
Abstract
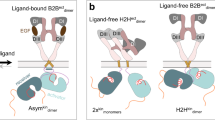
The epidermal growth factor receptor (EGFR) is frequently mutated in human cancer1,2, and is an important therapeutic target. EGFR inhibitors have been successful in lung cancer, where mutations in the intracellular tyrosine kinase domain activate the receptor1, but not in glioblastoma multiforme (GBM)3, where mutations occur exclusively in the extracellular region. Here we show that common extracellular GBM mutations prevent EGFR from discriminating between its activating ligands4. Different growth factor ligands stabilize distinct EGFR dimer structures5 that signal with different kinetics to specify or bias outcome5,6. EGF itself induces strong symmetric dimers that signal transiently to promote proliferation. Epiregulin (EREG) induces much weaker asymmetric dimers that drive sustained signalling and differentiation5. GBM mutations reduce the ability of EGFR to distinguish EREG from EGF in cellular assays, and allow EGFR to form strong (EGF-like) dimers in response to EREG and other low-affinity ligands. Using X-ray crystallography, we further show that the R84K GBM mutation symmetrizes EREG-driven extracellular dimers so that they resemble dimers normally seen with EGF. By contrast, a second GBM mutation, A265V, remodels key dimerization contacts to strengthen asymmetric EREG-driven dimers. Our results argue for an important role of altered ligand discrimination by EGFR in GBM, with potential implications for therapeutic targeting.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
Atomic coordinates and structure factors for the refined structural models described in this paper have been deposited in the Protein Data Bank (PDB) under accession codes 7LEN (EREG:sEGFRR84K, crystallized with trehalose), 7LFR (EREG:sEGFRR84K, crystallized with spermine) and 7LFS (EREG:sEGFRA265V). Source data are provided with this paper.
References
Sharma, S. V., Bell, D. W., Settleman, J. & Haber, D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 (2007).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
An, Z., Aksoy, O., Zheng, T., Fan, Q. W. & Weiss, W. A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 37, 1561–1575 (2018).
Wilson, K. J., Gilmore, J. L., Foley, J., Lemmon, M. A. & Riese, D. J., II. Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacol. Ther. 122, 1–8 (2009).
Freed, D. M. et al. EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell 171, 683–695 (2017).
Macdonald-Obermann, J. L. & Pike, L. J. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem. 289, 26178–26188 (2014).
Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20, 555–572 (2020).
Eck, M. J. & Yun, C. H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 1804, 559–566 (2010).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Heimberger, A. B. et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 11, 1462–1466 (2005).
Eskilsson, E. et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 20, 743–752 (2018).
Lee, J. C. et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 3, e485 (2006).
Ng, P. K. et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell 33, 450–462 (2018).
Lu, C. et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 30, 5432–5443 (2010).
Garrett, T. P. J. et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 110, 763–773 (2002).
Ferguson, K. M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 37, 353–373 (2008).
Ogiso, H. et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 (2002).
Liebschner, D. et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73, 148–157 (2017).
Diwanji, D. et al. Structures of the HER2–HER3–NRG1β complex reveal a dynamic dimer interface. Nature 600, 339–343 (2021).
Alvarado, D., Klein, D. E. & Lemmon, M. A. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 142, 568–579 (2010).
Binder, Z. A. et al. Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell 34, 163–177 (2018).
Orellana, L. et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl Acad. Sci. USA 116, 10009–10018 (2019).
Fan, Q. W. et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 24, 438–449 (2013).
An, Z. et al. EGFR cooperates with EGFRvIII to recruit macrophages in glioblastoma. Cancer Res. 78, 6785–6794 (2018).
Friedmann-Morvinski, D. et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012).
Del Vecchio, C. A. et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 32, 2670–2681 (2013).
Emlet, D. R. et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 74, 1238–1249 (2014).
Alcantara Llaguno, S. et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 22, 545–555 (2019).
Jaiswal, B. S. et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617 (2013).
Hopkins, J. B., Gillilan, R. E. & Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Cryst. 50, 1545–1553 (2017).
