
Abstract
Public databases contain a planetary collection of nucleic acid sequences, but their systematic exploration has been inhibited by a lack of efficient methods for searching this corpus, which (at the time of writing) exceeds 20 petabases and is growing exponentially1. Here we developed a cloud computing infrastructure, Serratus, to enable ultra-high-throughput sequence alignment at the petabase scale. We searched 5.7 million biologically diverse samples (10.2 petabases) for the hallmark gene RNA-dependent RNA polymerase and identified well over 105 novel RNA viruses, thereby expanding the number of known species by roughly an order of magnitude. We characterized novel viruses related to coronaviruses, hepatitis delta virus and huge phages, respectively, and analysed their environmental reservoirs. To catalyse the ongoing revolution of viral discovery, we established a free and comprehensive database of these data and tools. Expanding the known sequence diversity of viruses can reveal the evolutionary origins of emerging pathogens and improve pathogen surveillance for the anticipation and mitigation of future pandemics.
Similar content being viewed by others

Main
Viral zoonotic disease has had a major impact on human health over the past century, with notable examples including the 1918 Spanish influenza, AIDS, SARS, Ebola and COVID-19. There are an estimated 3 × 105 mammalian virus species from which infectious diseases in humans may arise2, of which only a fraction are known at present. Global surveillance of virus diversity is required for improved prediction and prevention of future epidemics, and is the focus of international consortia and hundreds of research laboratories3,4.
Pioneering works expanding the virome of the Earth have each uncovered thousands of novel viruses, with the rate of virus discovery increasing exponentially and driven largely by the increased availability of high-throughput sequencing5,6,7,8,9,10,11. Sequence analysis remains computationally expensive, in particular the assembly of short reads into contigs, which limits the breadth of samples analysed. Here we propose an alternative alignment-based strategy that is considerably cheaper than assembly and enables processing of massive datasets.
Petabases (1 × 1015 bases) of sequencing data are freely available in public databases such as the Sequence Read Archive (SRA)1, in which viral nucleic acids are often captured incidental to the goals of the original studies12. To catalyse global virus discovery, we developed the Serratus cloud computing infrastructure for ultra-high-throughput sequence alignment, screening 5.7 million ecologically diverse sequencing libraries or 10.2 petabases of data.
Identification of Earth’s virome is a fundamental step in preparing for the next pandemic. We lay the foundations for future research by enabling direct access to 883,502 RNA-dependent RNA polymerase (RdRP)-containing sequences, which include the RdRP from 131,957 novel RNA viruses (sequences with greater than 10% divergence from a known RdRP), including 9 novel coronaviruses. Altogether this captures the collective efforts of over a decade of sequencing studies in a free repository, available at https://serratus.io.
Accessing the planetary virome
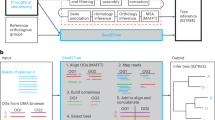
Serratus is a free, open-source cloud-computing infrastructure optimized for petabase-scale sequence alignment against a set of query sequences. Using Serratus, we aligned more than one million short-read sequencing datasets per day for less than 1 US cent per dataset (Extended Data Fig. 1). We used a widely available commercial computing service to deploy up to 22,250 virtual CPUs simultaneously (see Methods), leveraging SRA data mirrored onto cloud platforms as part of the NIH STRIDES initiative13.
Our search space spans data deposited over 13 years from every continent and ocean, and all kingdoms of life (Fig. 1). We applied Serratus in two of many possible configurations. First, to identify libraries that contain known or closely related viruses, we searched 3,837,755 (around May 2020) public RNA sequencing (RNA-seq), meta-genome, meta-transcriptome and meta-virome datasets (termed sequencing runs1) against a nucleotide pangenome of all coronavirus sequences and RefSeq vertebrate viruses. We then aligned 5,686,715 runs (January 2021) against all known viral RdRP amino acid sequences using a specially optimized version of DIAMOND v2 (ref. 14, Methods); this search was completed within 11 days, at a cost of US$23,980 (Fig. 1a, Methods).

a, Total bases searched from the 5,686,715 SRA sequencing runs analysed in the viral RdRP search grouped by sample taxonomy, where available (see Extended Data Figs. 1, 3, Supplementary Table 1). A total of 8,871 out of 15,016 (59%) of known RdRP sOTUs were observed in the SRA, and 131,957 unique and novel RdRP sOTUs were identified (see Extended Data Fig. 2). sOTUs identified in multiple taxonomic groups are counted in each group separately; numbers shown indicate the number of novel sOTUs in each group. WGS, whole-genome sequencing. b, Release dates of the runs included in the analysis reflecting the growth rate of available data. c, Sample locations for 635,656 RdRP-containing contigs (27.8% of samples lacked geographical metadata). The high density of RdRP seen in North America, western Europe and eastern Asia reflects the substantial acquisition bias for samples originating from these regions. Interactive RdRP map is available at https://serratus.io/geo.
Previous approaches for identifying sequences across the entire SRA rely on pre-computed indexes15,16 that require exact substring or hash-based matches, which limits their sensitivity to diverged sequences (Extended Data Fig. 1f). Pre-assembled reads (for example, the NCBI Transcriptome Shotgun Assembly database) enable efficient alignment-based searches5, but are at present available for only a small fraction of the SRA. Serratus aligns a query of up to hundreds of megabytes against unassembled libraries, achieving greater sensitivity to diverged viruses compared to substring (k-mer) indexes while using far fewer computational resources than de novo assembly (Fig. 1g, Methods).
A sketch of RdRP
Viral RdRP is a hallmark gene of RNA viruses that lack a DNA stage of replication17. We identified RdRP by a well-conserved amino acid sub-sequence that we call the ‘palmprint’. Palmprints are delineated by three essential motifs that together form the catalytic core in the RdRP structure18 (Fig. 2). We constructed species-like operational taxonomic units (sOTUs) by clustering palmprints at a threshold of 90% amino acid identity, chosen to approximate taxonomic species18.

a, The RdRP palmprint is the protein sequence spanning three well-conserved sequence motifs (A, B and C), including intervening variable regions, exemplified within the full-length poliovirus RdRP structure with essential aspartic acid residues (asterisks) (Protein Data Bank code: 1RA649). Conservation was calculated from RdRP alignment in a previous study19, trimmed to the poliovirus sequence; motif sequence logos are shown below. aa, amino acids. b, Per-phylum histogram of amino acid identity of novel sOTUs aligned to the NCBI non-redundant protein database. Extended Data Figure 3c shows the per-order distribution. Inset, Preston plot and linear regression of palmprint abundances indicates that singleton palmprints (that is, observed in exactly one run) occur within 95% confidence intervals of the value predicted by extrapolation from high-abundance palmprints (linear regression applied to log-transformed data), and this distribution is consistent through time (Extended Data Fig. 2). NA, not applicable; uncl, unclassified.
A total of 3,376,880 (59.38%) sequencing runs contained one or more reads that mapped to the RdRP query (E-value ≤ 1 × 10−4). We assembled RdRP aligned reads from each library (and their mate-pairs when available), which yielded 4,261,616 ‘microassembly’ contigs. Of these, 881,167 (20.7%) contained a high-confidence palmprint identified by Palmscan (false discovery rate = 0.001)18, representing 260,808 unique palmprints. Applying Palmscan to reference databases1,7,19, we obtained 45,824 unique palmprints, which clustered into 15,016 known sOTUs. If a newly acquired palmprint aligned to a known palmprint at an identityof 90% or greater, it was assigned membership to that reference sOTU; otherwise, it was designated as novel. We clustered novel palmprints at 90% identity and obtained 131,957 novel sOTUs, representing an increase in the number of known RNA viruses by a factor of 9.8. Clustering novel palmprints at genus-like 75% and family-like 40% thresholds yielded 78,485 and 3,599 novel OTUs, which represent increases of 8.0× and 1.9×, respectively (Fig. 2b).
We extracted host, geospatial and temporal metadata for each biological sample when available (Fig. 1c), noting that the majority (88%) of novel RdRP sOTUs were observed from metagenomic or environmental runs in which accurate host inference is challenging. Mapping observations of virus marker genes across time and space suggests ecological niches for these viruses, and improved characterization of sequence diversity can improve PCR primer design for in situ virus identification.
We estimate that around 1% of sOTUs are endogenous virus elements (EVEs); that is, viral RdRPs that have reverse-transcribed into a host germline. We did not attempt to systematically distinguish EVEs from viral RdRPs, noting that EVEs with intact catalytic motifs are likely to be recent insertions that can serve as a representative sequence for related exogenous viruses. Most (60.5%) recovered palmprints were found in exactly one run (singletons), and are observed within the expected frequency range predicted by extrapolating from more abundant sequences (Fig. 2b).
The abundance distribution of distinct palmprints is consistent with log-log-linear for each year from 2015 to 2020 (Extended Data Fig. 2e), and over time, singletons are confirmed by subsequent runs at an approximately constant rate (Extended Data Fig. 2g). The majority of novel viruses will be singletons until the diversity represented by the search query and the fraction of the planetary virome sampled in the SRA both approach saturation. Extrapolating one year forward, by when the SRA is expected to have doubled in size, we predict that 430,000 (95% confidence interval [330,000, 561,000]) additional unique palmprints could be identified by running Serratus with its current query (Fig. 2b).
