid lysosome repair. Schematic illustration of the assay is shown on the left. Lyso-pHluorin was stably expressed in various U2OS cells with indicated genetic modifications. Cells were treated with 1 mM LLOME for 10 min and then gently washed twice with pre-warmed culture media; fluorescent images were taken at the indicated time points. See quantification in Fig. 1h. Bar, 10 μm. e, Quantification of the lysosomal repair half-life of different cell lines using the Lyso-pHluosin assay. Data were derived from the experiment in (d). Mean ± sem; n = 3. f, The EGFP-Galectin3 rapid lysosomal repair assay demonstrates a role for PI4K2A in protecting lysosomes from severe damage. Schematic illustration of the assay is shown on the left. U2OS cells stably expressing EGFP-Galectin3 were treated with 1 mM LLOME for 60 min to continuously damage lysosomes. Fluorescence microscopic images were taken using live cells. Note, brighter Galectin3 puncta indicates larger and greater unrepaired lysosomal membrane pores. See quantification in Fig. 1i. Bar, 10 μm. g, Rapid lysosomal repair in PI4K2A-KO cells is rescued by expressing LAMP1-mCherry-PI4K2B but not its kinase dead mutant. Cells stably expressing indicated proteins were treated as in (f) and EGFP-Galectin3 punctate intensities were quantified. Mean ± sem; n = 3. Bar, 10 μm. h, Knockout of PI4K2A accelerates tau fibril spreading in cell culture. Top, Schematic illustration of the tau fibril spreading assay. Bottom, cells were incubated with sonicated, non-fluorescent tau fibrils. The percentage of cells showing bright tau-K18-mRuby puncta were examined on a daily basis. See quantification in Fig. 1j. Bar, 60 μm. i, Deletion of PI4K2A exacerbates lipofuscin accumulation in CLN3-knockout fibroblasts. Left, BJ human skin fibroblasts were infected with CRISPR lentivirus for the deletion of CLN3 alone or double deletion of PI4K2A and CLN3. CLN3-sg#1 and PI4K2A-sg#2 were used to obtain the knockout pools. Six days after infection, cells were fixed directly for lipofuscin detection. Right, quantification of lipofuscin intensities. 10–20 random cells per condition; Mean ± sem; n = 3. Bar, 10 μm. DAPI stains the nuclei. NS, not significant. Uncropped western blot images are provided in Supplementary Fig. 1. Source data for graphs in this Figure are provided.
Extended Data Fig. 5 PI4K2A-mediated PtdIns4P signaling recruits ORP9, ORP10, and ORP11 to damaged lysosomes.
a, Antibody verification by western blot using CRISPR knockout pools. Asterisk indicates a nonspecific band. b, ORP10 is mainly found in the P20 fraction and absent from the S20 fraction. P20, pellet after 20,000 xg centrifugation. S20, supernatant after 20,000 xg centrifugation. Asterisk indicates nonspecific bands. c, LMP induces new puncta of EGFP-ORP9/11 on damaged lysosomes. EGFP-ORP9 and -ORP11 are recruited to puncta outside of the Golgi complex upon lysosomal damage by LLOME (top). LMP-induced EGFP-ORP9 puncta colocalize with LAMP1 and IST1 (bottom). d, Endogenous ORP9 and stably expressed EGFP-ORP11 completely colocalize when forming puncta in response to lysosomal damage by LLOME. e, EGFP-ORP11 colocalizes with both PI4K2A and LAMP1 upon lysosomal damage. f, ORP9, but not EGFP-ORP3, forms puncta upon lysosomal damage. g, EGFP-ORP5 is not recruited to damaged lysosomes. Note that the ORP5 punctate structures observed here are not random diffuse signals; they are morphologically distinct from the diffuse signal of ORP3 in (f). When the confocal images were captured at different planes of the cell, the ORP5 structures appear dramatically different31. If the focus plane is away from the bottom of the cell, ORP5 appears as punctate structures on the PM with only diffuse signal in the cytosol. However, when the plane is near the bottom of the cell, as is the case here, ORP5 appears as extensive puncta throughout the cells corresponding to ER-plasma membrane contact sites at the bottom. h, i, Knockdown of PI4K2A by RNA interference (RNAi) blocks the recruitment of EGFP-ORP9 (h) and EGFP-ORP11 (i) to damaged lysosomes. U2OS cells