Manalastas-Cantos, K. et al. ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 54, 343–355 (2021).
Lemmon, M. A. et al. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 16, 281–294 (1997).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Smart, O. S. et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 (2012).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Winn, M. D., Isupov, M. N. & Murshudov, G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 (2001).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Dawson, J. P. et al. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol. Cell. Biol. 25, 7734–7742 (2005).
Ferguson, K. M., Darling, P. J., Mohan, M. J., Macatee, T. L. & Lemmon, M. A. Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 19, 4632–4643 (2000).
Essletzbichler, P. et al. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 24, 2059–2065 (2014).
Kiyatkin, A., van Alderwerelt van Rosenburgh, I. K., Klein, D. E. & Lemmon, M. A. Kinetics of receptor tyrosine kinase activation define ERK signaling dynamics. Sci. Signal. 13, eaaz5267 (2020).
Aksamitiene, E., Hoek, J. B. & Kiyatkin, A. Multistrip western blotting: a tool for comparative quantitative analysis of multiple proteins. Methods Mol. Biol. 1312, 197–226 (2015).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Therneau, T. M. A package for survival analysis in R. https://CRAN.R-project.org/package=survival (2020).
Kohsaka, S. et al. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci. Transl. Med. 9, eaan6566 (2017).
Neelam, B. et al. Structure–function studies of ligand-induced epidermal growth factor receptor dimerization. Biochemistry 37, 4884–4891 (1998).
Thompson, S. A., Harris, A., Hoang, D., Ferrer, M. & Johnson, G. R. COOH-terminal extended recombinant amphiregulin with bioactivity comparable with naturally derived growth factor. J. Biol. Chem. 271, 17927–17931 (1996).
Adam, R. et al. Modulation of the receptor binding affinity of amphiregulin by modification of its carboxyl terminal tail. Biochim. Biophys. Acta 1266, 83–90 (1995).
Bessman, N. J., Bagchi, A., Ferguson, K. M. & Lemmon, M. A. Complex relationship between ligand binding and dimerization in the epidermal growth factor receptor. Cell Rep. 9, 1306–1317 (2014).
Yu, S. et al. The non-small cell lung cancer EGFR extracellular domain mutation, M277E, is oncogenic and drug-sensitive. Onco Targets Ther. 10, 4507–4515 (2017).
Liu, P. et al. A single ligand is sufficient to activate EGFR dimers. Proc. Natl Acad. Sci. USA 109, 10861–10866 (2012).
Bessman, N. J., Freed, D. M. & Lemmon, M. A. Putting together structures of epidermal growth factor receptors. Curr. Opin. Struct. Biol. 29, 95–101 (2014).
Singh, B., Carpenter, G. & Coffey, R. J. EGF receptor ligands: recent advances. F1000Res. 5, 2270 (2016).
Macdonald, J. L. & Pike, L. J. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl Acad. Sci. USA 105, 112–117 (2008).
Ferguson, K. M., Hu, C. & Lemmon, M. A. Insulin and epidermal growth factor receptor family members share parallel activation mechanisms. Protein Sci. 29, 1331–1344 (2020).
Ferguson, K. M. et al. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 (2003).
Ramamurthy, V. et al. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 20, 259–269 (2012).
Matsuda, T. et al. Cell-free synthesis of functional antibody fragments to provide a structural basis for antibody-antigen interaction. PLoS ONE 13, e0193158 (2018).
Li, S. et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7, 301–311 (2005).
Lee, J. J. et al. Enzymatic prenylation and oxime ligation for the synthesis of stable and homogeneous protein–drug conjugates for targeted therapy. Angew. Chem. Int. Ed. Engl. 54, 12020–12024 (2015).
Alvarado, D., Klein, D. E. & Lemmon, M. A. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 461, 287–291 (2009).