RNA viruses have highly divergent sequences, even within the conserved RdRP17. Amino acid sequence alignment can recover the majority of RdRP short reads above 60% identity, but sensitivity falls as sequences diverge further (Extended Data Fig. 2f). Subsequent microassembly fragmentation can in part account for the decreased abundance of novel sOTUs below 60% identity (Fig. 2b); thus, the sensitivity to highly diverged (less than 50% identity) RdRP sequences is limited in the present study. Saturation of virus discovery within the SRA is far from complete, even if data-growth rates are ignored. Intensive searches for so-called highly diverged or ‘dark’ viruses20, in combination with iterative reanalysis (conceptually similar to PSI-BLAST21), are likely to yield further expansion of the known virome.
The total number of virus species is estimated to be 108 to 1012 (ref.22), so our data captured at most 0.1% of the global virome. However, if exponential data growth combined with increased search sensitivity continues, we are at the cusp of identifying a notable fraction of Earth’s total genetic diversity with tools such as Serratus.
Expanding known Coronaviridae
The SARS-CoV-2 pandemic has severely affected human society. We further exemplify the potential of Serratus for virus discovery with the Coronaviridae (CoV) family, including a recently proposed subfamily23 that contains a CoV-like virus, Microhyla alphaletovirus 1 (MLeV), in the frog Microhyla fissipes, and Pacific salmon nidovirus (PsNV) described in the endangered Oncorhynchus tshawytscha24.
First, we identified 52,772 runs that contain 10 or more CoV-aligned reads or 2 or more CoV k-mers (32-mer,16). These runs were de-novo-assembled with a new version of synteny-informed SPAdes called coronaSPAdes25. This yielded 11,120 identifiable CoV contigs that we annotated for a comprehensive assemblage of Coronaviridae in the SRA (see Methods for discussion). With these training data we defined a scoring function to predict the subsequent success of assembly (Extended Data Fig. 3b).
CoV and neighbouring palmprints comprise 70 sOTUs, 44 of which are described in public databases. Seventeen CoV sOTUs contained partial RdRP (inclusive of full palmprint) from an amplicon-based virus discovery study for which the data had not been publicly deposited at the time of writing26. The remaining nine sOTUs are novel viruses, with protein domains consistent with a CoV or CoV-like genome organization (Extended Data Fig. 4).
We operationally designate MLeV, PsNV and the nine novel viruses broadly as group E, noting that all were found in samples from non-mammalian aquatic vertebrates (Fig. 3). Notably, Ambystoma mexicanum (axolotl) nidovirus (AmexNV) was assembled in 18 runs, 11 of which yielded common contigs of approximately 19 kb. Easing the criteria of requiring an RdRP match in a contig, 28 out of 44 (63.6%) of the runs from the associated studies were AmexNV-positive27,28. Consistent assembly break points in AmexNV, PsNV and similar viruses suggest that the viral genomes of this clade of CoV-like viruses are organized in at least two segments, one containing ORF1ab with RdRP, and a shorter segment containing a lamin-associated domain protein, spike and N’ accessory genes (Fig. 3). An assembly gap with common break points is present in the published PsNV genome24. Together, these seven monophyletic species possibly represent a distinct clade of segmented CoV-like nidoviruses, although molecular validation of this hypothesis is required.

a, Phylogram for group-E sequences. Six viruses were similar to PsNV in Ambystoma mexicanum (axolotl; AmexNV), Puntigrus tetrazona (tiger barb; PtetNV), Hippocampus kuda (seahorse; HkudNV), Syngnathus typhle (broad-nosed pipefish; StypNV), Takifugu pardalis (fugu fish; TparNV) and the Acanthemblemaria sp. (blenny; AcaNV). More-distant members identified were in Hypomesus transpacificus (the endangered delta smelt; HtraNV), Silurus sp. (catfish; SilNV) and Monopterus albus (asian swamp eel; MalbNV). b, Unrooted phylogram for Coronaviridae annotated with genera (Greek letters) and group-E CoV-like nidoviruses (see also Extended Data Fig. 4). Maximum likelihood tree generated by clustering the RdRP amino acid sequences at 97% identity to show sub-species variability. c, Genome structure of AmexNV and the contigs recovered from group-E CoV-like viruses annotated with HMM matches. AmexNV contigs contain an identical 129-nt trailing sequence (Tr). All the putatively segmented CoV-like are monophyletic with PsNV. A gap in the PsNV reference sequence24 is shown with circles, overlapping the common contig ends seen in these viruses.
While our manuscript was under review, public transcriptome screening by Miller et al.29 identified three group-E CoV sequences that are not included in our sOTU analysis. One CoV+ library had failed at the alignment step, and microassembly from two others yielded incomplete palmprint sub-sequences and therefore lacked the required specificity for the systematic palmprint classification. A high-sensitivity reanalysis of microassemblies for any group-E RdRP sequence fragment captured the two CoV sequences that we missed from the Miller et al. study29, and found another approximately 25 putative-novel CoV species from 53 fragmented contigs (Supplementary Table 1e).
In addition to identifying genetic diversity within CoV, we cross-referenced CoV+ library metadata to identify possible zoonoses and vectors of transmission. Discordant libraries—ones in which a CoV is identified and the viral expected host30 does not match the sequencing library source taxa—were rare, accounting for only 0.92% of cases (Supplementary Table 1f).
An important limitation for these analyses is that the nucleic acid reads do not prove that viral infection has occurred in the nominal host species. For example, we identified five libraries in which a porcine, avian, or bat coronavirus was found in plant samples. The parsimonious explanation is that CoV was present in faeces or fertilizer originating from a mammalian or avian host applied to these plants. However, this exemplifies a merit of exhaustive search in identifying transmission vectors and for monitoring the geotemporal distribution of viruses.
Rapid expansion into the viral unknowns
The global mortality from viral hepatitis exceeds that of HIV/AIDS, tuberculosis or malaria31. Hepatitis delta virus (HDV) has a small circular RNA genome (around 1,700 nucleotides (nt)) that folds into a rod-like shape and encodes three genes: a delta antigen protein, and two self-cleaving delta ribozymes (drbz)32.
Before 2018, HDV was the sole known member of its genus; 13 drbz-containing members have since been characterized33,34,35,36,37,38, and recently a second class of ribozyme (known as hammerhead or hhrbz) characteristic of plant viroids was identified in delta-like viruses that we refer to as epsilon viruses39. By sequence search for the delta antigen protein and ribozymes, we identified 14 delta viruses, 39 epsilon viruses and 311 enigmatic sequences with delta-virus-like synteny that we term zeta viruses (Fig. 4, Extended Data Fig. 5). The evolutionary histories of these mammalian delta viruses are explored further elsewhere37.

a, Genome structure for the Marmota monax delta virus (MmonDV) and a delta-virus-like genome detected in an environmental dataset, each containing a negative-sense delta-antigen (δAg) ORF; two delta ribozymes (drbz); and characteristic rod-like folding, where each line shows the predicted base-pairing within the RNA genome, coloured by base-pairing confidence score (p-num)50. b, Similar genome structure for the Sulabanus spp. epsilon virus-like (SulaEV) and an epsilon-virus-like genome from an environmental dataset, each containing a negative-sense epsilon-antigen (εAg) ORF; two hammerhead ribozymes (hhrbz); and rod-like folding. c, Example of the compact genome structure of a Zeta virus-like from an environmental dataset containing two predicted zeta-antigen (ζAg+/−; protein alignment is shown in the outer circles) ORFs without stop codons; two hhrbzs overlapping with the ORFs; and rod-like folding. Further novel genomes are shown in Extended Data Figs. 5, 6. d, Maximum-likelihood phylogenetic tree of delta viruses derived from a delta-antigen protein alignment with bootstrap values. Two divergent environmental delta viruses could not yet be placed. e, Tree showing huge phage clade expansion. Black dots indicate branches with bootstrap values greater than 90. Outer ring indicates genome or genome fragment length: grey are sequences from Al-Shayeb et al.42 and reference sequences, shadings indicate previously defined clades of phages with very large genomes (200–735 kb). The Kabirphages (light purple) are shown in expanded view in Extended Data Fig. 7.
The zeta virus circular genomes are highly compressed, ranging from 324 to 789 nt and predicted to fold into rod-like structures. They contain a hhrbz in each orientation and encode two open reading frames (ORFs), one sense and one anti-sense. Both ORFs generally lack stop codons and encompass the entire genome, potentially producing an endless tandem repeat of antigen. The atypical coiled-coil domain of the HDV antigen40 is conserved in the antigens of new delta and epsilon viruses, whereas epsilon and zeta genomes show analogous hhrbzs (Extended Data Fig. 6), suggesting that these sequences share common ancestry. These abundant elements may help to solve a long-standing question about the origins of circular RNA subviral agents in higher eukaryotes (Extended Data Fig. 6), historically regarded as molecular fossils of a prebiotic RNA world41.
