
Abstract
Frontotemporal lobar degeneration (FTLD) is the third most common neurodegenerative condition after Alzheimer’s and Parkinson’s diseases1. FTLD typically presents in 45 to 64 year olds with behavioural changes or progressive decline of language skills2. The subtype FTLD-TDP is characterized by certain clinical symptoms and pathological neuronal inclusions with TAR DNA-binding protein (TDP-43) immunoreactivity3. Here we extracted amyloid fibrils from brains of four patients representing four of the five FTLD-TDP subclasses, and determined their structures by cryo-electron microscopy. Unexpectedly, all amyloid fibrils examined were composed of a 135-residue carboxy-terminal fragment of transmembrane protein 106B (TMEM106B), a lysosomal membrane protein previously implicated as a genetic risk factor for FTLD-TDP4. In addition to TMEM106B fibrils, we detected abundant non-fibrillar aggregated TDP-43 by immunogold labelling. Our observations confirm that FTLD-TDP is associated with amyloid fibrils, and that the fibrils are formed by TMEM106B rather than TDP-43.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
Data availability
Cryo-EM maps and atomic models of proteins from FTLD-TDP donor 1 have been deposited in the Protein Data Bank (PDB) and the Electron Microscopy Data Bank (EMDB) with accession codes 7SAQ and EMD-24953 for PM1, 7SAR and EMD-24954 for PM2, and 7SAS and EMD-24955 for PM3, respectively. Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the MassIVE partner repository with the dataset identifier PXD029876. Any other relevant data are available from the corresponding author upon reasonable request.
Code availability
Energetic calculations were performed using custom written software; the code is available at https://doi.org/10.5281/zenodo.6321286.
References
Mohandas, E. & Rajmohan, V. Frontotemporal dementia: an updated overview. Indian J. Psychiatry 51, S65–S69 (2009).
Neary, D. et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554 (1998).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Van Deerlin, V. M. et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239 (2010).
Chiti, F. & Dobson, C. M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68 (2017).
Benson, M. D. et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid Int. J. Exp. Clin. Invest. 27, 217–222 (2020).
Eisenberg, D. & Jucker, M. The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Falcon, B. et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140 (2018).
Falcon, B. et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423 (2019).
Kollmer, M. et al. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 10, 4760 (2019).
Schweighauser, M. et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469 (2020).
Zhang, W. et al. Novel tau filament fold in corticobasal degeneration. Nature 580, 283–287 (2020).
Ratnavalli, E., Brayne, C., Dawson, K. & Hodges, J. R. The prevalence of frontotemporal dementia. Neurology 58, 1615–1621 (2002).
Goldman, J. S. et al. Frontotemporal dementia: genetics and genetic counseling dilemmas. Neurologist 10, 227–234 (2004).
Mackenzie, I. R. A. et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113 (2011).
Lee, E. B. et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 134, 65–78 (2017).
Lashley, T., Rohrer, J. D., Mead, S. & Revesz, T. Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 41, 858–881 (2015).
Hasegawa, M. et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 (2008).
Lu, R.-C., Wang, H., Tan, M.-S., Yu, J.-T. & Tan, L. TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer’s disease in Han Chinese. J. Neural Transm. 121, 283–287 (2014).
Rutherford, N. J. et al. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79, 717–718 (2012).
Vass, R. et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–380 (2011).
Lang, C. M. et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 287, 19355–19365 (2012).
Brady, O. A., Zhou, X. & Hu, F. Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J. Biol. Chem. 289, 19670–19680 (2014).
Sawaya, M. R., Hughes, M. P., Rodriguez, J. A., Riek, R. & Eisenberg, D. S. The expanding amyloid family: structure, stability, function, and pathogenesis. Cell 184, 4857–4873 (2021).
Shi, Y. et al. Structure-based classification of tauopathies. Nature 598, 359–363 (2021).
Cruchaga, C. et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 68, 581–586 (2011).
Nicholson, A. M. et al. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J. Neurochem. 126, 781–791 (2013).
Inukai, Y. et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 582, 2899–2904 (2008).