stably expressing EGFP-ORP9 or EGFP-ORP11 were transfected with indicated siRNAs. After 72 h, cells were treated or not with 1 mM LLOME for 10 min and fixed for co-staining of endogenous LAMP1 and IST1. See Supplementary Table 4 for siRNA sequences. j–l, The kinase activity of PI4K2A is required for the recruitment of EGFP-ORP9/10/11 to damaged lysosomes. EGFP-ORP9 (j), EGFP-ORP10 (k), or EGFP-ORP11 (l) were stably expressed in wild type (WT) and PI4K2A knockout (PI4K2A-KO) cells, as well as in PI4K2A-KO cells re-expressing WT (KO + WT) or kinase dead PI4K2A (KO + KDAA). Cells were treated or not with 1 mM LLOME for 10 min and fixed for immunostaining of endogenous IST1. Note, in the absence of PI4K2A activity, all cells lost the recruitment of ORP9/10/11 to damaged lysosomes, as exemplified by the quantification of endogenous ORP9/11 in Fig. 2c. m, LMP-induced puncta of endogenous ORP9 and ORP11 are dependent on PI4K2A and its kinase activity. Indicated cell lines were treated with LLOME for 10 min and fixed for co-staining of endogenous ORP9 and ORP11. See Fig. 2c for quantification. n, o, ORP9 binding to PtdIns4P through its PH domain and to VAPA/B through its FFAT motif are both necessary for its efficient recruitment/accumulation on damaged lysosomes. n, schematic illustration of ORP9 mutants. o, indicated ORP9 mutants no longer accumulate on damaged lysosomes. Note, despite the presence of intact PH domain for PtdIns4P binding, the ORP9-FYAA mutant is no longer efficiently recruited to damaged lysosomes, suggesting that PtdIns4P-binding alone is insufficient for stable ORP9 association with lysosomes. p, The LMP-induced lysosomal recruitment of ORP9/10/11 requires PtdIns4P-binding through their PH domain. RE indicates the point mutation of a conserved arginine residue required for PtdIns4P-binding to the PH domain of all the three ORPs. U2OS cells stably expressing mCherry-ORP9, ORP10, or ORP11 or their RE mutants were treated with LLOME for 10 min and then fixed for immunostaining of endogenous LAMP1. Note, while all RE mutants lost lysosomal targeting downstream of LMP, ORP9/10-RE but not ORP11-RE can still be localized to the Golgi complex in resting conditions, indicating different mechanisms for their basal Golgi targeting. See Supplementary Results and Supplementary Table 3 for more details regarding the Golgi and lysosomal targeting of the ORPs. DAPI stains the nuclei. Bar, 10 μm. Uncropped western blot images are provided in Supplementary Fig. 1.
Extended Data Fig. 6 The redundancy of ORP9/10/11/OSBP in their recruitment to damaged lysosomes and in the subsequent establishment of extensive ER-lysosome contacts.
a, The recruitment of EGFP-VAPA to damaged lysosomes is dependent on PI4K2A and its kinase activity. Indicated cells stably expressing EGFP-VAPA were treated or not with LLOME for 10 min and fixed for immunostaining of endogenous LAMP1. The percentages of cells showing extensive VAPA-LAMP1 contacts were quantified in Fig. 2e. b, An EGFP-VAPA mutant that no longer binds to FFAT motifs is poorly recruited to ORP9 puncta induced by lysosomal damage. U2OS cells stably expressing EGFP-VAPA WT or the K94D/M96D mutant were treated with LLOME for 10 min and then fixed for immunostaining of endogenous ORP9. c, Left: ORP9 knockout diminishes ORP11 recruitment to damaged lysosomes, but not vice versa. Wild-type (WT), ORP9 knockout (ORP9-KO), and ORP11-KO U2OS cells were treated or not with 1 mM LLOME for 10 min and then fixed for co-staining of endogenous ORP9 and ORP11. Right: ORP9 knockout does not affect ORP10 recruitment to damaged lysosomes. WT and ORP9-KO cells stably expressing EGFP-ORP10 were treated or not with 1 mM LLOME and fixed for staining of endogenous ORP9. d, ORP10 knockout does not affect the recruitment of ORP9 or ORP11 to damaged lysosomes. WT and ORP10-KO cells were treated or not with 1 mM LLOME for 10 min and then fixed for co-staining of endogenous ORP9 and ORP11. e, ORP9/10/11 triple knockout (TKO) cells show rapid and augmented OSBP recruitment to damaged lysosomes. Wild type U2OS cells