To evaluate the feasibility of applying Serratus in the context of microbiome research, we sought to locate bacteriophages that are related to recently reported huge phages42, searching for terminase amino acid sequences. Targeted assembly of 287 high-scoring runs returned 252 terminase-containing contigs of greater than 140 kb. Phylogenetics of these sequences resolved new groups of phages with large genomes (Fig. 4e). Although most phages were from a single animal genus, we identified closely related phages that crossed animal orders, including related phages in a human from Bangladesh (ERR866585) and in groups of cats (PRJEB9357) and dogs (PRJEB34360) from England, sampled five years apart. Similarly, we recovered two approximately 554-kb Lak megaphage genomes (among the largest animal microbiome phages reported so far) that are extremely closely related to sequences previously reported from pigs, baboons and humans43 (Extended Data Fig. 7). These two genomes were circularized and manually curated to completion. The large carrying capacity of such phages and broad distribution underlines their potential for extensive lateral gene transfer amongst animal microbiomes and modification of host bacterial function. These sequences substantially expand the inventory of phages with genomes whose length range overlaps with those of bacteria.
Discussion
Since the completion of the human genome, the growth of DNA sequencing databases has outpaced Moore’s Law. Serratus provides rapid and focused access to genomic sequences captured over more than a decade by the global research community, which would otherwise be inaccessible in practice. This work and further extensions of petabase-scale genomics15,16,44 are shaping a new era in computational biology, enabling expansive gene discovery, pathogen surveillance and pangenomic evolutionary analyses.
Optimal translation of such massive datasets into meaningful biomedical advances requires free and open collaboration among scientists45. The current pandemic underscores the need for prompt, unrestricted and transparent data sharing. With these goals in mind, we deposited 7.3 terabytes of virus alignments and assemblies into an open-access database that can be explored via a graphical web interface at https://serratus.io or programmatically through the Tantalus R package and its PostgreSQL interface.
The ‘metagenomics revolution’ of virus discovery is accelerating7,11. Innovative fields such as high-throughput viromics46 can leverage vast collections of virus sequences to inform policies that predict and mitigate emerging pandemics47. Combining ecoinformatics with virus, host and geotemporal metadata offers a proof-of-concept for a global pathogen surveillance network, arising as a by-product of centralized and open data sharing.
Human population growth and encroachment on animal habitats is bringing more species into proximity, leading to an increased rate of zoonosis2 and accelerating the Anthropocene mass extinction48. While Serratus enhances our capability to chronicle the full genetic diversity of our planet, the genetic diversity of the biosphere is diminishing. Thus, investment in the collection and curation of biologically diverse samples, with an emphasis on geographically underrepresented regions, has never been more pressing—if not for the conservation of endangered species, then to better conserve our own.
Methods
Serratus alignment architecture
Serratus (v0.3.0) (https://github.com/ababaian/serratus) is an open-source cloud-infrastructure designed for ultra-high-throughput sequence alignment against a query sequence or pangenome (Extended Data Fig. 1). Serratus compute costs are dependent on search parameters (expanded discussion available: https://github.com/ababaian/serratus/wiki/pangenome_design). The nucleotide vertebrate viral pangenome search (bowtie2, database size: 79.8 MB) reached processing rates of 1.29 million SRA runs in 24 h at a cost of US$0.0062 per dataset (Extended Data Fig. 1). The translated-nucleotide RdRP search (DIAMOND14; database size: 7.1 MB) reached processing rates exceeding 0.5 million SRA runs in 12 h at a cost of US$0.0042 per dataset. All 5,686,715 runs analysed in the RdRP search were completed within 11 days for a total cost of US$23,980 or around US$2,350 per petabase. For a detailed breakdown of Serratus project costs and recommendations for managing cloud-computing costs, see Serratus wiki: https://github.com/ababaian/serratus/wiki/budget. Tutorials on how to find particular novel viruses using Serratus data are available at https://github.com/ababaian/serratus /wiki/Find_novel_viruses.
Computing cluster architecture
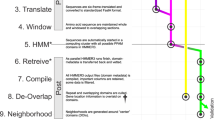
The processing of each sequencing library is split into three modules: ‘dl’ (download), ‘align’ and ‘merge’. The dl module acquires compressed data (.sra format) via prefetch (v2.10.4), from the Amazon Web Services (AWS) Simple Storage Service (S3) mirror of the SRA, decompresses to FASTQ with fastq-dump (v2.10.4) and splits the data into chunks of 1 million reads or read-pairs (‘fq-blocks’) into a temporary S3 cache bucket. To mitigate excessive disk usage caused by a few large datasets, a total limit of 100 million reads per dataset was imposed. The align module reads individual fq-blocks and aligns to an indexed database of user-provided query sequences using either bowtie2 (v2.4.1, --very-sensitive-local)51 for nucleotide search, or DIAMOND (v2.0.6 development version, --mmap-target-index --target-indexed --masking 0 --mid-sensitive -s 1 -c1 -p1 -k1 -b 0.75)14 for translated-protein search. Finally, the merge module concatenates the aligned blocks into a single output file (.bam for nucleotide, or .pro for protein) and generates alignment statistics with a Python script (see details about Summarizer in ‘Generating viral summary reports’ below).
Computing resource allocation
Each component is launched from a separate AWS autoscaling group with its own launch template, allowing the user to tailor instance requirements per task. This enabled us to minimize the use of costly block storage during compute-bound tasks such as alignment. We used the following Spot instance types; dl: 250 GB SSD block storage, 8 virtual CPUs (vCPUs), 32 GB RAM (r5.xlarge) around 1,300 instances; align: 10 GB SSD block storage, 8 vCPUs, 8 GB RAM (c5.xlarge) around 4,300 instances; merge: 150 GB SSD block storage, 4 vCPUs, 4 GB RAM (c5.large) around 60 instances. Users should note that it may be necessary to submit a service ticket to access more than the default EC2 instance limit.
AWS Elastic Compute Cloud (EC2) instances have higher network bandwidth (up to 1.25 GB s−1) than block storage bandwidth (250 MB s−1). To exploit this, we used S3 buckets as a data buffering and streaming system to transfer data between instances following methods developed in a previous cloud architecture (https://github.com/FredHutch/sra-pipeline). This, combined with splitting of FASTQ files into individual blocks, effectively eliminated file input/output (i/o) as a bottleneck, as the available i/o is multiplied per running instance (conceptually analogous to a RAID0 configuration or a Hadoop distributed file system52).
Using S3 as a buffer also allowed us to decouple the input and output of each module. S3 storage is cheap enough that in the event of unexpected issues (for example, exceeding EC2 quotas) we could resolve system problems in real time and resume data processing. For example, shutting down the align modules to hotfix a genome indexing problem without having to re-run the dl modules, or if an alignment instance is killed by a Spot termination, only that block needs to be reprocessed instead of the entire sequencing run.
Work queue and scheduling
The Serratus scheduler node controls the number of desired instances to be created for each component of the workflow, based on the available work queue. We implemented a pull-based work queue. After boot-up, each instance launches a number of ‘worker’ threads equal to the number of CPUs available. Each worker independently manages itself via a boot script, and queries the ‘scheduler’ for available tasks. Upon completion of the task, the worker updates the scheduler of the result: success, or fail, and queries for a new task. Under ideal conditions, this allows for a worst-case response rate in the hundreds of milliseconds, keeping cluster throughput high. Each task typically lasts several minutes depending on the pangenome.
The scheduler itself was implemented using Postgres (for persistence and concurrency) and Flask (to pool connections and translate REST queries into SQL). The Flask layer allowed us to scale the cluster past the number of simultaneous sessions manageable by a single Postgres instance. The work queue can also be managed manually by the user, to perform operations such as re-attempting the downloading of an SRA accession after a failure or to pause an operation while debugging. Up to 300,000 SRA jobs can be processed in the work queue per batch process.
The system is designed to be fully self-scaling. An ‘autoscaling controller’ was implemented, which scales-in or scales-out the desired number of instances per task every five minutes on the basis of the work queue. As a backstop, when all workers on an instance fail to receive work instructions from the scheduler, the instance self shuts-down. Finally a ‘job cleaner’ component checks the active jobs against currently running instances. If an instance has disappeared owing to SPOT termination or manual shutdown, it resets the job allowing it to be processed up by the next available instance.
To monitor cluster performance in real-time, we used Prometheus (v2.5.0) and node exporter to retrieve CPU, disk, memory and networking statistics from each instance, to expose performance information about the work queue, and Python exporter to export information from the Flask server. This allowed us to identify and diagnose performance problems within minutes to avoid costly overruns.
Generating viral summary reports
We define a viral pangenome as the entire collection of reference sequences belonging to a taxonomic viral family, which may contain both full-length genomes and sequence fragments such as those obtained by RdRP amplicon sequencing.
We developed a Summarizer module written in Python to provide a compact, human- and machine-readable synopsis of the alignments generated for each SRA dataset. The method was implemented in Serratus_summarizer.py for nucleotide alignment and Serratus_psummarizer.py for amino acid alignments. Reports generated by the Summarizer are text files with three sections described in detail online (https://github.com/ababaian/serratus/wiki/.summary-Reports). In brief, each contains a header section with alignment metadata and one-line summaries for each virus family pangenome, reference sequence and gene, respectively, with gene summaries provided for protein alignments only.
