
Abstract
The development of catalytic chemical processes that enable the revalorization of nitrous oxide (N2O) is an attractive strategy to alleviate the environmental threat posed by its emissions1,2,3,4,5,6. Traditionally, N2O has been considered an inert molecule, intractable for organic chemists as an oxidant or O-atom transfer reagent, owing to the harsh conditions required for its activation (>150 °C, 50‒200 bar)7,8,9,10,11. Here we report an insertion of N2O into a Ni‒C bond under mild conditions (room temperature, 1.5–2 bar N2O), thus delivering valuable phenols and releasing benign N2. This fundamentally distinct organometallic C‒O bond-forming step differs from the current strategies based on reductive elimination and enables an alternative catalytic approach for the conversion of aryl halides to phenols. The process was rendered catalytic by means of a bipyridine-based ligands for the Ni centre. The method is robust, mild and highly selective, able to accommodate base-sensitive functionalities as well as permitting phenol synthesis from densely functionalized aryl halides. Although this protocol does not provide a solution to the mitigation of N2O emissions, it represents a reactivity blueprint for the mild revalorization of abundant N2O as an O source.
Similar content being viewed by others
Main
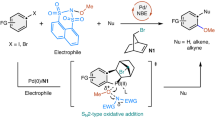
The increasing emission of greenhouse gases represents a global environmental threat, and strategies to address this issue have been the focus of intense research in recent times1,2. From the sustainability point of view, the development of chemical processes that extend beyond the traditional degradations and repurpose such gaseous by-products as useful synthons to produce valuable chemical feedstocks is highly desirable. Whereas the revalorization of CO2 or CH4 as carbon sources for organic synthesis through catalytic strategies has received a great deal of attention12,13, much less interest has been focused on the chemical transformation of another major contributor to the global warming: N2O. Governmental reports and recent scientific evidence both warn of the consequences that result from the increasing presence of this undervalued gas in the atmosphere3,4,5. N2O exhibits a global warming potential >300 times that of CO2, with a decomposition half-time in the atmosphere of >100 years6. Human activities have accelerated the emissions, with an estimated rate of increase for N2O of 2% per decade. Yet, a detailed analysis through the lens of sustainable synthesis presents a unique opportunity for N2O revalorization, as it represents an excellent O-atom source: it is readily available, non-toxic (laughing gas) and releases benign N2 as a by-product on O removal. Conversely, N2O is an inert gas, whose activation requires high temperatures (140–350 °C) and pressures (50–200 bar), resulting in limited applications as an oxidant for organic synthesis7,8,14. Yet, the structure of N2O has captivated chemists, who studied in detail coordination modes and the reactivity thereof in a plethora of metal complexes9. However, few reports focused on its activation towards the formation of C‒O bonds10,15 (arguably among the most valuable bonds in organic synthesis), as it would permit access to highly coveted alcohols, ethers, epoxides and so on. Still, these few examples rely on traditional metal–oxo reactivity, which requires high temperatures (100–200 °C) and pressures (10 bar)11 or long reaction times (1 ton per week)16 (Fig. 1a). In a groundbreaking report17, a fundamentally different outcome was observed: on exposure to a N2O atmosphere, the O atom could be inserted into a Hf‒Ph bond of complex 1 forging the desired Hf‒O‒Ph (2) with extrusion of N2. However, regioselectivity issues arose, as the O atom was competitively transferred to the hydride ligand, thus also producing a Hf‒O‒H complex (3). Mechanistic studies on the O insertion step into various M‒C(sp2) bonds using N2O, peroxides or oxygen suggest that an organometallic Baeyer–Villiger (OMBV)-type mechanism is operating, whereby the anionic carbon migrates to the coordinated O atom, forging the M‒O‒C bond18,19,20,21,22,23. On the basis of this reactivity, we aimed at unlocking the potential of N2O as an O-atom source in a fundamentally different catalytic synthesis of phenols. In the canonical transition-metal-catalysed phenol synthesis from aryl halides24,25,26,27,28, th‑e C(sp2)‒O bond-forming step proceeds through the well-established ligand exchange with a nucleophilic O source. On reductive elimination with the aryl group, the desired C(sp2)‒O bond is formed while the metal center is twofold-reduced (Fig. 1b, left). The source of O in these cases is usually H2O or a protic O-based nucleophile in combination with a base that lead to the corresponding phenol29. In the alternative catalytic cycle proposed herein, the fundamental step for C(sp2)‒O bond formation capitalizes on the OMBV-type mechanism: on N2O coordination to the metal centre, the electrophilic O is