or three independent ORP-TKO clones were stimulated with 1 mM LLOME for 10 min and then fixed and stained for endogenous OSBP and LAMP1. Note that OSBP recruitment in wild type cells appeared rather weak, which is likely due to competition from ORP9/10/11, the majorly recruited PtdIns4P effectors. This is likely also the reason why the PtdIns4P probe OSBP-PH-GFP was only substantially recruited after 20–30 min of LLOME treatment (Fig. 1c, d, Supplementary Video 1), whereas PI4K2A and ORP9/10/11 recruitment appear saturated within 10 min (Figs. 1e, 2b, Extended Data Figs. 3, 5). See also Supplementary Results. f, Western blot analysis of indicated protein expression in ORP knockout cells. ORP-QKO indicates knockout of all the ORP proteins recruited to damaged lysosomes identified in mass spectrometry, including ORP9, ORP10, ORP11, and OSBP. Asterisk indicates a nonspecific band. g, The recruitment of PI4K2A and the ESCRT subunit IST1 to damaged lysosomes are unaffected in ORP-QKO cells. h, ORP-QKO cells produce higher levels of PtdIns4P on damaged lysosomes compared with wild type cells. Wild-type (WT) and three independent ORP-QKO cell lines stably expressing the PtdIns4P probe OSBP-PH-GFP were pretreated or not with Brefeldin A to disassemble the Golgi complex, followed by 1 mM LLOME treatment for 30 min. The cells were then fixed to stain for endogenous LAMP1. Note, higher basal lysosomal PtdIns4P levels were also detected in ORP-QKO cells, consistent with previous findings that OSBP plays a role in removing endolysosomal PtdIns4P in resting conditions47. i, Quantification of OSBP-PH-GFP/LAMP1 colocalization in WT or ORP-QKO U2OS cells following LLOME treatment. Note, within 2 h a decrease in colocalization was observed in WT but not ORP-QKO cells. Mean ± sem; n = 15 cells over three trials for each condition. j, Reconstituted ORP9 was robustly recruited to damaged lysosomes in ORP9/11-DKO cells, but not in ORP-TKO or ORP-QKO cells, suggesting that lysosomal recruitment of ORP9 requires the presence of either ORP10 or ORP11. k, The recruitment of reconstituted ORP9, ORP10, ORP11, and OSBP to damaged lysosomes in ORP-QKO cells. When re-expressed in ORP-QKO cells, ORP10 alone, but not ORP9 or ORP11, can be recruited to damaged lysosomes. However, when any two of the three proteins were introduced back to the ORP-QKO cells, both proteins were recruited to damaged lysosomes, consistent with the previously reported hetero-dimerization between ORP9 and ORP1115. Different from the other ORPs, re-expressed OSBP alone was robustly recruited to lysosomes upon LLOME treatment. l, Re-expressing ORP proteins in ORP-QKO cells rescues VAPA recruitment to damaged lysosomes. Left: ORP-QKO cells stably expressing EGFP-VAPA and the indicated mCherry-tagged ORP protein(s) were stimulated with LLOME for 10 min and then fixed for confocal microscopy. See Fig. 2g for quantifications. m, Co-immunoprecipitation experiment showing heterodimerization of Flag-ORP9 with either EGFP-ORP10 or -ORP11 but the absence of ORP9 homodimerization. Whole cell lysates from 293T cells stably expressing indicated proteins were subject to immunoprecipitation using indicated antibodies, followed by immunoblotting. n, OSBP forms homodimers shown by co-immunoprecipitation (co-IP) between EGFP-OSBP and Flag-OSBP. The whole cell lysates from 293T cells stably expressing EGFP-OSBP and Flag-OSBP were subjected to immunoprecipitation using control IgG, Flag, or GFP antibodies. The presence of the two OSBP fusion proteins in each IP samples were analyzed. Note that ORP9 does not form homodimers in similar assays in (m). o, Rescuing lysosomal repair by re-expressing ORP proteins in ORP-QKO cells. Cells stably expressing EGFP-Galectin3 and the indicated mCherry-ORPs were treated continuously with 1 mM LLOME for 60 min. Fluorescence images were taken with live cells. Quantification is shown in Fig. 2i. DAPI stains the nuclei. Bar, 10 μm. NS, not significant. Uncropped western blot images are provided in Supplementary Fig. 1. Source data for graphs in this Figure are provided.