For each summary line we include descriptive statistics gathered from the alignment data such as the number of aligned reads, estimated read depth, mean alignment identity and coverage; that is, the distribution of reads across each reference sequence or pangenome. Coverage is measured by dividing a reference sequence into 25 equal bins and depicted as an ASCII text string of 25 symbols, one per bin; for example oaooomoUU:oWWUUWOWamWAAUW. Each symbol represents log2(n + 1), where n is the number of reads aligned to a bin in this order _.:uwaomUWAOM^. Thus, ‘_’ indicates no reads, ‘.’ exactly one read, ‘:’ two reads, ‘u’ 3–4 reads, ‘w’ 5–7 reads and so on; ‘^’ represents >213 = 8,192 reads in the bin. For a pangenome, alignments to its reference sequences are projected onto a corresponding set of 25 bins. For a complete genome, the projected pangenome bin number 1, 2, …, 25 is the same as the reference sequence bin number. For a fragment, a bin is projected onto the pangenome bin implied by the alignment of the fragment to a complete genome. For example, if the start of a fragment aligns halfway into a complete genome, bin 1 of the fragment is projected to bin floor(25/2) = 12 of the pangenome. The introduction of pangenome bins was motivated by the observation that bowtie2 selects an alignment at random when there are two or more top-scoring alignments, which tends to distribute coverage over several reference sequences when a single viral genome is present in the reads. Coverage of a single reference genome may therefore be fragmented, and binning to a pangenome better assesses coverage over a putative viral genome in the reads while retaining pangenome sequence diversity for detection.
Identification of viral families within a sequencing dataset
The Summarizer implements a binary classifier predicting the presence or absence of each virus family in the query on the basis of pangenome-aligned short reads. For a given family F, the classifier reports a score in the range [0,100] with the goal of assigning a high score to a dataset if it contains F and a low score if it does not. Setting a threshold on the score divides datasets into disjoint subsets representing predicted positive and negative detections of family F. The choice of threshold implies a trade-off between false positives and false negatives. Sorting by decreasing score ranks datasets in decreasing order of confidence that F is present in the reads.
Naively, a natural measure of the presence of a virus family is the number of alignments to its reference sequences. However, alignments may be induced by non-homologous sequence similarity, for example low-complexity sequence.
The score for a family was therefore designed to reflect the overall coverage of a pangenome because coverage across all or most of a pangenome is more likely to reflect true homology; that is, the presence of a related virus. Ideally, coverage would be measured individually for each base in the reference sequence, but this could add undesirable overhead in compute time and memory for a process that is executed in the Linux alignment pipe (FASTQ decompression → aligner → Summarizer → alignment file compression). Coverage was therefore measured by binning as described above, which can be implemented with minimal overhead.
A virus that is present in the reads with coverage too low to enable an assembly may have less practical value than an assembled genome. Also, genomes with lower identity to previously known sequences will tend to contain more novel biological information than genomes with high identity but highly diverged genomes will tend to have fewer aligned reads. With these considerations in mind, the classifier was designed to give higher scores when coverage is high, read depth is high and/or identity is low. This was accomplished as follows. Let H be the number of bins with at least 8 alignments to F, and L be the number of bins with from 1 to 7 alignments. Let S be the mean alignment percentage identity, and define the identity weight w = (S/100)−3, which is designed to give higher weight to lower identities, noting that w is close to 1 when identity is close to 100% and increases rapidly at lower identities. The classification score for family F is calculated as ZF = max(w(4H + L)),100). By construction, ZF has a maximum of 100 when coverage is consistently high across a pangenome, and is also high when identity is low and coverage is moderate, which may reflect high read depth but many false negative alignments due to low identity. Thus, ZF is greater than zero when there is at least one alignment to F and assigns higher scores to SRA datasets that are more likely to support successful assembly of a virus belonging to F.
Sensitivity to novel viruses as a function of identity
We aimed to assess the sensitivity of our pipeline as a function of sequence identity by asking what fraction of novel viruses is detected at increasingly low identities compared to the reference sequences used for the search. Several variables other than identity affect sensitivity, including read length, whether reads are mate-paired, sequencing error rate, coverage bias and the presence of other similar viruses that may cause some variants to be unreported in the contigs. Coverage bias can render a virus with high average read depth undetectable, in particular if the query is RdRP-only and the RdRP gene has low coverage or is absent from the reads. Successful detection might be defined in different ways, depending on the goals of the search; for example, a single local alignment of a reference to a read (maximizing sensitivity, but not always useful in practice); a microassembled palmprint; a full assembly contig that contains a complete palmprint or otherwise classifiable fragment of a marker gene; or an assembly of a complete genome. We assessed alignment sensitivity of bowtie2 --very-sensitive-local and Serratus-optimized DIAMOND14 as a function of identity by simulating typical examples in a representative scenario: unpaired reads of length 100 with a base call error rate of 1%. We manually selected test-reference pairs of RefSeq complete Ribovirus genomes at RdRP amino acid identities 100%, 95% … 20%, generating simulated length-100 reads at uniformly distributed random locations in the test genome with a mean coverage of 1,000×. For bowtie2, the complete reference genome was used as a reference; for DIAMOND the reference was the translated amino acid sequence of the RdRP gene (400 amino acids), which was identified by aligning to the ‘wolf18’ dataset. These choices model the coronavirus pangenome used as a bowtie2 query and the rdrp1 protein reference used as a DIAMOND query, respectively. Sensitivity was assessed as the fraction of reads aligned to the reference. With bowtie2, the number of unmapped reads reflects a combination of lack of alignment sensitivity and divergence in gene content as some regions of the genome may lack homology to the reference. With DIAMOND, the number of unmapped reads reflects a combination of lack of alignment sensitivity and the fraction of the genome that is not RdRP, which varies by genome length 1g. They show that the fraction of aligned reads by bowtie2 drops to around 2% to 4% at 90% RdRP amino acid identity, and maps no reads for most of the lower identity test–reference pairs. DIAMOND maps around 5% to 10% of reads down to 50% RdRP amino acid identity, then less than 1% at lower identities; around 30% to 35% is the lower limit of practical detection.
Defining viral pangenomes and the SRA search space
Nucleotide search pangenomes
To create a collection of viral pangenomes, a comprehensive set of complete and partial genomes representing the genetic diversity of each viral family, we used two approaches.
For Coronaviridae, we combined all RefSeq (n = 64) and GenBank (n = 37,451) records matching the NCBI Nucleotide53 server query “txid11118[Organism:exp]” (date accessed: 1 June 2020). Sequences of fewer than 200 nt were excluded as well as sequences identified to contain non-CoV contaminants during preliminary testing (such as plasmid DNA or ribosomal RNA fragments). Remaining sequences were clustered at 99% identity with UCLUST (USEARCH: v11.0.667)54 and masked by Dustmasker (ncbi-blast:2.10.0) (--window 30 and --window 64)55. The final query contained 10,101 CoV sequences (accessions in Supplementary Table 1a; masked coordinates in Supplementary Table 1b). SeqKit (v0.15) was used for working with fasta files56.
For all other vertebrate viral family pangenomes, RefSeq sequences (n = 2,849) were downloaded from the NCBI Nucleotide server with the query "Viruses[Organism] AND srcdb refseq[PROP] NOT wgs[PROP] NOT cellular organisms[ORGN] NOT AC 000001:AC 999999[PACC] AND ("vhost human"[Filter] AND "vhost vertebrates"[Filter])" (date accessed: 17 May 2020). Retroviruses (n = 80) were excluded as preliminary testing yielded excessive numbers of alignments to transcribed endogenous retroviruses. Each sequence was annotated with its taxonomic family according to its RefSeq record; those for which no family was assigned by RefSeq (n = 81) were designated as ‘unknown’.
The collection of these pangenomes was termed ‘cov3m’, and was the nucleotide sequence reference used for this study.
Amino acid viral RdRP search panproteome
For the translated-nucleotide search of viral RNA-dependent RNA polymerase (RdRP; hereinafter viral RdRP is implied) we combined sequences from several sources. (1) The ‘wolf18’ collection is a curated snapshot (around 2018) of RdRP from GenBank (ref. 19 accessed: ftp://ftp.ncbi.nlm.nih.gov/pub/wolf/_suppl/rnavir18/RNAvirome.S2.afa). (2) The ‘wolf20’ collection is RdRPs from assembled from marine metagenomes (ref. 7 accessed: ftp://ftp.ncbi.nlm.nih.gov/pub/wolf/_suppl/yangshan/gb_rdrp.afa). (3) All viral GenBank protein sequences were aligned with DIAMOND --ultra-sensitive14 against the combined wolf18 and wolf20 sequences (E-value < 1 × 10−6). These produced local alignments that contained truncated RdRP, so each RdRP-containing GenBank sequence was then re-aligned to the wolf18 and wolf20 collection to ‘trim’ them to ‘wolf’ RdRP boundaries. (4) The above algorithm was also applied to all viral GenBank nucleotide records to capture additional RdRP not annotated as such by GenBank. A region of HCV capsid protein shares similarity to HCV RdRP; sequences annotated as HCV capsid were therefore removed. Eight novel coronavirus RdRP sequences identified in a pilot experiment were added manually. The combined RdRP sequences from the above collections were clustered (UCLUST) at 90% amino acid identity and the resulting representative sequences (centroids, n = 14,653) used as the rdrp1 search query.
In addition, we added delta virus antigen proteins from NC 001653, M21012, X60193, L22063, AF018077, AJ584848, AJ584847, AJ584844, AJ584849, MT649207, MT649208, MT649206, NC 040845, NC 040729, MN031240, MN031239, MK962760, MK962759 and eight additional homologues we identified in a pilot experiment.