Acknowledgements
We thank members of the Lemmon and Ferguson laboratories for discussions and comments on the manuscript. This work was supported by NCI grant R01-CA198164 (to M.A.L. and K.M.F.). Crystallographic data were collected at GM/CA@APS, funded by NCI (ACB-12002) and NIGMS (AGM-12006). The Eiger 16M detector at GM/CA-XSD was funded by NIH grant S10 OD012289. This research also used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The survival analysis of patients with GBM used data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Author information
Authors and Affiliations
Contributions
C.H., A.K., K.M.F. and M.A.L. conceived the project. C.H. performed all protein production, purification, crosslinking, analytical ultracentrifugation and binding studies, with assistance from C.A.L. C.H. and S.E.S. performed all SAXS studies. C.H., K.M.F., C.A.L. and S.E.S. performed and/or interpreted all crystallographic analysis. A.K. performed cell signalling studies. Z.Y. and C.H. analysed TCGA data. M.A.L. and K.M.F. supervised the project. C.H. and M.A.L. drafted the manuscript, and all authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Michael Eck, Steven Hubbard and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Ligand-dependence of GBM-mutated EGFR.
a, Full-length human EGFR – WT or harbouring an R84K, T239P or A265V mutation – was stably expressed in the engineered haploid eHAP cell line43, which has negligible endogenous EGFR (undetectable by western blotting). Stably transfected cells were serum-starved overnight and either left unstimulated or stimulated with EGF (100 ng ml-1) for the indicated times. Levels of phosphorylated EGFR (pY845: CST #2231, used at 1:1000) and ERK1/2 (pT202/pY204: CST #9106, used at 1:1000), were then detected by immunoblotting of whole cell lysates, also blotting for total EGFR and ERK (see Methods) and Grb2 as loading control5,45. EGFR phosphorylation and ERK phosphorylation are both ligand-dependent in all cases, with no evidence for constitutive activation of the mutated receptors. Representative blots are shown for three biologically independent experiments. See Supplementary Fig. 1 for gel source data. b, IL-3-dependent Ba/F3 cells were stably transfected with WT EGFR or with variants harbouring an R84K or A265V mutation. Cells were either left untreated or were treated with IL-3 (2 ng ml-1) for 72 h, after which a CyQuant Direct proliferation assay was used to detect the number of viable cells in each condition. Resulting fluorescence signals were normalized to that seen with untreated WT and shown as mean ± s.d. (n = 3 experiments for ‘test’ samples). IL-3 treatment promotes robust proliferation in all cases (as positive control: n = 3 for WT, n = 2 for R84K and A265V). As previously reported12,13,48, the mutated EGFRs promoted statistically significant increases in viable cell numbers compared with WT EGFR in the absence of ligand (or IL-3), but effects were very small at these expression levels – with the number of viable cells increased (compared with WT) by just 1.31-fold (P = 0.0002) for R84K and 1.35-fold for A265V (P = 0.0003). P values are for unpaired two-tailed Student’s t-tests. c, Chemiluminescence signals for phosphorylated EGFR in ligand dose-response experiments (see Fig. 1b, c) were quantitated and normalized for Grb2 signals either on different gels as sample processing controls (for gel source data, see Supplementary Fig. 1) or for Grb2 loaded on the same gel using the Multistrip western blotting approach45, with both giving identical results. The resulting pEGFR/Grb2 ratios were plotted (mean ± s.d., n = 3 biologically independent experiments) here and in Fig. 1d as dose-response curves. Responses were normalized to 100% (maximum value) and 0% (without ligand) and the resulting curves were individually fit to a simple model: pEGFR = (100 x [ligand])/(EC50 + [ligand]) for each experiment. Mean EC50 values (± s.d.) are listed in Fig. 1d. d, Ba/F3 cells stably transfected with WT EGFR or variants harbouring R84K or A265V mutations were sorted by flow cytometry (see Methods) to yield cell populations with similar levels of cell surface EGFR. Cells were either left untreated or were treated with noted doses of EGF or EREG for 72 h. A CyQuant Direct proliferation assay was used to measure the number of viable cells in each condition. Results were divided by the signal from untreated cells to give ‘fold increase’ in cell number, and are plotted across all experiments (mean ± s.d., n = 3 biologically independent experiments). EC50 values for each ligand were calculated individually for each experiment using the equation: cell# = (max x [ligand])/(EC50 + [ligand]), and mean values (n = 3) are listed (± s.d.) on the graphs. EC50 values for EGF and EREG are not significantly different for R84K (P = 0.2072) or A265V (P = 0.7915), reflecting loss of ligand discrimination. P values are for unpaired two-tailed Student’s t-tests.