eventually inserted into the M‒C bond, with concomitant formation of N2 (Fig. 1b, right). In contrast to the traditional synthesis of phenols, after C‒O bond formation the oxidation state of the metal centre remains intact, thus requiring an external reductant to close the cycle. To orchestrate this reductive process, we focused our attention on Ni and its demonstrated ability to manoeuvre between different oxidation states through single-electron transfer30,31,32. Here we demonstrate that a mechanistically guided approach for the activation of N2O with organometallic complexes results in the development of a mild and selective catalytic synthesis of high-value phenols from aryl halides using N2O as an electrophilic O source33. The mild conditions (25 °C and 1.5–2 atm) allow the accommodation of a variety of functional groups, including base-sensitive moieties, thus providing an orthogonal strategy to the current technologies (Fig. 1c).

a, Reactivity of N2O and transition metals in the formation of C–O bonds. The right panel is based on the work described in ref. 17; OAT, oxygen atom transfer; Cp*, pentamethylcyclopentadienyl anion b, Comparison between traditional C(sp2)–O bond formation via reductive elimination (left panel) and through O insertion (right panel; this work) for the synthesis of phenols. Red., reduction.; OA, oxidative addition; RE, reductive elimination; LM, ligand metathesis; c, Activation of N2O in a Ni reductive catalytic cross-electrophile phenol synthesis, (this work).
To investigate the feasibility of the M‒C(sp2) oxidation, we drew inspiration from previous work17, in which N2O was demonstrated to react with certain phosphine–Ni(II) complexes34,35. To this end, we synthesized the product of oxidative addition 4, and studied its reactivity with N2O (Fig. 2a). As expected, 4 rapidly decomposes mainly towards homocoupling (5) when dissolved in DMA under argon, with only traces of protodemetallation (6) detected (path a). This reactivity is exacerbated by the presence of reducing agents such as Zn (path b). Yet, when the argon atmosphere is replaced by N2O, the bright red colour of the solution of 4 remains, thus pointing towards a slower decomposition rate. After acidic workup, a 15% yield of phenol 7 is observed. However, when the same reaction is performed in the presence of a reducing agent, substantially higher yields of 7 were observed, with a 73% yield obtained when a combination of Zn and NaI was used (path d). These results point to the feasibility of developing a reductive catalytic protocol based on Ni catalysis using aryl halide precursors. From our extensive ligand survey (see Supplementary Information), it was evident that tridentate nitrogenated ligands with the general pattern of 2-substituted bipyridine were crucial to obtain catalytic activity, with terpyridine (L18) and 6-pyrazolyl-2,2'-bipyridine (L50) affording the highest yields of 9 (Fig. 2b and Supplementary Information). Analysis of the R group revealed three key features of the ligand for catalytic activity: replacing the N atom with C‒H or S prevents catalytic activity (L48 and L61); steric encumbrance next to the N of the pyrazole unit inhibits productive catalysis (L55 and L58); and electron-deficient substituents on the pyrazole markedly reduce the yield of phenol. To confirm Ni–ligand ligation in the reaction mixture, complex 10 was prepared and structurally characterized. A 75% yield of 9 was obtained using 10 as a catalyst, thus confirming that the pre-ligated complex is catalytically competent. L50 represents a new ligand platform in Ni catalysis, with virtually no examples. As Fig. 2a, b suggests that formal Ni(I)‒C(sp2) species might be involved, we prepared terpyridine–Ni derivatives such as 11 (Fig. 2c), as greater stability of the corresponding (terpy)Ni(I)‒Ar has been noted36,37,38. As for 4, reaction of 11 in the absence of reducing agent led to no phenol formation. Yet, despite the presence of two Me groups in ortho, reaction performed under N2O in the presence of Zn and NaI afforded the desired mesitol (12) in 49% yield on acidic workup. To further confirm the involvement of a formal Ni(I)‒C(sp2) in the tridentate system, the (tBu-terpy)Ni(I)‒I (13) was reacted with Ph2Zn under N2O. Despite the reported instability of (terpy)Ni(I)‒Ph (ref. 37), a 20% yield of phenol (14) was obtained. These findings suggest that reduction of Ni(II) species to formal Ni(I) and the presence of iodide salts in the system are of importance to forge the desired C(sp2)‒O bond. Mechanistic investigations on M‒Ar oxy insertions for late transition metals (Pd, Ni and Fe) reveal that subtle changes in the ligand environment also lead to differences between the metal‒oxo/oxyl or concerted Baeyer-Villiger pathways for Ar‒O bond formation18,19,20,21,22,23. In this case, we suggest that in the continuum between the two extreme possibilities offered in the OMBV reaction, the oxy insertion of N2O in a d9 complex lies towards the formation of the M‒O bond and N2, before Ar migration39.