SRA search space and queries
To run Serratus, a target list of SRA run accessions is required. We defined 11 (not-mutually exclusive) queries as our search space, which were named human, mouse, mammal, vertebrate, invertebrate, eukaryotes, prokaryotes/others, bat (including genomic sequences), virome, metagenome and mammalian genome (Supplementary Table 1c). Our search was restricted to Illumina sequencing technologies and to RNA-seq, meta-genomic and meta-transcriptome library types for these organisms (except for the mammalian genome query, which was genome or exome). Before each Serratus deployment, target lists were depleted of accessions already analysed. Reprocessing of a failed accession was attempted at least twice. In total, we aligned 3,837,755/4,059,695 (94.5%) of the runs in our nucleotide-pangenome search (around May 2020) and 5,686,715/5,780,800 (98.37%) of the runs in our translated-nucleotide RdRP search (around January 2021).
User interfaces for the Serratus databases
We implemented an on-going, multi-tiered release policy for code and data generated by this study, as follows. All code, electronic notebooks and raw data are immediately available at https://github.com/ababaian/serratus and on the s3://serratus-public bucket, respectively. Upon completion of a project milestone, a structured data release is issued containing raw data into our viral data warehouse s3://lovelywater/. For example, the .bam nucleotide alignment files from 3.84 million SRA runs are stored in s3://lovelywater/bam/X.bam; and the protein .summary files are in s3://lovelywater/psummary/X.psummary, where X is a SRA run accession. These structured releases enable downstream and third-party programmatic access to the data.
Summary files for every searched SRA dataset are parsed into a publicly accessible AWS Relational Database (RDS) instance that can be queried remotely via any PostgreSQL client. This enables users and programs to perform complex operations such as retrieving summaries and metadata for all SRA runs matching a given reference sequence with above a given classifier score threshold. For example, one can query for all records containing at least 20 aligned reads to hepatitis delta virus (NC 001653.2) and the associated host taxonomy for the corresponding SRA datasets:
SELECT sequence_accession, run_id, tax_id, n_reads FROM nsequence JOIN srarun ON (nsequence.run_id = srarun.run) WHERE n_reads >= 20
For users unfamiliar with SQL, we developed Tantalus (https://github.com/serratus-bio/tantalus, an R programming-language package that directly interfaces the Serratus PostgreSQL database to retrieve summary information as data-frames. Tantalus also offers functions to explore and visualize the data.
Finally, the Serratus data can be explored via a graphical web interface by accession, virus or viral family at https://serratus.io/explorer. Under the hood, we developed a REST API to query the database from the website. The website uses React+D3.js to serve graphical reports with an overview of viral families found in each SRA accession matching a user query.
All four data access interfaces are under ongoing development, receiving community feedback via their respective GitHub issue trackers to facilitate the translation of this data collection into an effective viral discovery resource. Documentation for data access methods is available at https://serratus.io/access.
Geocoding BioSamples
To generate the map in Fig. 1c, we parsed and extracted geographical information from all 16 million BioSample XML submissions57. Geographic information is either in the form of coordinates (latitude and longitude) or freeform text (for example, ‘France’, ‘Great Lakes’). For each BioSample, coordinate extraction was attempted using regular expressions. If that failed, text extraction was attempted using a manually curated list of keywords that capture BioSample attribute names that are likely to contain geographical information. If that failed, then we were unable to extract geographical information for that BioSample. Geocoding the text to coordinates was done using Amazon Location Service on a reduced set of distinct filtered text values (52,028 distinct values from 2,760,241 BioSamples with potential geographical text). BioSamples with geocoded coordinates were combined with BioSamples with submitted coordinate information to form a set of 5,325,523 geospatial BioSamples. This is then cross-referenced with our subset of SRA accessions with an RdRP match to generate the figure.
All intermediate and resulting data from this step are stored on the SQL database described above. Development work is public at https://github.com/serratus-bio/biosample-sql.
Viral alignment, assembly and annotation
Upon identification of CoV reads in a run from alignment, we assembled 52,772 runs containing at least 10 reads that aligned to our CoV pangenome or at least 2 reads with CoV-positive k-mers16. A total of 11,120 of the resulting assemblies contained identifiable CoV contigs, of which only 4,179 (37.58%) contained full-length CoV RdRP (Supplementary Table 1d). The discrepancy between alignment-positive, assembly-positive and RdRP-positive libraries arises owing to random sampling of viral reads and assembly fragmentation. In this respect, alignment or k-mer based methods are more sensitive than assembly in detecting for the presence of low-abundance viruses (genome coverage < 1) with high identity to a reference sequence. Scoring libraries for genome coverage and depth is a good predictor of ultimate assembly success (Extended Data Fig. 3); thus, it can be used to efficiently prioritize computationally expensive assembly in the future, as has been previously demonstrated for large-scale SRA alignment analyses58.
DIAMOND optimization and output
To optimize DIAMOND14 for small (<10 MB) databases such as the RdRP search database, we built a probabilistic hash set that stores 8-bit hash values for the database seeds, using SIMD instructions for fast probing. This index is loaded as a memory mapped file to be shared among processes and allows us to filter the query reads for seeds contained in the database, thus omitting the full construction of the query seed table. We also eliminated the overhead of building seed distribution histograms that is normally required to allocate memory and construct the query table in a single pass over the data using a deque-like data structure. In addition, query reads were not masked for simple repeats, as the search database is already masked. These features are available starting from DIAMOND v2.0.8 with the command line flags --target-indexed --masking 0. In a benchmark of 4 sets of 1 million reads from a bat metagenome (ERR2756788), the implemented optimization produced a speed-up of ×1.47 and reduced memory use by 64%, compared to the public unmodified DIAMOND v2.0.6, using our optimized set of parameters in both cases (see 1.1.1). Together, the optimized parameters and implementation reduced DIAMOND runtime against RdRP search from 197.96 s (s.d. = 0.18 s), to 21.29 s (s.d. = 0.23 s) per million reads, a speed-up of a factor of 9.3. This effectively reduced the computational cost of translated-nucleotide search for Serratus from US$0.03 to US$0.0042 per library.
DIAMOND output files (we label .pro) were specified with the command -f 6 qseqid qstart qend qlen qstrand sseqid sstart send slen pident evalue cigar qseq_translated full_qseq full_qseq_mate.
coronaSPAdes
RNA viral genome assembly faces several distinct challenges stemming from technical and biological bias in sequencing data. During library preparation, reverse transcription introduces 50 end coverage bias, and GC-content skew and secondary structures lead to unequal PCR amplification59. Technical bias is confounded by biological complexity such as intra-sample sequence variation due to transcript isoforms and/or to the presence of multiple strains.
To address the assembly challenges specific to RNA viruses, we developed coronaSPAdes (v3.15.3), which is described in detail in a companion manuscript25. In brief, rnaviralSPAdes and the more specialized variant, coronaSPAdes, combines algorithms and methods from several previous approaches based on metaSPAdes60, rnaSPAdes61 and metaviralSPAdes62 with a HMMPathExtension step. coronaSPAdes constructs an assembly graph from an RNA-seq dataset (transcriptome, meta-transcriptome, and meta-virome are supported), removing expected sequencing artifacts such as low complexity (poly-A/poly-T) tips, edges, single-strand chimeric loops or double-strand hairpins61 and subspecies-bases variation62.
To deal with possible misassemblies and high-covered sequencing artefacts, a secondary HMMPathExtension step is performed to leverage orthogonal information about the expected viral genome. Protein domains are identified on all assembly graphs using a set of viral hidden Markov models (HMMs), and similar to biosyntheticSPAdes63, HMMPathExtension attempts to find paths on the assembly graph that pass through significant HMM matches in order.
coronaSPAdes is bundled with the Pfam SARS-CoV-2 set of HMMs64, although these may be substituted by the user. This latter feature of coronaSPAdes was used for HDV assembly, in which the HMM model of HDAg, the hepatitis delta antigen, was used instead of the Pfam SARS-CoV-2 set. Note that despite the name, the HMMs from this set are quite general, modelling domains found in all coronavirus genera in addition to RdRP, which is found in many RNA virus families. Hits from these HMMs cover most bases in most known coronavirus genomes, enabling the recovery of strain mixtures and splice variants.
Microassembly of RdRP-aligned reads
Reads aligned by DIAMOND14 in the translated-nucleotide RdRP search are stored in the .pro alignment file. All sets of mapped reads (3,379,127 runs) were extracted, and each non-empty set was assembled with rnaviralSPAdes (v3.15.3)25 using default parameters. This process is referred to as ‘microassembly’, as a collection of DIAMOND hits is orders of magnitude smaller than the original SRA accession (40 ± 534 KB compressed size, ranging from a single read up to 53 MB). Then bowtie251 (default parameters) was used to align the DIAMOND read hits of an accession back to the microassembled contigs of that accession. Palmscan (v1.0.0, -rdrp -hicon)18 was run on microassembled contigs, resulting in high-confidence palmprints for 337,344 contigs. Finally mosdepth (v0.3.1)65 was used to calculate a coverage pileup for each palmprint hit region within microassembled contigs.