Extended Data Fig. 2 Cross-linking studies of ligand-induced sEGFR dimerization.
a, Quantitation and summary of SAXS I(0)/c measurements reported in Fig. 2 across multiple repeats. For sEGFRWT, only EGF (black) doubles the I(0) value, representing selective EGF-induced dimerization. By contrast, both EREG (magenta) and EGF (black) induce dimerization of sEGFR harbouring L38R (red), R84K (green), A265V (blue) or A265T (gold) mutations – with EREG-induced dimerization of A265 variants appearing slightly less robust. Data represent mean I(0)/c ± s.d. for 10 repeats (WT + EGF), 6 repeats (WT + EREG), 4 repeats (A265V + EGF and A265V + EREG), 3 repeats (L38R + EREG, R84K + EGF, R84K + EREG, A265T + EGF, A265T + EREG), and 2 repeats (L38R + EGF) – where a repeat corresponds to a biologically independent sample. An additional single experiment was undertaken for T239P + EREG, which showed an elevation of I(0)/c by 1.44 fold. The degree of sEGFR dimerization for EGF and EREG is significantly different only for WT (P < 0.0001) and A265T (P < 0.0001). P values are from unpaired two-tailed Student’s t-tests. We estimate based on the SAXS data in Fig. 2a–e that the GBM mutations studied here strengthen dimerization of EREG-bound sEGFR by several hundred fold, based on the following considerations. Since sEGFRWT at 70 μM shows no dimerization when saturated with EREG, the dissociation constant (Kd) for dimers of the EREG:sEGFR complex must be >450 µM (assuming that we could detect a minimum of 10% dimer by SAXS). By contrast, the complete dimerization seen for the EREG:sEGFR complex with mutated variants (when corrected for differences in ligand-binding affinities) places a lower limit of ~0.7 μM on Kd for these dimers. Thus, GBM mutations must enhance dimerization of the EREG:sEGFR complex by at least ~650-fold. b, Representative crosslinking analysis of sEGFR dimerization (n = 3 biologically independent samples for each mutated variant). Different sEGFR variants at 5 μM were incubated alone or with the noted ligand (EGF or EREG) at 6 μM, and subjected to 100 μM DSS for 30 min (see Methods). Samples were then subjected to SDS-PAGE and stained with Coomassie blue. Dimer and monomer bands are marked (note the shift in monomer band position following ligand cross-linking). EGF promotes dimerization of all variants. EREG fails to increase sEGFRWT dimerization above that seen without ligand, but detectably enhances dimerization of all variants with GBM mutations, consistent with the SAXS data shown in Fig. 2. See Supplementary Fig. 1 for gel source data, and (c) for quantitation and reproducibility information. c, Quantitation of data in (b), including additional repeats for each variant. For sEGFRWT, EGF induces substantially more dimerization than EREG (P < 0.0001), whereas the difference between EGF and EREG is not significant for L38R (P = 0.0522) or A265T (P = 0.0577), and only just reaches statistical significance for R84K (P = 0.0410) and A265V (P = 0.0377). P values are for unpaired two-tailed Student’s t-tests. Data in the graph represent mean ± s.d. for 6 repeats (WT + EGF and WT + EREG), or 3 repeats (A265V + EGF, A265V + EREG, R84K + EGF, R84K + EREG, A265T + EGF, A265T + EREG, L38R + EGF, and L38R + EREG), where repeats refer to biologically independent samples. d, Top panel: sEGFRWT and sEGFRR84K (5 μM) were crosslinked alone or with the noted ligands (EGF, AREG, EREG, or EPGN) at 6 μM. Middle panel: as in Top panel, but with 60 μM ligand. Bottom panel: Crosslinking studies were performed with 60 μM ligand added to sEGFRWT and 6 μM added to sEGFRR84K, to account for affinity differences. Data are representative of three biologically independent samples. See Supplementary Fig. 1 for gel source data. e, As for (d), but using sEGFRA265V with 6 μM or 60 μM ligand as marked, for 5 biologically independent samples of sEGFRA265V with EGF and EREG, but n = 2 for AREG and EPGN. See Supplementary Fig. 1 for gel source data.