Extended Data Fig. 7 ORP9/10/11 transport phosphatidylserine (PS) to damaged lysosomes.
a, Alignment of human ORPs with yeast Osh6 which specifically transports phosphatidylserine (PS) but not sterols. Bold residues are direct PS contact sites of Osh6. The AAA mutants are designed to lose PS-binding, which have been confirmed in this study. b, Coomassie blue staining of purified ORP9 and ORP11. c, FRET-based assay demonstrating higher lipid transport activity by ORP9/11 (50 nM/50 nM = 100 nM in total) in the presence of PtdIns4P in the acceptor liposome. Note that there was a slight increase in NBD fluorescence in the absence of acceptor liposomes (donor only), which is consistent with ORP9/11-mediated NBD-PS extraction from the donor membranes without further delivery to acceptor membranes. d, ORP9/11 monomers and heterodimers show similar PS transport activity in vitro. ORP9/11 (50 nM/50 nM) heterodimers or monomers (100 nM each) were added to donor and acceptor liposome mixtures, and the changes in NBD fluorescence was monitored over time. The acceptor liposomes in all reactions contained 5% PtdIns4P. See also Supplementary Results. e, The PS probe GFP-Lact-C2 is quickly recruited to damaged lysosomes. U2OS cells stably expressing GFP-Lact-C2 were treated with 1 mM LLOME for the indicated time periods and then briefly washed with 0.1% Triton-X100 (detergent wash, see Methods) to remove background PS signals from the cytosol. The cells were then immediately fixed. f, The recruitment of GFP-Lact-C2 to damaged lysosomes is dependent on PI4K2A and its kinase activity, as well as the ORPs enriched on damaged lysosomes. Cells were treated with LLOME and detergent-washed before fixation and immunostaining. The colocalization of GFP-Lact-C2 and LAMP1 was quantified in Fig. 3d. g, ORP9 is recruited to damaged lysosomes earlier than the PS probe GFP-Lact-C2. U2OS Cells stably expressing GFP-Lact-C2 were treated with LLOME and detergent-washed before fixation and immunostaining of endogenous ORP9 and LAMP1. Arrow indicates ORP9 puncta negative for GFP-Lact-C2. h, The colocalization between LAMP1 and ORP9 or GFP-Lact-C2 in (g) was quantified. Data show mean ± sem of Pearson’s correlation coefficient; n = 15 cells over three trials for each condition. i, LMP-induced lysosomal PS transport in ORP-QKO cells can be rescued by the reconstitution of any two of ORP9/10/11. Cells were treated with 1 mM LLOME for 20 min and detergent-washed before being fixed for immunostaining of LAMP1. j, The colocalization of GFP-Lact-C2 and LAMP1 in (i) was quantified. Data show mean ± sem of Pearson’s correlation coefficient; n = 30 cells for each condition. k, ORP-QKO cells stably expressing GFP-Lact-C2 and mCherry-ORP9/11 or their mutants were treated with LLOME for 20 min and detergent-washed before being fixed for immunostaining of LAMP1. See Fig. 3k for quantification. l, The LMP-induced lysosomal recruitment of VAPA in ORP-QKO cells can be strongly rescued by the AAA mutants (loss of PS binding to the lipid transport domain) of ORP9/10 or ORP9/11, weakly rescued by ORP10/11 or the HHAA mutants of ORP9/11, and cannot be rescued by the RE mutants of ORP9/11. See Fig. 3e for details of the mutants. ORP-QKO cells stably expressing EGFP-VAPA and the indicated mCherry-ORP proteins were treated with LLOME for 10 min before fixed for confocal microscopy. m, PIP strips assay showing specific binding of purified EGFP-Lact-C2 to PS. n, Schematic illustration for lysosomal purification, lipid extraction, PVDF spotting, and PS detection. Wild type or ORP-QKO cells stably expressing LAMP1-GFP-twin-Strep were treated or not with 1 mM LLOME for 30 min, followed by lysosomal purification using sptreptavidin beads. Lipids were extracted from lysosomes on beads and dropped onto PVDF membrane for PS detection by purified EGFP-Lact-C2. On the right are representative images of the lipid/protein overlay assay for lysosomal PS detection with LAMP2 immunoblots as total membrane input controls. DAPI stains the nuclei. Bar, 10 μm. NS, not significant. Uncropped western blot images are provided in Supplementary Fig. 1. Source data for graphs in this Figure are provided.