Classification of assembled RdRP sequences
Our methods for RdRP classification are described and validated in a companion paper18. In brief, we defined a barcode sequence, the polymerase palmprint (PP),as an approximately 100-amino-acid segment of the RdRP palm subdomain delineated by well-conserved catalytic motifs. We implemented an algorithm, Palmscan, to identify palmprint sequences and discriminate RdRPs from reverse transcriptases. The combined set of RdRP palmprints from public databases and our assemblies was classified by clustering into operational taxonomic units (OTUs) at 90%, 75% and 40% identity, giving species-like, genus-like and family-like clusters (sOTUs, gOTUs and fOTUs), respectively. Tentative taxonomy of novel OTUs was assigned by aligning to palmprints of named viruses and taking a consensus of the top hits above the identity threshold for each rank.
Quality control of assembled RdRP sequences
Our goal was to identity novel viral RdRP sequences and novel sOTUs in SRA libraries. From this perspective, we considered the following to be erroneous to varying degrees: sequences that are (a) not polymerases; (b) not viral; (c) with differences due to experimental artefacts; or (d) with sufficient differences to cause a spurious inference of a novel sOTU. We categorized potential sources of such errors and implemented quality control procedures to identify and mitigate them, as follows.
Point errors are single-letter substitution and indel errors that may be caused by PCR or sequencing per se. Random point errors are not reproduced in multiple non-PCR duplicate reads and are unlikely to assemble because such errors almost always induce identifiable structures in the assembly graph (tips and bubbles) that are pruned during graph simplification. In rare cases, a contig may contain a read with random point errors. Such contigs will have low coverage of around 1, and we therefore recorded coverage as a quality control metric and assessed whether low-coverage assemblies were anomalous compared to high-coverage assemblies by measures such as the frequencies with which they are reproduced in multiple libraries compared to exactly one library, finding no noticeable difference when coverage is low.
Chimeras of polymerases from different species could arise from PCR amplification or assembly. We used the UCHIME2 (usearch v8.0.1623) algorithm66 to screen assembled palmprint sequences, finding no high-scoring putative chimeras. Mosaic sequences formed by joining a polymerase to unrelated sequence would either have an intact palmprint, in which case the mosaic would be irrelevant to our analysis, or would be rejected by Palmscan owing to the lack of delimiting motifs.
Reverse transcriptases are homologous to RdRP. Retroviral insertions into host genomes induce ubiquitous sequence similarity between host genomes and viral RdRP. Palmscan was designed to discriminate RdRP from sequences of reverse transcriptase origin. Testing on a large decoy set of non-RdRP sequences with recognizable sequence similarity showed that the Palmscan false discovery rate for RdRP identification is 0.001. We estimated the probability of false positive matches in unrelated sequence by generating sufficient random nucleotide and amino acid sequences to show that the expected number of false positive palmprint identifications is zero in a dataset of comparable size to our assemblies. We also regard the low observed frequency of palmprints in DNA whole-genome sequencing data (in 2.6 Pbp or 25.8% of reads, accounted for 100 known palmprints and 95 novel palmprints or 0.13% of the total identified) as a de facto confirmation of the low probability false positives in unrelated sequence.
Endogenous viral elements (EVEs; that is, insertions of viral sequence into host genomes that are potentially degraded and non-functional) cannot be distinguished from viral genomes on the basis of the palmprint sequence alone. To assess the frequency of EVEs in our data, we re-assembled 890 randomly chosen libraries yielding one or more palmprints using all reads, extracted the 23,530 resulting contigs with a positive palmprint hit by Palmscan, and classified them using Virsorter2 (v2.1)67. Of these contigs, 11,914 were classified as viral, confirming the Palmscan identification; 49 as Viridiplantae (green plants); 46 as Metazoa; 25 as Fungi and the remainder were unclassified. Thus, 120/12,034 = 1% of the classified contigs were predicted as non-viral, suggesting that the frequency of EVEs in the reported palmprints is around 1%.
Annotation of CoV assemblies
Accurate annotation of CoV genomes is challenging owing to ribosomal frameshifts and polyproteins that are cleaved into maturation proteins68, and thus previously annotated viral genomes offer a guide to accurate gene-calls and protein functional predictions. However, although many of the viral genomes we were likely to recover would be similar to previously annotated genomes in Refseq or GenBank, we anticipated that many of the genomes would be taxonomically distant from any available reference. To address these constraints, we developed an annotation pipeline called DARTH (version maul)69 which leverages both reference-based and ab initio annotation approaches.
In brief, DARTH consists of the following phases: standardize the ordering and orientation of assembly contigs using conserved domain alignments, perform reference-based annotation of the contigs, annotate RNA secondary structure, ab initio gene-calling, generate files for aiding assembly and annotation diagnostics, and generate a master annotation file. It is important to put the contigs in the ‘expected’ orientation and ordering to facilitate comparative analysis of synteny and as a requirement for genome deposition. To perform this standardization, DARTH generates the six-frame translation of the contigs using the transeq (EMBOSS:6.6.0.0)70 and uses HMMER3 (v3.3.2)71 to search the translations for Pfam domain models specific to CoV64. DARTH compares the Pfam accessions from the HMMER alignment to the NCBI SARS-CoV-2 reference genome (NCBI Nucleotide accession NC_045512.2) to determine the correct ordering and orientation, and produces an updated assembly FASTA file. DARTH performs reference-based annotation using VADR (v1.1)72, which provides a set of genome models for all CoV RefSeq genomes73. VADR provides annotations of gene coordinates, polyprotein cleavage sites, and functional annotation of all proteins. DARTH supplements the VADR annotation by using Infernal74 to scan the contigs against the SARS-CoV-2 Rfam release75 which provides updated models of CoV 50 and 30 untranslated regions (UTRs) along with stem-loop structures associated with programmed ribosomal frame-shifts. Although VADR provides reference-based gene-calling, DARTH also provides ab initio gene-calling by using FragGeneScan (v1.31)76, a frameshift-aware gene caller. DARTH also generates auxiliary files that are useful for assembly quality and annotation diagnostics, such as indexed BAM files created with SAMtools (v1.7)77 representing self-alignment of the trimmed reads to the canonicalized assembly using bowtie251, and variant-calls using bcftools from SAMtools. DARTH generates these files so that the can be easily loaded into a genome browser such as JBrowse78 or IGV79. As the final step DARTH generates a single Generic Feature Format (GFF) 3.0 file80 containing combined set of annotation information described above, ready for use in a genome browser, or for submitting the annotation and sequence to a genome repository.
Phage assembly
Each metagenomic dataset was individually de-novo-assembled using MEGAHIT (v1.2.9)81, and filtered to remove contigs smaller than 1 kb in size. ORFs were then predicted on all contigs using Prodigal (v2.6.3)82 with the following parameters: -m -p meta. Predicted ORFs were initially annotated using USEARCH54 to search all predicted ORFs against UniProt83, UniRef90 and KEGG84. Sequencing coverage of each contig was calculated by mapping raw reads back to assemblies using bowtie251. Terminase sequences from Al-Shayeb et al.42 were clustered at 90% amino acid identity to reduce redundancy using CD-HIT (v4.8.1)85, and HMM models were built with hmmbuild (from the HMMER3 suite71) from the resulting set. Terminases in the assemblies from Serratus were identified using hmmsearch, retaining representatives from contigs greater than 140 kb in size. Some examples of prophage and large phages that did not co-cluster with the sequences from Al-Shayeb et al. were also recovered because they were also present in a sample that contained the expected large phages. The terminases were aligned using MAFFT (v7.407)86 and filtered by TrimAL (v1.14)87 to remove columns comprising more than 50% gaps, or 90% gaps, or using the automatic gappyout setting to retain the most conserved residues. Maximum likelihood trees were built from the resulting alignments using IQTREE (v1.6.6)88.
Deploying the assembly and annotation workflow
The Serratus search for known or closely related viruses identified 37,131 libraries (14,304 by nucleotide and 23,898 by amino acid) as potentially positive for CoV (score ≥ 20 and ≥10 reads). To supplement this search we also used a recently developed index of the SRA called STAT16, which identified an additional 18,584 SRA datasets not in the defined SRA search space. The STAT BigQuery (accessed 24 June 2020) was: WHERE tax id=11118 AND total count >1.
We used AWS Batch to launch thousands of assemblies of NCBI accessions simultaneously. The workflow consists of four standard parts: a job queue, a job definition, a compute environment, and finally, the jobs themselves. A CloudFormation template (https://gitlab.pasteur.fr/rchikhi_pasteur/serratus-batch-assembly/-/blob/10934001/template/template.yaml) was created for building all parts of the cloud infrastructure from the command line. The job definition specifies a Docker image, and asks for 8 virtual CPUs (vCPUs, corresponding to threads) and 60 GB of memory per job, corresponding to a reasonable allocation for coronaSPAdes. The compute environment is the most involved component. We set it to run jobs on cost-effective Spot instances (optimal setting) with an additional cost-optimization strategy (SPOT_CAPACITY_OPTIMIZED setting), and allowing up to 40,000 vCPUs total. In addition, the compute environment specifies a launch template which, on each instance, (i) automatically mounts an exclusive 1 TB EBS volume, allowing sufficient disk space for several concurrent assemblies, and (ii) downloads the 5.4 GB CheckV (v0.6.0)89 database, to avoid bloating the Docker image.