a, Stoichiometric reactivity using bipyridine-supported Ni(II) oxidative addition complexes. conv., conversion; equiv., equivalents. b, Key electronic and structural features of the ligand in the catalytic synthesis of phenols from aryl halides and N2O. Details on the complete optimization of the reaction conditions can be found in the Supplementary Information. c, Reactivity studies of tridentate terpyridine-supported Ni(II) and Ni(I) complexes with N2O, pointing to the involvement of Ni(I)–C(sp2) species. Electron paramagnetic resonance (EPR) details and discussion for 13 can be found in the Supplementary Information. aUsing 2 atm of N2O. bWith 5 mol% extra L50. cYields using NiBr2(diglyme).
With the optimized catalytic system in hand, a preliminary scope of the aryl halide counterpart was interrogated. As shown in Fig. 3, aryl iodides bearing other halogens in both para (9, 15 and 16) and meta (17–19) positions smoothly afforded the corresponding phenol in excellent yields. The presence of electron-withdrawing groups such as CF3 (7), ketone (20), ester (21 and 24) or nitrile (22–23) posed no difficulty for the C‒O bond formation. Electron-donating substituents such as alkyl (25), aryl (26), or even methoxy and thiomethyl (27 and 28) delivered phenol in good yields. Moreover, a fluorene derivative (29) featuring benzylic C‒H bonds was also amenable for phenol synthesis, albeit in 38% yield. A classical feature of reductive couplings is that steric hindrance at the ortho position can impede reactivity. Indeed, C‒O bond formation from indanone (31) and 1-chloro-2-iodobenzene (30) derivatives afforded slightly diminished yields. In contrast to 30, 32 was obtained in 79% yield, illustrating a possible beneficial chelating effect of the ortho OMe and the Ni centre. A silylated benzylic alcohol (33) or a diethylphosphonate (34) were also tolerated in this protocol. Heterocycles such as indole (35), quinoline (36), carbazole (37) or dibenzothiophene (38) also afforded the corresponding phenol in good yields. Substrates prone to rapid oxidation after C‒O bond formation could be further functionalized in situ, as exemplified by the 68% yield obtained for the pivaloyl derivative 39. An iodide derivative of the biologically active agent clofibrate could be converted to phenol 40 in 78% yield despite the presence of a tertiary α-oxy ester. This protocol does not require the use of nucleophilic alkoxy surrogates, and hence base-sensitive functionalities such as esters or sensitive amides, can be tolerated. An example of this chemoselectivity is observed in the derivatization of a substrate containing pinacol boronate. In this case, phenol 41 was still obtained in 56% yield, thus providing an orthogonal tool to classical oxidation. The observation of 7% yield of sulfoxide in the reaction of 28 (ref. 40), and the low yield obtained for fluorenol 29, suggest that the oxy-insertion step lies towards the oxo/oxyl–pathway in the continuum postulated for OMBV-type reactions. N2 was detected using a gas chromatography–thermal conductivity detector in the headspace after the reaction had finished for 7, 9, 18, 25 and 34 (Fig. 3). When the oxygen on the solvent was labelled ([18O]DMF, 25% 18O), no 18O was incorporated in 9. On the other hand, when N15N18O was used (ca. 23% 18O), 22% ± 1 of the O in 9 was labelled (Supplementary Information). Together, these data point to N2O as the source of O.