Extended Data Fig. 8 In parallel with ORP9/10/11-mediated PS transport, OSBP drives lysosomal cholesterol transfer as an auxiliary mechanism for rapid lysosomal repair.
a, Rapid lysosomal repair in ORP-QKO cells was only rescued by re-expressing wild type ORP9/11 but not their mutants defective in PS-transport. Cells stably expressing EGFP-Galectin3 and indicated mCherry-ORPs were treated with 1 mM LLOME for 60 min to continuously damage lysosomes. Images were taken using live cells. See image quantification in Fig. 3h. b, Reconstitution of OSBP alone in ORP-QKO cells fully rescues ER tethering to damaged lysosomes. ORP-QKO cells stably expressing EGFP-VAPA and the indicated mCherry-OSBP proteins were treated with LLOME for 10 min before being fixed for confocal microscopy. Similar to ORP mutations in Extended Data Fig. 7k, l, OSBP-2RE loses lysosomal recruitment; OSBP-HHAA does not perform PtdIns4P/cholesterol exchanges between membranes, despite its strong lysosomal recruitment. c, OSBP chimeric proteins rescues ER tethering to damaged lysosomes in ORP-QKO cells. See Fig. 3i for the details of the chimeric proteins. OSBP-Kes1 is constructed similarly to OSBP-Osh6. Cells stably expressing EGFP-VAPA and mCherry-OSBP-X chimeric proteins were stimulated with LLOME for 10 min and then fixed for the staining of endogenous LAMP1. Note, all cells expressing mCherry chimeric proteins showed extensive EGFP-VAPA recruitment to lysosomes, with three-channel colocalization. d, Rapid lysosomal repair in ORP-QKO cells was rescued by re-expressing the indicated chimeric proteins but not the OSBP-Osh6-HHAA mutant defective in PtdIns4P/PS counter transport. Cells stably expressing EGFP-Galectin3 and indicated chimeric proteins were treated as in (a). See image quantification in Fig. 3j. e, LLOME triggers PI4K2A- and OSBP-dependent lysosomal transport of cholesterol. U2OS cells with indicated genetic modifications were stimulated with LLOME for 60 min and then fixed for the staining of endogenous IST1. KDAA is a kinase dead mutant of PI4K2A. 2RE is a mutant of OSBP that is no longer recruited to damaged lysosomes due to loss of PtdIns4P binding to its PH domain. f, The colocalization of the cholesterol probe GFP-D4H and IST1 in (e) was quantified. Data show mean ± sem of Pearson’s correlation coefficient; n = 25 cells over 3 trials for each condition. Note that the PS transporters ORP9/11 cannot rescue lysosomal cholesterol accumulation. g, Reconstitution of wild type OSBP, but not its cholesterol transport defective mutants, appears to rescue rapid lysosomal repair in ORP-QKO cells. The same OSBP mutants in (e) and (f) were tested here. Cells stably expressing EGFP-Galectin3 and mCherry-OSBP were continuously treated with 1 mM LLOME and the EGFP-Galectin3 puncta in live cells were analyzed in the right panel. About 50–100 random cells were quantified for each condition. Mean ± sem; n = 3. h, Similar to OSBP-Osh6 in panel c, d, OSBP-Kes1 also rescues rapid lysosomal repair, whereas the cholesterol transport defective HHAA mutant does not. Left, cells stably expressing EGFP-Galectin3 and mCherry-OSBP-Kes1 were continuously treated with 1 mM LLOME and the EGFP-Galectin3 puncta were analyzed with live cells. Right, quantification of the Galectin3 intensities above threshold. About 50–100 random cells were quantified for each condition. Mean ± sem; n = 3. DAPI stains the nuclei. Bar, 10 μm. NS, not significant. Source data for graphs in this Figure are provided.