The peak AWS usage of our Batch infrastructure was around 28,000 vCPUs, performing around 3,500 assemblies simultaneously. A total of 46,861 accessions out of 55,715 were assembled in a single day. They were then analysed by two methods to detect putative CoV contigs. The first method is CheckV89, followed selecting contigs associated to known CoV genomes. The second method is a custom script (https://gitlab.pasteur.fr/rchikhi_pasteur/serratus-batch-assembly/-/blob/10934001/stats/bgc_parse_and_extract.py) that parses coronaSPAdes BGC candidates and keeps contigs containing CoV domain(s). For each accession, we kept the set of contigs obtained by the first method (CheckV) if it is non-empty, and otherwise we kept the set of contigs from the second method (BGC).
A majority (76%) of the assemblies were discarded for one of the following reasons: (i) no CoV contigs were found by either filtering method; (ii) reads were too short to be assembled; (iii) Batch job or SRA download failed; or (iv) coronaSPAdes ran out of memory. A total of 11,120 assemblies were considered for further analysis.
The average cost of assembly was between US$0.30 and US$0.40 per library, varying depending on library type (RNA-seq versus metagenomic). This places an estimate of 46–95-fold higher cost for assembly alone compared to a cost of US$0.0042 or US$0.0065 for an alignment-based search.
Taxonomic and phylogenetic analyses
Taxonomy prediction for coronavirus genomes
We developed a module, SerraTax, to predict taxonomy for CoV genomes and assemblies (https://github.com/ababaian/serratus/tree/master/containers/serratax). SerraTax was designed with the following requirements in mind: provide taxonomy predictions for fragmented and partial assemblies in addition to complete genomes; report best-estimate predictions balancing over-classification and under-classification (too many and too few ranks, respectively); and assign an NCBI Taxonomy Database90 identifier (TaxID).
Assigning a best-fit TaxID was not supported by any previously published taxonomy prediction software to the best of our knowledge; this requires assignment to intermediate ranks such as sub-genus and ranks below species (commonly called strains, but these ranks are not named in the Taxonomy database), and to unclassified taxa, for example, TaxID 2724161, unclassified Buldecovirus, in cases in which the genome is predicted to fall inside a named clade but outside all named taxa within that clade.
SerraTax uses a reference database containing domain sequences with TaxIDs. This database was constructed as follows. Records annotated as CoV were downloaded from UniProt83, and chain sequences were extracted. Each chain name, for example Helicase, was considered to be a separate domain. Chains were aligned to all complete coronavirus genomes in GenBank using UBLAST (usearch: v11.0.667)54 to expand the repertoire of domain sequences. The reference sequences were clustered using UCLUST54 at 97% sequence identity to reduce redundancy.
For a given query genome, ORFs are extracted using the getorf (EMBOSS:6.6.0) software70. ORFs are aligned to the domain references and the top 16 reference sequences for each domain are combined with the best-matching query ORF. For each domain, a multiple alignment of the top 16 matches plus query ORF is constructed on the fly by MUSCLE (v3.8.3191) and a neighbour-joining tree is inferred from the alignment, also using MUSCLE. Finally, a consensus prediction is derived from the placement of the ORF in the domain trees. Thus, the presence of a single domain in the assembly suffices to enable a prediction; if more domains are present they are combined into a consensus.
Taxonomic assignment by phylogenetic placement
To generate an alternate taxonomic annotation of an assembled genome, we created a pipeline based on phylogenetic placement, SerraPlace.
To perform phylogenetic placement, a reference phylogenetic tree is required. To this end, we collected 823 reference amino acid RdRP sequences, spanning all Coronaviridae. To this set we added an outgroup RdRP sequence from the Torovirus family (NC 007447). We clustered the sequences to 99% identity using USEARCH (ref. 54, UCLUST algorithm, v11.0.667), resulting in 546 centroid sequences. Subsequently, we performed multiple sequence alignment on the clustered sequences using MUSCLE. We then performed maximum likelihood tree inference using RAxML-NG (ref. 92, ‘PROTGTR+FO+G4’, v0.9.0), resulting in our reference tree.
To apply SerraPlace to a given genome, we first use HMMER (ref. 71, v3.3) to generate a reference HMM, based on the reference alignment. We then split each contig into ORFs using esl-translate, and use hmmsearch (P value cut-off 0.01) and seqtk (commit 7c04ce7) to identify those query ORFs that align with sufficient quality to the previously generated reference HMM. All ORFs that pass this test are considered valid input sequences for phylogenetic placement. This produces a set of likely placement locations on the tree, with an associated likelihood weight. We then use Gappa (v0.6.1,93) to assign taxonomic information to each query, using the taxonomic information for the reference sequences. Gappa assigns taxonomy by first labelling the interior nodes of the reference tree by a consensus of the taxonomic labels of all descendant leaves of that node. If 66% of leaves share the same taxonomic label up to some level, then the internal node is assigned that label. Then, the likelihood weight associated with each sequence is assigned to the labels of internal nodes of the reference tree, according to where the query was placed.
From this result, we select that taxonomic label that accumulated the highest total likelihood weight as the taxonomic label of a sequence. Note that multiple ORFs of the same genome may result in a taxonomic label, in which case, we select the longest sequence as the source of the taxonomic assignment of the genome.
Phylogenetic inference
We performed phylogenetic inferences using a custom snakemake (v6.6.0) pipeline (available at https://github.com/lczech/nidhoggr), using ParGenes (v1.1.2)94. ParGenes is a tree search orchestrator, combining ModelTestNG (v0.1.3)95 and RAxML-NG, and enabling higher levels of parallelization for a given tree search.
To infer the maximum likelihood phylogenetic trees, we performed a tree search comprising 100 distinct starting trees (50 random, 50 parsimony), as well as 1,000 bootstrap searches. We used ModelTest-NG to automatically select the best evolutionary model for the given data. The pipeline also automatically produces versions of the best maximum likelihood tree annotated with Felsenstein’s Bootstrap96 support values, and Transfer Bootstrap Expectation values97.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All Serratus data, raw and processed, are released into the public domain immediately in accordance with the Bermuda Principles and freely available at https://serratus.io/access. Assembled genomes for this study are available on GenBank under project PRJEB44047.
Code availability
Serratus (v0.3.0) is available at https://github.com/ababaian/serratus. Archival copies of all code and software generated for this study are freely available at https://github.com/serratus-bio. Electronic notebooks for experiments are available at https://github.com/ababaian/serratus.
References
Leinonen, R., Sugawara, H. & Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 39, D19–D21 (2011).
Anthony, S. J. et al. A strategy to estimate unknown viral diversity in mammals. mBio 4, e00598-13 (2013).
Johnson, C. K. et al. Global shifts in mammalian population trends reveal key predictors of virus spillover risk. Proc. R. Soc. B 287, 20192736 (2020).
Carroll, D. et al. The Global Virome Project. Science 359, 872–874 (2018).
Shi, M. et al. The evolutionary history of vertebrate RNA viruses. Nature 556, 197–202 (2018).
Wahba, L. et al. An extensive meta-metagenomic search identifies SARS-CoV-2-homologous sequences in pangolin lung viromes. mSphere 5, 00160-20 (2020).
Wolf, Y. I. et al. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 5, 1262–1270 (2020).
Mitchell, A. L. et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578 (2020).
Chen, I.-M. A. et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49, D751–D763 (2021).
Camarillo-Guerrero, L. F., Almeida, A., Rangel-Pineros, G., Finn, R. D. & Lawley, T. D. Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109 (2021).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Moore, R. A. et al. The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS One 6, e19838 (2011).
NIH. STRIDES Initiative—Data Science at NIH https://datascience.nih.gov/strides (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Karasikov, M. et al. MetaGraph: indexing and analysing nucleotide archives at petabase-scale. Preprint at https://www.biorxiv.org/content/10.1101/2020.10.01.322164v2 (2020).
Katz, K. S. et al. STAT: a fast, scalable, MinHash-based k-mer tool to assess Sequence Read Archive next-generation sequence submissions. Genome Biol. 22, 270 (2021).
Koonin, E. V. & Dolja, V. V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. Rev. 78, 278–303 (2014).
Babaian, A. & Edgar, R. C. Ribovirus classification by a polymerase barcode sequence. Preprint at https://www.biorxiv.org/content/10.1101/2021.03.02.433648v1 (2021).
Wolf, Y. I. et al. Origins and evolution of the global RNA virome. mBio 9, e0239-18 (2018).
Obbard, D. J., Shi, M., Roberts, K. E., Longdon, B. & Dennis, A. B. A new lineage of segmented RNA viruses infecting animals. Virus Evol. 6, vez061 (2020).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Koonin, E. V. et al. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 84, e00061-19 (2020).
Bukhari, K. et al. Description and initial characterization of metatranscriptomic nidovirus-like genomes from the proposed new family Abyssoviridae, and from a sister group to the Coronavirinae, the proposed genus Alphaletovirus. Virology 524, 160–171. (2018).
Mordecai, G. J. et al. Endangered wild salmon infected by newly discovered viruses. eLife 8. e47615 (2019).
Meleshko, D., Hajirasouliha, I. & Korobeynikov, A. coronaSPAdes: from biosynthetic gene clusters to RNA viral assemblies. Bioinformatics 38, 1–8 (2022).
Tao, Y. et al. Broad-range virus detection and discovery using microfluidic PCR coupled with high-throughput sequencing. Preprint at https://www.biorxiv.org/content/10.1101/2020.06.10.145052v1 (2020).