Extended Data Fig. 3 Kinetics of EGFR and ERK activation by AREG (98aa) and EGF in MCF7 cells.
a, b, Representative time courses (of 3 biologically independent repeats with similar results in each case) of EGFR phosphorylation at Y1173 and Y1068, as well as ERK phosphorylation, in parental MCF-7 cells induced by saturating levels of (a) AREG (1 μM) or (b) EGF (16 nM), exactly as described5. Cells were starved overnight, and were then left unstimulated or stimulated with ligand for the noted times at 37 °C, using growth factor ligands added to starvation medium. Analysis by immunoblotting was then performed exactly as described5,44. Note that the commercially available 98aa form of AREG used here appears to elicit sustained signalling and weak dimerization of sEGFR in these studies – consistent with other work6. We previously reported that a shorter in-house-produced 90aa form of AREG induces more transient (EGF-like) signalling and stronger sEGFR dimerization5. Different signalling properties of AREG forms with different carboxy termini have been reported by others49,50,51, and will be the subject of separate detailed studies. See Supplementary Fig. 1 for gel source data.
Extended Data Fig. 4 SE-AUC studies of TGFα-induced sEGFR dimerization.
a, The noted sEGFR variants were subjected to sedimentation equilibrium analytical ultracentrifugation (SE-AUC) as described in Methods, with or without adding a 1.2-fold excess of TGFα. The low UV absorbance of TGFα allows precise quantitation of TGFα-induced sEGFR dimerization using SE-AUC as described41. Representative data are shown (n = 3 biologically independent samples) for 10 μM sEGFR at 6,000 r.p.m., with the natural logarithm of the absorbance, ln(A280), at radial distance r plotted against (r2 − r02)/2. This transformation of the data gives a straight line for a single species, with slope proportional to molecular weight. Expected data for pure monomer and pure sEGFR–TGFα dimer are shown as dotted and solid grey lines (marked). Data points are colour coded for the different variants as described in the key, with filled circles representing data with added ligand and unfilled circles without. No dimerization was seen in the absence of ligand for any variant under these conditions, consistent with the SAXS studies shown in Fig. 2 and our previous work52. Kd values for each sEGFR–TGFα complex are listed below the graph, determined by global fit of SE-AUC data as described previously41 and in Methods. Mean values ± s.d. from 3 biologically independent samples are reported for all cases except L38R (where n = 1). Whereas sEGFRR84K dimerizes with essentially the same Kd as sEGFRWT following TGFα binding, sEGFRA265V and sEGFRA265T dimerize slightly more weakly (P = 0.006 and 0.005 respectively, for unpaired two-tailed Student’s t-tests). b, Effects on ligand-dependent dimerization also appear specific for GBM extracellular mutations. A rare extracellular EGFR lung cancer mutation (M253E)53, not seen in GBM, instead enhances both ligand-independent (confirmed by crosslinking) and ligand-induced sEGFR dimerization. SE-AUC analysis of sEGFR harbouring the M253E mutation is shown. Unlike GBM variants, M253E-mutated sEGFR dimerizes constitutively, being substantially dimeric in the absence of ligand. TGFα-bound M253E sEGFR also appears to form species larger than dimers, with an estimated Kd in the range of 0.33 μM. M253E-mutated sEGFR was used at 10 μM, and the sample was spun at 6,000 r.p.m. This experiment was only done with one biologically independent sample of sEGFRM253E (in triplicate with- and without TGFα), as the protein is difficult to produce in sufficient quantities.