Laferrière, F. et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 22, 65–77 (2019).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
O’Brien, R. J. & Wong, P. C. Amyloid precursor protein processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 34, 185–204 (2011).
Nonaka, T. et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 (2013).
Arseni, D. et al. Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601, 139–143 (2022).
Schmidt, M. et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 10, 5008 (2019).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Wagner, T. et al. Two particle-picking procedures for filamentous proteins: SPHIRE–crYOLO filament mode and SPHIRE-STRIPER. Acta Crystallogr. D 76, 613–620 (2020).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
He, S. & Scheres, S. H. W. Helical reconstruction in RELION. J. Struct. Biol. 198, 163–176 (2017).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Terwilliger, T. C. Automated side-chain model building and sequence assignment by template matching. Acta Crystallogr. D 59, 45–49 (2003).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P. & Eisenberg, D. S. Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 27, 653–659 (2020).
Thevis, M., Ogorzalek Loo, R. R. & Loo, J. A. In-gel derivatization of proteins for cysteine-specific cleavages and their analysis by mass spectrometry. J. Proteome Res. 2, 163–172 (2003).
McConnell, S. A. et al. Protein labeling via a specific lysine–isopeptide bond using the pilin polymerizing sortase from Corynebacterium diphtheriae. J. Am. Chem. Soc. 140, 8420–8423 (2018).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Acknowledgements
We thank Z. H. Zhou for the use of Electron Imaging Center for Nanomachines (EICN) instruments supported by the NIH (1S10RR23057 and IS10OD018111), NSF (DBI-1338135) and California NanoSystems Institute at UCLA. We thank S. Scheres and M. Goedert for discussion of TMEM106B antibodies, and L. Salwinski for evaluation of the significance of sequence matching. The authors acknowledge NIH AG 054022, NIH AG061847, DOE DE-FC02-02ER63421, NIH GM103479, NIH GM007185, NSF DGE-1650604, ADRC (P30 AG062677), Einstein Aging Study (P01 AG003949), ALLFTD (U19 AG063911), C9ORF72 P01 (P01 NS084974), and the National Facility for Translational Medicine (Shanghai) for support.
Author information
Authors and Affiliations
Contributions
Y.X.J. and Q.C. extracted fibrils from the material donated by patients with FTLD-TDP, prepared cryo-EM grids, and collected and processed cryo-EM data. Y.X.J., Q.C. and M.R.S. built the atomic models. P.G. assisted in cryo-EM data collection. Y.X.J. and R.A. performed western blotting and immunolabelling assays. Y.X.J. performed patient genotyping. J.Y.F., R.R.O.L. and J.A.L. performed mass spectrometry. M.D. and D.W.D. prepared frozen brain samples and performed immunohistochemistry staining. Q.C. and M.R.S. performed solvation energy calculations. All authors analysed the results and wrote the manuscript. D.S.E. supervised the project.
Corresponding author
Ethics declarations
Competing interests
D.S.E. is an advisor and equity shareholder in ADRx, Inc. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Henning Stahlberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Comparison of FTLD-TDP and non-FTLD-TDP donors.
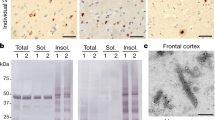
a, Age distribution of FTLD-TDP donors (F, n = 40) and non-FTLD-TDP controls (N, n = 8). Donors with and without fibrils detected under negative stain EM are coloured black and blue, respectively. P-value of 0.53 (n.s., not significant) from an unpaired, two-tailed t-test suggests that the presence of fibrils is disease-dependent but not age-dependent. b, Western blots of sarkosyl-insoluble fractions from FTLD-TDP (F) and non-FTLD-TDP donors (N) probed by TMEM106B antibody. The ~35 kDa TMEM106B-positive band was found in none of the non-FTLD-TDP donors (N5 shown in Fig. 1b as donor 5) and all of the FTLD-TDP donors except F35, F3, and F27. P-value of less than 0.0001 was obtained from an unpaired, two-tailed t-test comparing presence of the ~35kDa band (value of “1” for present, “0” for absent) in non-FTLD-TDP donors (0 out of 8, n = 8) and FTLD-TDP donors (22 out of 25, n = 25). Fibrils were not observed in F3 and F27 by negative stain EM, consistent with the western blot. Fibrils were observed in F35, which suggests that western blot may not always be accurate in detecting TMEM106B aggregation. Western blot membranes were prepared in parallel and exposed with equal time. The original, uncropped blots are shown in Supplementary Fig. 3.