Scope of aryl iodides. [N2], N2 detected by a gas chromatography–thermal conductivity detector at the end of the reaction. All yields are of isolated pure material. Yield in brackets: 1H NMR yield calculated using dibromomethane as an internal standard. incorp., incorporated. See the Supplementary Information for details of the procedures. aUse of L18 as the ligand instead of L50. bOwing to the rapid oxidation of the free alcohol, 39 was obtained after quenching with Piv2O.
The same optimized reaction conditions for aryl iodides permitted C‒O bond formation of more accessible and commercially available aryl bromides. Yet, electron-withdrawing substituents were required to allow C(sp2)‒Br cleavage to occur. In this sense, phenols bearing CF3 (7), Ac (20) and CN (22) in the para position, as well as paraben (21), could be obtained in high yields (Fig. 4a). Medicinally relevant phthalide was also smoothly converted to the phenol (42), thus providing a method to synthesize this building block with three fewer steps compared with the reported method41. Phenols derived from π-extended or conjugated systems such as naphthoate 43 or cinnamate 44 were also obtained in 65% and 78% yields, respectively. In contrast to current light-mediated processes, no isomerization of the double bond in 44 was observed30. Finally, another base-sensitive group such as the aryl methyl sulfone could be tolerated and the corresponding phenol 45 was obtained in 82% yield. Heterocyclic bromides are not compatible with the current protocol.

a, Catalytic synthesis of phenols from aryl bromides using N2O. b, Exploring the insertion of O atoms into densely functionalized aryl halides (starting from Ar–X). c, Using greenhouse gases as building blocks for the synthesis of biologically relevant molecules. All yields are of isolated pure material. See Supplementary Information for experimental details. aUse of L18 as the ligand instead of L50. bYields determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
Complex aryl halides functionalized with sensitive moieties were then tested. For example, an empagliflozin derivative, which contains a plethora of weak C‒H bonds prone to HAT, was smoothly converted to the corresponding phenol (46) in excellent yield (Fig. 4b). An ester derivative of the natural product eugenol afforded the desired phenol (47) in an 84% yield, highlighting the high chemoselectivity of this process over alternative oxidation through metal–oxo pathways. Despite the triphasic nature of the protocol, synthesis of 47 could be scaled up to 5 mmol with only a slight reduction in the yield (66%). Substrates containing saturated N-heterocycles such as piperazinone 48, azetidine 49, pyrrolidinone (aniracetam intermediate) 50 and nortropinone derivative 51 are well tolerated. The requirement of an electron-withdrawing group to activate the aryl bromide can be turned into a synthetic advantage, thus permitting regioselective control on the activated aryl bromide (52, 78%). Finally, a derivative ezetimibe, the drug used to treat high blood cholesterol, could be smoothly converted into the corresponding phenol (53), without altering the chiral and unprotected secondary alcohol, the ester and the strained β-lactam. Similar chemoselectivity can be observed in the conversion of paroxetine derivative 54. Finally, Fig. 4c illustrates a proof-of-concept of the potential for the revalorization of greenhouse gases for organic synthesis. It is now possible to combine N2O and CO2 revalorization strategies and obtain metaxolone (59), in which 66% of the oxygen atoms originate from waste gaseous feedstock42. A more striking example is illustrated in the synthesis of bazedoxifene (68), a drug candidate against breast and pancreatic cancer. The three phenolic building blocks could be rapidly obtained from the parent halides in good yields (64–66). Subsequent Fischer-indole synthesis allows access to indole 67, enabling the synthesis of bazedoxifene (68) with all O atoms originating from N2O (refs. 43,44,45,46). Whereas a 42% yield could be obtained for precursor 63 with 1 mol% catalyst loading, a <10% yield of 14 was observed with the same catalyst loading, with substantial protodehalogenation of the parent iodide 61, which highlights the subtle differences between aryl iodides and aryl bromides in this system.