Extended Data Fig. 5 Symmetry of the EREG-induced sEGFRR84K dimer and implications for negative cooperativity in EGFR.
a, Overlay of the EREG-induced sEGFRR84K dimer (dark green ribbons) with the symmetric dimers of sEGFRWT induced by TGFα (1MOX15, gold ribbons) or EGF (3NJP14,17, grey ribbons). EREG, TGFα and EGF are coloured magenta, orange, and black respectively. b, Schematic of half-of-the-sites negative cooperativity in ligand binding to WT EGFR20,54,55 for any ligand (blue). As we previously described in detail for the Drosophila EGFR20, and as also seems to apply to human EGFR54, binding of a single ligand can promote formation of asymmetric sEGFR dimers (left-hand side of cartoon) with autoinhibitory domain I–domain II interactions broken (unfilled red star) only in one protomer. This asymmetric dimerization is driven by contacts involving N-terminal regions of domain II as well as altered dimer arm docking5,20 – together restraining domain II in the unliganded protomer. When a second ligand binds to this dimer, it must ‘wedge’ apart the two ligand-binding domains (I and III) in the right-hand protomer to drive formation of the symmetric dimer (top right in cartoon). This requires disruption of autoinhibitory domain I–domain II interactions in both molecules (both red stars are unfilled). It also requires disruption of domain II dimer interface contacts – with a resulting bend in domain II (Extended Data Fig. 6d) – giving rise to the symmetric 2:2 dimer. This is readily achieved by high-affinity ligands such as EGF and TGFα, but low-affinity EGFR ligands like EREG56 cannot disrupt the autoinhibitory domain I–domain II interactions or bend the restrained domain II to optimize dimer arm contacts. As a consequence, low-affinity ligands fail to wedge apart domains I and III in the right-hand protomer – instead binding to an unaltered asymmetric dimer (lower right in cartoon) through a compromised set of ligand–receptor interactions (i.e. a remodeled binding site5: see Extended Data Fig. 8d). The R84K mutation lowers this barrier to dimer ‘symmetrization’ by weakening autoinhibitory domain I–domain II interactions so that the second ligand-binding event more readily bends domain II and symmetrizes dimers. This appears to be the origin of the R84K mutation’s ability to selectively stabilize dimers induced by low-affinity EGFR ligands. Weakening of autoinhibitory domain I–domain II interactions may also explain the enhanced ligand-binding affinity seen for R84K EGFR (Extended Data Fig. 7b). The ability of the R84K mutation to equalize the two EREG-binding sites in a dimer, and to increase EREG affinity also argues that this mutation removes a barrier to ligand binding, and may diminish the half-of-the-sites negative cooperativity seen in wild-type EGFR20,54,57,58.
Extended Data Fig. 6 Autoinhibitory domain I–domain II interactions in different sEGFR structures.
a, As expected for autoinhibitory interactions, R84–A265 and L38–F263 interactions are broken in ‘active’ symmetric dimers of sEGFR induced upon activation with TGFα (1MOX15) or EGF (3NJP14,17). This configuration is represented by unfilled red stars, as in Fig. 3. b, Disposition of key GBM-mutated residues (L38, R84 and A265, coloured red) for comparison in ‘inactive’ configurations of the EGFR extracellular region. The positions of these side-chains are shown in monomeric tethered forms of sEGFR59,60,61,62,63. In each case, the R84 side-chain directly contacts that of A265, and the L38 side-chain is in van der Waal’s contact with that of F263 (grey spheres: not mutated in GBM). These represent autoinhibitory interactions between domains I and II as described in the text and pointed out in our previous studies64. This configuration is represented as a filled red star. c, Importantly, the autoinhibitory R84–A265 and L38–F263 interactions are also retained in the ligand-bound monomer observed when EPGN binds to sEGFRWT (5WB85). Moreover, as shown in the lower two panels, these autoinhibitory interactions are retained in the right-hand molecule of the asymmetric EREG-induced dimer of sEGFRWT (5WB7: Fig. 3a) – see filled red star – but are lost in the left-hand molecule (unfilled red star). d, Comparison of the ‘bend’ in domain II in inactive monomeric forms of sEGFR (light green) and active dimeric forms (grey) – colours corresponding to those used for sEGFR chains in Fig. 3a. The structures of unliganded monomeric sEGFR (PDBID: 1NQL59) and an EGF-induced WT sEGFR dimer (PDBID: 3NJP14) were used. Only residues 187–310 of domain II are shown. In the left-hand panel, the two structures are overlaid using residues 187–238 as reference. In the right-hand panel, residues 238–310 are used as reference. This analysis reveals that the two structures differ by a bend at residue D238 (marked as ‘Curvature defining point’). The approximate direction of curvature is shown by green and grey brush strokes on each structure. The dimer arm is labelled, as are disulphide-bonded modules 2–8 of domain II59. This figure is based on one by Ferguson16.