Extended Data Fig. 2 Fibril screen of FTLD-TDP and non-FTLD-TDP donors by negative stain EM.
Negative stain EM images of sarkosyl-insoluble fractions from all donors. Scale bar 200 nm. Donors 1–5 (F26, F36, F17, F40 and N5, respectively) are also shown in Fig. 1a. Fibrils with similar morphologies were found in all FTLD-TDP donors except F3 and F27. No fibrils were found in any non-FTLD-TDP donors. P-value of less than 0.0001 was obtained from an unpaired, two-tailed t-test comparing fibrils detected by EM (value of 1 for present, 0 for absent) in FTLD-TDP donors (38 out of 40, n = 40) and non-FTLD-TDP donors (0 out of 8, n = 8).
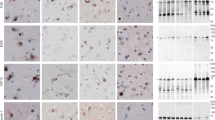
Extended Data Fig. 3 Neuropathological diagnosis of donors 1–4 as FTLD-TDP.
Immunohistochemistry staining using a phosphor-Ser409/410 TDP-43 antibody was performed for brain sections from donors 1–4. All four donors were confirmed to be FTLD-TDP cases, representing the 4 subtypes A to D, respectively. For all figures, scale bar 20 μm. a, b, c, Donor 1 is FTLD-TDP type C, with long, thick neurites (arrows in b) and 'Pick-body like NCI' in dentate fascia (c). d, e, f, Donor 2 is FTLD-TDP type B, displaying characteristic granular cytoplasmic NCI in cortex (d), hippocampus (e) and dentate fascia (f). g, h, i, Donor 3 is FTLD-TDP type A, exhibiting small dense neuronal cytoplasmic inclusions (NCI), sparse neuronal intranuclear inclusions (NII, arrow in g), and perivascular glial inclusions (arrow in h). j, k, l, Donor 4 is FTLD-TDP type D, shown by frequent NII (arrows in j, k and l) and small NCI and neurites. a, b, d, e, g, h, i, j, k, l are brain sections from the temporal cortex. c and f are brain sections from dentate fascia.
Extended Data Fig. 4 Cryo-EM data processing of amyloid fibrils from FTLD-TDP donors 1 to 4.
a, Representative micrographs of PM1-3 from FTLD-TDP donor 1. Scale bar 500 Å. b, Representative 2D classes of PM1-3 from FTLD-TDP donors 1 to 4. 2D classes are stitched together to show a full cross-over of each morphology. c, 3D reconstructions of PM1-3 from the four FTLD-TDP donors. d, Distributions of the three fibril polymorphs in the four FTLD-TDP donors.
Extended Data Fig. 5 Cryo-EM maps and FSC curves of FTLD-TDP donor 1.
a, Views of the cryo-EM maps from FTLD-TDP donor 1 with five layers shown. b, FSC curves between two half-maps (left) and the cryo-EM reconstruction and refined atomic model (middle) of each polymorph PM1 (blue), PM2 (green), and PM3 (pink). FSC curves between cryo-EM reconstruction and the query model (red, Direction 1 chain of Extended Data Fig. 5a) and the atomic model of PM1 from FTLD-TDP donor 1 (blue) are compared on the right.
Extended Data Fig. 6 Cryo-EM maps of TMEM106B fibrils from FTLD-TDP donors 1 to 4.
The models of PM1-3 from donor 1 were rigid body fitted into the maps of PM1-3 from donors 2 to 4. These cryo-EM structures reveal that the polymorphs from all four donors share the same protofilament fold. Two subtypes of PM2 are exhibited by donors 1 and 4 (light green) and donors 2 and 3 (dark green), respectively.