Conclusions
Through a distinct fundamental organometallic step, a catalytic protocol for the revalorization of N2O as a green, mild and chemoselective O-atom insertion reagent for organic synthesis has been unlocked. Mechanistically guided insights into the reactivity of N2O with Ni complexes point to formally low-valent Ni(I)‒aryl permitting the O insertion in an efficient manner. The inert N2O molecule succumbs to activation under mild conditions for the selective synthesis of phenols from aryl halides. The catalytic system features an electronically asymmetric tridentate bipyridine-based ligand (L50) for the Ni centre, which enables selective C‒O bond formation. The reported conditions are simple and robust, allowing phenol formation in densely functionalized molecules. Whereas other catalytic protocols capitalize on nucleophilic HO‒ counterparts, this method represents a unique example of catalytic C‒O bond formation with an electrophilic O-atom source, which in turn can accommodate base-sensitive functionalities. Furthermore, this protocol demonstrates the feasibility of accessing relevant drugs for which N2O is the sole source of oxygen atoms.
Data availability
Details on the procedures, optimization, characterization and mechanisms, including spectra of new compounds and compounds made using the reported method, are available in the Supplementary Information. Crystallographic data for compound 10 can be obtained free of charge from www.ccdc.cam.ac.uk under reference number 2114695.
Change history
21 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-022-05064-7
References
Kyoto Protocol Reference Manual on Accounting of Emissions and Assigned Amount (UNFCCC; 2008); https://unfccc.int/resource/docs/publications/08_unfccc_kp_ref_manual.pdf
Fawzy, S., Osman, A. I., Doran, J. & Rooney, D. W. Strategies for mitigation of climate change: a review. Environ. Chem. Lett. 18, 2069–2094 (2020).
Crutzen, P. J. The influence of nitrogen oxides on the atmospheric ozone content. Q. J. R. Meteorol. Soc. 96, 320–325 (1970).
Ravishankara, A. R., Daniel, J. S. & Portmann, R. W. Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326, 123–125 (2009).
Davidson, E. A. Representative concentration pathways and mitigation scenarios for nitrous oxide. Environ. Res. Lett. 7, 024005 (2012).
Tian, H. et al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature 586, 248–256 (2020).
Parmon, V. N., Panov, G. I., Uriarte, A. & Noskov, A. S. Nitrous oxide in oxidation chemistry and catalysis: application and production. Catal. Today 100, 115–131 (2005).
Teles, J. H., Roessler, B., Pinkos, R., Genger, T. & Preiss, T. Method for producing a ketone. US patent US7449606 (2008).
Tolman, W. B. Binding and activation of N2O at transition-metal centers: recent mechanistic insights. Angew. Chem. Int. Ed. 49, 1018–1024 (2010).
Severin, K. Synthetic chemistry with nitrous oxide. Chem. Soc. Rev. 44, 6375–6386 (2015).
Groves, J. T. & Roman, J. S. Nitrous oxide activation by a ruthenium porphyrin. J. Am. Chem. Soc. 117, 5594–5595 (1995).
Aresta, M., Dibenedetto, A. & Angelini, A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 114, 1709–1742 (2014).
Sun, L., Wang, Y., Guan, N. & Li, L. Methane activation and utilization: current status and future challenges. Energy Technol. 8, 1900826 (2020).
Corona, T. & Company, A. Nitrous oxide activation by a cobalt(ii) complex for aldehyde oxidation under mild conditions. Dalton Trans. 37, 14530–14533 (2016).
Gianetti, T. L. et al. Nitrous oxide as a hydrogen acceptor for the dehydrogenative coupling of alcohols. Angew. Chem. Int. Ed. 55, 1854–1858 (2015).
Yonke, B. L., Reeds, J. P., Zavalij, P. Y. & Sita, L. R. Catalytic degenerate and nondegenerate oxygen atom transfers employing N2O and CO2 and a MII/MIV cycle mediated by Group 6 MIV terminal oxo complexes. Angew. Chem. Int. Ed. 50, 12342–12346 (2011).