Extended Data Fig. 9 Independent of macroautophagy, ATG2 mediates rapid lysosomal repair through its lipid transport activity stimulated by PS.
a, Immunoblotting of ATG2A/B protein levels in wild type or ATG2A/B-DKO cells. Two individual clones from each set of CRISPR guides were used for further characterization. b, Double knockout of ATG2A/B causes robust defects of rapid lysosomal repair as shown by the EGFP-Galectin3 assay. The same knockout clones from (a) were used for this assay. Cells were continuously challenged with LLOME and live cell images were captured at indicated time points. Note that re-expression of ATG2A was sufficient to rescue rapid lysosomal repair in the DKO cells. See Fig. 4c for quantifications. c, Immunoblotting of OSBP levels in indicated CRISPR pools. d, Further deletion of OSBP in ATG2A/B-DKO cells causes dramatic defects in rapid lysosomal repair at an early time point. Note that loss of OSBP or ATG2A/B alone does not cause apparent defects within 30 min of LLOME treatment, indicating functional redundancy. e, quantification of the early time point Galectin3 intensities above threshold in (d). About 50–100 random cells were quantified for each condition. Mean ± sem; n = 3. f, Four distinct ATG2A lipid transport mutants are unable to rescue rapid lysosomal repair in ATG2A/B-DKO cells. Stable U2OS cell lines with indicated genetic modifications were continuously challenged with LLOME and live cell images were captured at indicated time points. See quantification in Fig. 4h. g, The ATG2A lipid transport mutants cannot restore autophagic turnover in ATG2A/B-DKO cells. Stable U2OS cell lines with indicated genetic modifications were directly harvested for whole cell lysate extraction followed by immunoblotting of indicated proteins. Data represent more than five experiments. h, EGFP-Galectin3 assay showing the defects of rapid lysosomal repair in wild type cells stably expressing LAMP1-CT but not mCherry-CT or LAMP1-mCherry. See quantification in Fig. 4j. i. Overexpression of different ATG2A-CT fusion proteins including LAMP1-CT does not block macroautophagy. Cells with indicated genetic modifications were treated with 100 nM Bafilomycin A1 for 4 h, followed by immunoblotting of indicated proteins. ATG2A/B-DKO cells served as a positive control for autophagy defects with marked accumulation of both p62 and LC3-II. Quantification of LC3-II intensities normalized to GAPDH is shown. Mean ± sem; n = 3. j, Liposome pull down assays testing the membrane binding capacity of purified MBP–CT or its 5E mutant. k, Highly conserved basic residues in ATG2A-CT. The residues mutated in 5E are in red and also shown in panel (l). l, AlphaFold structure of ATG2A-CT (amino acids 1754–1821). Six highly conserved basic residues are labeled, five of which were mutated in the 5E mutant. m, ATG2A-ΔCT and −5E mutants form dramatically reduced numbers of puncta in response to lysosomal damage. U2OS cells stably expressing EGFP-tagged ATG2A-WT, -ΔCT or −5E were stimulated with LLOME and the numbers of EGFP-ATG2A puncta 20 min after stimulation were determined using live cell imaging. Mean ± sem; n = 15 cells over three trials for each condition. n, EGFP-Galectin3 assay showing the failure of ATG2A-5E mutant in rescuing rapid lysosomal repair in ATG2A/B DKO cells. See quantification in Fig. 4m. o, Knockout of PI4K2A or ORPs does not affect LC3 turnover, indicating normal macroautophagy. Indicated cell lines were treated with 100 nM bafilomycin A1 for four hours followed by whole cell lysate harvest for immunoblotting. p, PI4K2A activity does not affect LLOME-induced LC3 lipidation. U2OS cells with indicated genetic modifications were treated with 1 mM LLOME for 15 to 60 min and then whole cell lysates were analyzed for the level of LC3. Asterisk indicates a nonspecific band. q, LLOME-induced LC3 lipidation is independent of PI4K2A, ORPs, ATG2, and ATG13. U2OS cells with indicated genetic modifications were treated with 1 mM LLOME for 30 min and then whole cell lysates were analyzed for the level of LC3. r, Immunoblotting shows the loss of ATG5 and ATG7 in relevant U2OS knockout cells. Asterisk indicates a nonspecific band. Note that ATG5 is conjugated to ATG12 in wild type cells, which shows a higher band compared with unconjugated ATG5 in ATG7-KO cells. s, ATG5-KO and ATG7-KO cells have normal rapid lysosomal repair, without increased EGFP-Galectin 3 puncta after continuous challenging with 1 mM LLOME. Bar, 10 μm. t, Quantification of EGFP-Galectin3 intensities above threshold in (s). 50–100 random cells were quantified for each condition. Mean ± sem; n = 3. u, Immunoblotting shows no evidence of degradation of either LAMP1 or LAMP2 within four hours of LLOME treatment. U2OS cells were treated with 1 mM LLOME for 1 to 4 h and whole cell lysates was then analyzed for levels of the indicated protein. DAPI stains the nuclei. Bar, 10 μm. Uncropped western blot images are provided in Supplementary Fig. 1. Source data for graphs in this Figure are provided.