Extended Data Fig. 7 Similarity of the two ligand-binding sites in EREG-induced sEGFRR84K dimers.
a, Comparison of the two EREG-binding sites in the symmetric dimer of R84K-mutated sEGFR, overlaid by superposition of the ligand chains. Chain A of sEGFRR84K (left in Fig. 3b) is shown in grey ribbons, and chain B (right in Fig. 3b) is shown in dark green ribbons – with the respective bound ligands coloured pink and magenta. Side-chains involved in direct EREG–sEGFR contacts are shown and labelled. Those in the ligands superimpose very well (see Y13, H16, M25, Y29, for example), with a few exceptions (e.g. R31 and F45). Similarly, sEGFR side-chains in the binding sites overlay well, including D22, R29, Y45, E90 and S99 in domain I and D355, L348, F357, and H409 in domain III. Accordingly, the root-mean-square deviation for all atoms in the 56 residues involved in ligand–receptor contacts (35 from sEGFR, 21 from EREG) is 1.4 Å. b, Comparison of EREG binding to different sEGFR variants as assessed using surface plasmon resonance (see Methods). Representative binding curves for one of the biological replicates are plotted on the left (n = 3 for WT, L38R, R84K, and n = 2 for A265V and A265T). The WT sEGFR construct used in these studies bound to immobilized EREG in surface plasmon resonance studies with a Kd value of 6.6 ± 0.6 μM (n = 3 biologically independent samples; mean ± s.d.). GBM mutations in domain I increased ligand-binding affinity by ~6 fold for L38R (P < 0.0001: n = 3) and almost 10-fold for R84K (P < 0.0001: n = 3), with Kd values respectively of 1.1 ± 0.4 μM and 0.7 ± 0.2 μM. Domain II GBM mutations increased ligand-binding affinity by 4.2-fold for A265V (n = 2) and just ~2-fold for A265T (n = 2), with Kd values respectively of 1.6 and 3.1 μM (no s.d. is quoted for n = 2). These smaller differences are consistent with the asymmetry retained in the A265V ligand binding sites. P values where quoted are for unpaired two-tailed Student’s t-tests.
Extended Data Fig. 8 Structural features of the asymmetric dimer of sEGFRA265V induced by EREG.