Vaughan, G. A., Rupert, P. B. & Hillhouse, G. L. Selective O-atom transfer from nitrous oxide to hydride and aryl ligands of bis(pentamethylcyclopentadienyl)hafnium derivatives. J. Am. Chem. Soc. 109, 5538–5539 (1987).
Figg, T. M., Cundari, T. R. & Gunnoe, T. B. Non-redox oxy-insertion via organometallic Baeyer–Villiger transformations: a computational Hammett study of platinum(II) complexes. Organometallics 30, 3779–3785 (2011).
Figg, T. M., Webb, J. R., Cundari, T. R. & Gunnoe, T. B. Carbon–oxygen bond formation via organometallic Baeyer–Villiger transformations: a computational study on the impact of metal identity. J. Am. Chem. Soc. 134, 2332–2339 (2012).
Cheng, M.-J. et al. The para-substituent effect and pH-dependence of the organometallic Baeyer–Villiger oxidation of rhenium–carbon bonds. Dalton Trans. 41, 3758–3763 (2012).
Mei, J. et al. Oxygen atom insertion into iron(II) phenyl and methyl bonds: a key step for catalytic hydrocarbon functionalization. Organometallics 33, 5597–5605 (2014).
Bischof, S. M. et al. Functionalization of rhenium aryl bonds by O-atom transfer. Organometallics 30, 2079–2082 (2011).
Sugimoto, H. et al. Oxygen atom insertion into the osmium–carbon bond via an organometallic oxido–osmium(V) intermediate. Organometallics 40, 102–106 (2021).
Liu, Y., Liu, S. & Xiao, Y. Transition-metal-catalyzed synthesis of phenols and aryl thiols. Beilstein J. Org. Chem. 13, 589–611 (2017).
Anderson, K. W., Ikawa, T., Tundel, R. E. & Buchwald, S. L. The selective reaction of aryl halides with KOH: synthesis of phenols, aromatic ethers, and benzofurans. J. Am. Chem. Soc. 128, 10694–10695 (2006).
Sambiagio, C., Marsden, S. P., Blacker, A. J. & McGowan, P. C. Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev. 43, 3525–3550 (2014).
Tlili, A., Xia, N., Monnier, F. & Taillefer, M. A very simple copper-catalyzed synthesis of phenols employing hydroxide salts. Angew. Chem. Int. Ed. 48, 8725–8728 (2009).
Zhao, D. et al. Synthesis of phenol, aromatic ether, and benzofuran derivatives by copper-catalyzed hydroxylation of aryl halides. Angew. Chem. Int. Ed. 48, 8729–8732 (2009).
Fier, P. S. & Maloney, K. M. Synthesis of complex phenols enabled by a rationally designed hydroxide surrogate. Angew. Chem. Int. Ed. 56, 4478–4482 (2017).
Terrett, J. A., Cuthbertson, J. D., Shurtleff, V. W. & MacMillan, D. W. C. Switching on elusive organometallic mechanisms with photoredox catalysis. Nature 524, 330–334 (2015).
Yang, L. et al. Synthesis of phenols: organophotoredox/nickel dual catalytic hydroxylation of aryl halides with water. Angew. Chem. Int. Ed. 57, 1968–1972 (2018).
Sun, R., Qin, Y. & Nocera, D. G. General paradigm in photoredox nickel-catalyzed cross-coupling allows for light-free access to reactivity. Angew. Chem. Int. Ed. 59, 9527–9533 (2020).
Li, Z. et al. A tautomeric ligand enables directed C‒H hydroxylation with molecular oxygen. Science 372, 1452–1457 (2021).
Koo, K., Hillhouse, G. L. & Rheingold, A. L. Oxygen-atom transfer from nitrous oxide to an organonickel(II) phosphine complex. Syntheses and reactions of new nickel(II) aryloxides and the crystal structure of [cyclic] (Me2PCH2CH2PMe2)Ni(O-o-C6H4CMe2CH2). Organometallics 14, 456–460 (1995).