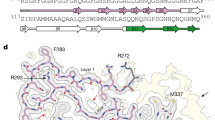
Extended Data Fig. 7 Identification of the TMEM106B molecule by atomic model building.
a, Atomic model building flowchart for PM1 of FTLD-TDP donor 1. In the sequence alignment (bottom right), lines indicate identical residues and two dots indicate similar residues. b, Comparison of query model (Direction 1 chain, orange) with final model built with TMEM106B sequence (blue). Four representative regions of the cryo-EM map are shown. Residues that show clear differences in side chain density fitting are labelled in both models.
Extended Data Fig. 8 Evidence of glycosylation and genetic polymorphism in maps of TMEM106B fibrils.
a, Three-dimensional reconstructions of PM1-3 from FTLD-TDP donor 1. White arrows point to the four glycosylation sites within the fibril core. Red arrows point to the residual densities outside the fibril core in PM1 and PM3, which may correspond to the binding of the same undefined, negatively charged ligand present in the PM2 dimer interface (Extended Data Fig. 9). b, Maps and models of the four glycosylation sites with or without the sugar group in PM1 (left), PM2 (middle), and PM3 (right) from FTLD-TDP donor 1. c, Position of Thr/Ser185 genetic polymorphism in the conserved fibril fold (left) and the map and model of the Thr/Ser185 environment (right, represented by PM1 from donor 1).
Extended Data Fig. 9 Diverse dimer interfaces of PM2 and PM3 of TMEM106B fibrils.
a, Dimer arrangements of PM2 and PM3 from FTLD-TDP donors 1 to 4. PM2 and PM3 from all donors are aligned at chain A (grey, represented by PM2 of donor 1). Chain B of PM2 (light green for donors 1 and 4, dark green for donors 2 and 3) and PM3 (pink) from each donor is shown. Residual densities in the PM2 dimer interfaces are shown as green ovals. The dimer arrangement of PM3 is consistent among all donors, whereas there are two subtypes of dimer arrangements for PM2. b, Atomic model and the residual density in the dimer interface of donor 1 (left, represents donors 1 and 4) or donor 2 (right, represents donors 2 and 3). In donors 1 and 4, Arg180 from each protofilament is on the opposite sides of an extra density in the middle of the PM2 dimer interface; in donors 2 and 3, the dimer interface is shifted to Lys178. Although two slightly different dimer interfaces were observed, we consider PM2 in all four FTLD-TDP donors to be the same morphology because of the similarity in dimer formation (see Discussion). c, Comparison of the residues near the PM3 interface (far left) from PM1 (blue), PM2 (green), PM3 (pink), and the superimposition of those residues from PM1-3 (far right). PM1-3 are all represented by FTLD-TDP donor 1.
Extended Data Fig. 10 Detection of TMEM106B peptides by mass spectrometry.
a, Sequence map of TMEM106B. Sequences in red indicate unique peptides detected by LC-MS/MS from excised gel bands. b, Fragmentation spectra (MS/MS) of detected peptides from TMEM106B. Detected fragment ions (b, y, and immonium ions) are labelled accordingly. The peptide modifications methionine oxidation (OX) and cysteine carbamidomethylation (CA) were observed. SDS-PAGE gel of sarkosyl-insoluble fraction of donor 1 shown as an insert. Box indicates the gel region, which corresponds to the ~35kDa band from the TMEM106B western blot (Fig. 1b), excised for LC-MS/MS analyses.
Supplementary information
Supplementary Figures
This file contains Supplementary Figs. 1–3.
Rights and permissions
About this article

Cite this article
Jiang, Y.X., Cao, Q., Sawaya, M.R. et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 605, 304–309 (2022). https://doi.org/10.1038/s41586-022-04670-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04670-9
This article is cited by
-
TAF15 amyloid filaments in frontotemporal lobar degeneration
Nature (2024)
-
Automated model building and protein identification in cryo-EM maps
Nature (2024)
-
Design of amyloidogenic peptide traps
Nature Chemical Biology (2024)
-
Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy
Nature Communications (2024)
-
TMEM106B coding variant is protective and deletion detrimental in a mouse model of tauopathy
Acta Neuropathologica (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