a, The asymmetric A265V-mutated (left) and WT (right) sEGFR dimers induced by EREG are compared, with disposition of the autoinhibitory domain I–domain II residues shown in the lower panels. Colours parallel those used in Figs. 3 and 4, with the right-hand molecule coloured slate blue for A265V and light green for WT sEGFR. Side-chain contacts between residues at positions 38 and 263 and between residues 84 and 265 are retained in the right-hand molecule (filled red star) but not the left-hand (unfilled red star) in each case. V265, corresponding to the GBM substitution (A265V), is coloured red. b, Local structural consequences of the A265V mutation in the domain I–domain II interface region of EREG-induced sEGFR dimers. The right-hand side of the EREG-bound WT and A265V-mutated sEGFR structures shown in (a) are superimposed using domain I as reference, with WT sEGFR shown in light green and A265V sEGFR in slate blue. Replacing A265 with a valine displaces the Cα position for residue 265 by ~1.2 Å (red arrow), and this is propagated to a shift in position of F263 by ~2.5 Å (red arrow). As a result of consequent small displacements in domain II constituents beyond this position, the locations of Y275 and R285 – which provide the docking side for the dimer arm Y251 residue in Fig. 4b – are altered, allowing remodeling of this binding site to enhance dimerization strength as described in the text. c, d, Comparison of the binding sites on the two sides of the EREG-induced dimer for A265V-mutated sEGFR (c) and WT sEGFR (d), illustrating that the differences seen between the two sites in the asymmetric WT dimer5 are retained in the A265V variant despite stronger dimerization and slightly stronger ligand binding. The regions corresponding to the two ligand-binding sites are superimposed with EREG as the reference. The left-hand molecule is coloured grey in each case, and the right-hand molecule slate blue in (c) for A265V and light green in (d) for WT. The pink ligand is bound to the left-hand (grey) sEGFR molecule, and the magenta ligand is bound to the right-hand sEGFR molecule. For clarity, ligand side-chains that are not substantially different in orientation are omitted – the exceptions being L15, Y29, and V39, which are consistently reoriented between the two sites. Contact side-chains in the receptor are shown, illustrating their substantial displacement with respect to the ligand in the two sites, with a contact residue root-mean-square deviation between the two sites of 3.0 Å in both A265V and WT (compared with 1.4 Å for the two sites in EREG-bound R84K). Examples include the ~10 Å displacement of D22 and R29, and ~7 Å displacements of L17 and F20 that are marked by red arrows in domain I. These changes are essentially the same in A265V and WT. Shifts in domain III are generally smaller, but are essentially the same in A265V and WT sEGFR. Thus, the compromised binding to the right-hand molecule previously reported5 is fully retained in the A265V variant despite stronger dimerization.
Extended Data Fig. 9 Kaplan-Meier estimates of overall survival in glioblastoma patients with different EGFR status from the TCGA database.
Two-sided log-rank tests were performed to assess the overall survival differences between different patient groups, with no corrections made for multiple pairwise comparisons. Patients with no EGFR alteration (n = 322 patients; red line) had median overall survival times of 14.4 months (95% CI, 12.7–15.8), which was not statistically significantly different from: a, Patients with any EGFR alteration (n = 270 patients; blue dashed line), who had median overall survival times of 14.3 months (95% CI, 13.3–15.6), P = 0.06. b, Patients with an EGFR mutation (n = 22 patients; blue dashed line), who had median overall survival times of 15.9 months (95% CI, 11.3-23.2), P = 0.6. c, Patients with an EGFR mutation and EGFR amplification (n = 58 patients; blue dashed line), who had median overall survival times of 13.9 months (95% CI, 11.2–17.5), P = 0.66. d, Patients with EGFR amplification (n = 85 patients; blue dashed line), who had median overall survival times of 14.3 months (95% CI, 11.8–16.8), P = 0.28.
Supplementary information
Supplementary Figure 1
This file contains the raw (uncropped) gel images for Figs. 1 and 2 and for Extended Data Figs. 1–3.
Rights and permissions
About this article

Cite this article
Hu, C., Leche, C.A., Kiyatkin, A. et al. Glioblastoma mutations alter EGFR dimer structure to prevent ligand bias. Nature 602, 518–522 (2022). https://doi.org/10.1038/s41586-021-04393-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04393-3
This article is cited by
-
Mechanistic insights and the clinical prospects of targeted therapies for glioblastoma: a comprehensive review
Experimental Hematology & Oncology (2024)
-
Tumor suppressor role of the complement inhibitor CSMD1 and its role in TNF-induced neuroinflammation in gliomas
Journal of Experimental & Clinical Cancer Research (2024)
-
Development of Cas13a-based therapy for cancer treatment
Molecular Biology Reports (2024)
-
Extrachromosomal circular DNA: biogenesis, structure, functions and diseases
Signal Transduction and Targeted Therapy (2022)
-
EGFR ligands dictate tumour suppression
Nature Cell Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