Matsunaga, P. T., Hillhouse, G. L. & Rheingold, A. L. Oxygen-atom transfer from nitrous oxide to a nickel metallacycle. Synthesis, structure, and reactions of [cyclic] (2,2'-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 115, 2075–2077 (1993).
Lin, C.-Y. & Power, P. P. Complexes of Ni(i): a “rare” oxidation state of growing importance. Chem. Soc. Rev. 46, 5347–5399 (2017).
Klein, A., Kaiser, A., Sarkar, B., Wanner, M. & Fiedler, J. The electrochemical behaviour of organonickel complexes: mono-, di- and trivalent nickel. Eur. J. Inorg. Chem. 2007, 965–976 (2007).
Hamacher, C., Hurkes, N., Kaiser, A., Klein, A. & Schüren, A. Electrochemistry and spectroscopy of organometallic terpyridine nickel complexes. Inorg. Chem. 48, 9947–9951 (2009).
Figg, T. M. & Cundari, T. R. Mechanistic study of oxy insertion into nickel-carbon bonds with nitrous oxide. Organometallics 31, 4998–5004 (2012).
Liang, Y. et al. Electrochemically induced nickel catalysis for oxygenation reactions with water. Nat. Catal. 4, 116–123 (2021).
Walsh, S. et al. Inhibitors of the renal outer medullary potassium channel. Patent WO2013039802 (A1) (2013).
Del Vecchio, A. et al. Audisio, carbon isotope labeling of carbamates by late-stage [11C], [13C] and [14C]carbon dioxide incorporation. Chem. Commun. 56, 11677–11680 (2020).
Solyev, P. N., Sherman, D. K., Novikov, R. A., Levina, E. A. & Kochetkov, S. N. Hydrazo coupling: the efficient transition-metal-free C–H functionalization of 8-hydroxyquinoline and phenol through base catalysis. Green Chem. 21, 6381–6389 (2019).
Kelly, P. M. et al. Synthesis, antiproliferative and pro-apoptotic activityof 2-phenylindoles. Bioorg. Med. Chem. 24, 4075–4099 (2016).
Wang, C., Sun, H., Fang, Y. & Huang, Y. General and efficient synthesis of indoles through triazene-directed C–H annulation. Angew. Chem. Int. Ed. 52, 5795–5798 (2013).
Hwang, S. G. et al. Methods of preparing bazedoxifene. Patent KR201895239 (A) (2018).
Acknowledgements
Financial support for this work was provided by Max-Planck-Gesellschaft, Max-Planck-Institut für Kohlenforschung, Fonds der Chemischen Industrie (FCI-VCI), the Swiss National Science Foundation (Early Mobility Postdoctoral Fellowship (grant number 184406), 2019–2021) (F.L.V.) and Universidad de Oviedo-Banco Santander (Mobility Fellowship S.G.-P.). We thank A. Fürstner for discussions and generous support. We also thank the members of the analytical department at MPI-Kohlenforschung for support in the characterization of compounds.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
J.C. and F.L.V. conceived the idea. F.L.V. designed the approach, optimized the process, performed the experiments, analysed the experimental data and prepared the Supplementary Information. S.G.-P. helped in the initial stages of the catalysis optimization. A.M.C. performed ligand syntheses and contributed in mechanistic studies. S.N. carried out the reactions with labelled compounds, and aided in the scope, scalability and limitations of the reported protocol. E.J.R. carried out EPR measurements and analysis. J.B. helped in the scale up of the optimized ligand and in starting materials syntheses for the scope. The manuscript was written by F.L.V. and J.C. The project was directed by J.C.
Corresponding author
Ethics declarations
Competing interests
Max-Planck-Institut für Kohlenforschung has filed a patent (EP21202163.8) on the procedure described in this article.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains general methods, chemical compounds, figures and references.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Le Vaillant, F., Mateos Calbet, A., González-Pelayo, S. et al. Catalytic synthesis of phenols with nitrous oxide. Nature 604, 677–683 (2022). https://doi.org/10.1038/s41586-022-04516-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04516-4
This article is cited by
-
Synthesis of meta-carbonyl phenols and anilines
Nature Communications (2024)
-
N2O revalorization
Nature Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