Extended Data Fig. 10 Summary illustration of the phosphoinositide-initiated membrane tethering and lipid transport (PITT) pathway for rapid lysosomal repair.
(1) LMP-induced Ca2+ release triggers the rapid lysosomal recruitment of PI4K2A that generates high levels of PtdIns4P on damaged lysosomes. (2) Lysosomal PtdIns4P in turn recruits and stimulates ORP9/10/11 to establish extensive ER-lysosome membrane contacts and mediate subsequent ER-to-lysosome phosphatidylserine (PS) transport. (3) Lysosomal accumulation of PS activates the lipid transporter ATG2 which delivers large amounts of lipids to lysosomes for direct membrane repair. (4) Downstream of PtdIns4P signaling and in parallel of ORP9/10/11, OSBP acts as a redundant membrane tether which transports cholesterol (Chol) rather than PS to damaged lysosomes. Due to the intrinsic capability of cholesterol to improve membrane rigidity and stability, lysosomal cholesterol accumulation might directly assist in membrane repair.
Supplementary information
Supplementary Information
This file contains Supplementary Results and Discussion, Supplementary Tables 1–6 and descriptions for Supplementary Videos 1–5.
Supplementary Fig. 1
Uncropped blots.
Supplementary Video 1
The PtdIns4P probe OSBP-PH-GFP is rapidly recruited to damaged lysosomes. U2OS cells stably expressing OSBP-PH-GFP and LAMP1-mCherry were stimulated with 1 mM LLOME and the trafficking of OSBP-PH-GFP was recorded by live cell imaging.
Supplementary Video 2
ORP9 is rapidly recruited to damaged lysosomes. U2OS cells stably expressing EGFP-ORP9 and LAMP1-mCherry were stimulated with 1 mM LLOME and the trafficking of EGFP-ORP9 was recorded by live cell imaging.
Supplementary Video 3
ORP11 is rapidly recruited to damaged lysosomes. U2OS cells stably expressing EGFP-ORP11 and LAMP1-mCherry were stimulated with 1 mM LLOME and the trafficking of EGFP-ORP11 was recorded by live cell imaging.
Supplementary Video 4
EGFP-ATG2A is rapidly and dynamically recruited to damaged lysosomes. U2OS cells stably expressing EGFP-ATG2A and LAMP1-mCherry were stimulated with 1 mM LLOME and the trafficking of EGFP-ATG2A was recorded by live cell imaging.
Supplementary Video 5
ML-SA1, an agonist of the main lysosomal calcium channel TRPML1, does not trigger lysosomal recruitment of ATG2A. U2OS cells stably expressing EGFP-ATG2A and LAMP1-mCherry were stimulated with 25 μM ML-SA1 and the trafficking of EGFP-ATG2A was recorded by live cell imaging..
Source data
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tan, J.X., Finkel, T. A phosphoinositide signalling pathway mediates rapid lysosomal repair. Nature 609, 815–821 (2022). https://doi.org/10.1038/s41586-022-05164-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05164-4
This article is cited by
-
A conserved ion channel function of STING mediates noncanonical autophagy and cell death
EMBO Reports (2024)
-
Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology
Nature Reviews Molecular Cell Biology (2024)
-
Discovering genetic mechanisms underlying the co-occurrence of Parkinson’s disease and non-motor traits
npj Parkinson's Disease (2024)
-
WIPI4 loss linked to ferroptosis
Nature Cell Biology (2024)
-
PRKAA2, MTOR, and TFEB in the regulation of lysosomal damage response and autophagy
Journal of Molecular Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