Tsai, S. L., Baselga-Garriga, C. & Melton, D. A. Midkine is a dual regulator of wound epidermis development and inflammation during the initiation of limb regeneration. eLife 9, e50765 (2020).
Sabin, K. Z., Jiang, P., Gearhart, M. D., Stewart, R. & Echeverri, K. AP-1 cFos/JunB /miR-200a regulate the pro-regenerative glial cell response during axolotl spinal cord regeneration. Commun. Biol. 2, 91 (2019).
Miller, A. K. et al. Slippery when wet: cross-species transmission of divergent coronaviruses in bony and jawless fish and the evolutionary history of the Coronaviridae. Virus Evol. 7. veab050 (2021).
Mukherjee, S. et al. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res. 49, D723–D733 (2021).
Stanaway, J. D. et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet 388, 1081–1088 (2016).
Taylor, J. M. Infection by hepatitis delta virus. Viruses 12, 648 (2020).
Szirovicza, L. et al. Snake deltavirus utilizes envelope proteins of different viruses to generate infectious particles. mBio 11, e03250-19 (2020).
Wille, M. et al. A divergent hepatitis D-like agent in birds. Viruses 12, 720 (2018).
Chang, W.-S. et al. Novel hepatitis D-like agents in vertebrates and invertebrates. Virus Evol. 5, vez021 (2019).
Paraskevopoulou, S. et al. Mammalian deltavirus without hepadnavirus coinfection in the neotropical rodent Proechimys semispinosus. Proc. Natl Acad. Sci. USA 117, 17977–17983 (2020).
Bergner, L. M. et al. Diversification of mammalian deltaviruses by host shifting. Proc. Natl Acad. Sci. USA 118, e2019907118 (2021).
Iwamoto, M. et al. Identification of novel avian and mammalian deltaviruses provides new insights into deltavirus evolution. Virus Evol. 7, veab003 (2021).
De la Peña, M., Ceprián, R., Casey, J. L. & Cervera, A. Hepatitis delta virus-like circular RNAs from diverse metazoans encode conserved hammerhead ribozymes. Virus Evol. 7, veab016 (2021).
Zuccola, H. J., Rozzelle, J. E., Lemon, S. M., Erickson, B. W. & Hogle, J. M. Structural basis of the oligomerization of hepatitis delta antigen. Structure 6, 821–830 (1998).
Flores, R., Gago-Zachert, S., Serra, P., Sanjuán, R. & Elena, S. F. Viroids: survivors from the RNA world? Annu. Rev. Microbiol. 68, 395–414 (2014).
Al-Shayeb, B. et al. Clades of huge phages from across Earth’s ecosystems. Nature 578, 425–431 (2020).
Devoto, A. E. et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 4, 693–700 (2019).
Bradley, P., Den Bakker, H. C., Rocha, E. P. C., McVean, G. & Iqbal, Z. Ultra-fast search of all deposited bacterial and viral genomic data. Nat. Biotechnol. 37, 152–159 (2019).
Baker, D. et al. No more business as usual: agile and effective responses to emerging pathogen threats require open data and open analytics. PLoS Pathog. 16, e1008643 (2020).
Letko, M., Seifert, S. N., Olival, K. J., Plowright, R. K. & Munster, V. J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 18, 461–471 (2020).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020).
Chase, J. M., Blowes, S. A., Knight, T. M., Gerstner, K. & May, F. Ecosystem decay exacerbates biodiversity loss with habitat loss. Nature 584, 238–243 (2020).
Thompson, A. A. & Peersen, O. B. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 23, 3462–3471 (2004).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Schatz, M. C. CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics 25, 1363–1369 (2009).
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46, D8–D13 (2018).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Morgulis, A., Gertz, E. M., Schäffer, A. A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962 (2016).
Courtot, M., Gupta, D., Liyanage, I., Xu, F. & Burdett, T. BioSamples database: FAIRer samples metadata to accelerate research data management. Nucleic Acids Res. https://doi.org/10.1093/nar/gkab1046 (2021).
Levi, K., Rynge, M., Abeysinghe, E. & Edwards, R. A. Searching the Sequence Read Archive using Jetstream and Wrangler. In Proc. Practice and Experience on Advanced Research Computing 1–7 (Association for Computing Machinery, 2021).
Hunt, M. et al. IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics 31, 2374–2376 (2015).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-seq data. GigaScience 8, giz100 (2019).
Antipov, D., Raiko, M., Lapidus, A. & Pevzner, P. A. metaviralSPAdes: assembly of viruses from metagenomic data. Bioinformatics 36, 4126–4129 (2020).
Meleshko, D. et al. BiosyntheticSPAdes: reconstructing biosynthetic gene clusters from assembly graphs. Genome Res. 29, 1352–1362 (2019).
Pfam team. Pfam SARS-CoV-2 Special Update (part 2) https://xfam.wordpress.com/2020/04/06/pfam-sars-cov-2-special-update-part-2/ (2020).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2018).
Edgar, R. C. UCHIME2: improved chimera prediction for amplicon sequencing. Preprint at https://doi.org/10.1101/074252 (2016).
Guo, J. et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 37 (2021).
Thiel, V. et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84, 2305–2315 (2003).
Altman, T. DARTH Coronavirus Annotation Pipeline https://bitbucket.org/tomeraltman/DARTH/src/master/ (2020).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 7, e1002195 (2011).
Schäffer, A. A. et al. VADR: validation and annotation of virus sequence submissions to GenBank. BMC Bioinformatics 21, 211 (2020).
Nawrocki, E. Coronavirus Annotation using VADR https://github.com/nawrockie/VADR/wiki/Coronavirus-annotation#build (2020).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Rfam team. Rfam Coronavirus Special Release https://xfam.wordpress.com/2020/04/27/rfam-coronavirus-release/ (2020).
Rho, M., Tang, H. & Ye, Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38, e191 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Buels, R. et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 66 (2016).
Robinson, J. T., Thorvaldsdóttir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant review with the Integrative Genomics Viewer. Cancer Res. 77, e31–e34 (2017).
Eilbeck, K. et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 6, R44 (2005).
Li, D. et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Hyatt, D., LoCascio, P. F., Hauser, L. J. & Uberbacher, E. C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230 (2012).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Altman, T., Travers, M., Kothari, A., Caspi, R. & Karp, P. D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinformatics 14, 112 (2013).
Li, W., Fu, L., Niu, B., Wu, S. & Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinformatics 13, 656–668 (2012).
Nakamura, T., Yamada, K. D., Tomii, K. & Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585 (2020).
Schoch, C. L. et al. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database 2020, baaa062 (2020).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Czech, L., Barbera, P. & Stamatakis, A. Genesis and Gappa: processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 36, 3263–3265 (2020).
Morel, B., Kozlov, A. M. & Stamatakis, A. ParGenes: a tool for massively parallel model selection and phylogenetic tree inference on thousands of genes. Bioinformatics 35, 1771–1773 (2018).
Darriba, D. et al. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2019).
Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 (1985).
Lemoine, F. et al. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556, 452–456 (2018).
Crisci, M. A. et al. Wide distribution of alternatively coded Lak megaphages in animal microbiomes. Preprint at https://www.biorxiv.org/content/10.1101/2021.01.08.425732v1 (2021).
Rapid reconstitution of the fecal microbiome after extended diet-induced changes indicates a stable gut microbiome in healthy adult dogs. Appl. Environ. Microbiol. 86, e00562-20 (2020).
Acknowledgements
The Serratus project is an initiative of the hackseqRNA genomics hackathon (https://www.hackseq.com). We thank the many contributors for code snippets and bioinformatic discussion (E. Erhan, J. Chu, S. Jackman, I. Birol, K. Wellman, O. Fornes, C. Xu, M. Huss, K. Ha, M. Krzywinski, E. Nawrocki, R. McLaughlin, C. Morgan-Lang, C. Blumberg and the J. Brister laboratory); A. Rodrigues, S. McMillan, V. Wu, C. Kennett, K. Chao, and N. Pereyaslavsky for AWS support; the J. Joy laboratory, G. Mordecai, J. Taylor, S. Roux, N. Kyrpides, E. Jan, T. Reddy, L. Bergner, R. Orton and D. Streicker for virology discussions; and H.-G. Drost and D. Weigel for supporting the adoption of DIAMOND v2 for Serratus protein alignments as part of an extended feature request. We are grateful to the entire team managing the NCBI SRA and the biology community for data sharing, with particular thanks to the E. Brodie, E. Lilleskov and E. Young laboratories. T.A. thanks Advanced Research Computing resource at the University of British Columbia and B.B. thanks the Max Plank Society for financial support. P.B. was financially supported by the Klaus Tschira Foundation; R.C. by ANR Transipedia, Inception and PRAIRIE grants (PIA/ANR16-CONV-0005, ANR-18-CE45-0020, ANR-19-P3IA-0001); and M.d.l.P. by the Ministerio de Economía y Competitividad of Spain and FEDER grants (BFU2017-87370-P and PID2020-116008GB-I00). A.K. and D.M. were supported by the Russian Science Foundation (grant 19-14-00172) and computation was carried out in part by Resource Centre ‘Computer Centre of SPbU’. A.K. and D.M. are grateful to Saint Petersburg State University for the overall support of this work. Project support and computing resources were provided by the University of British Columbia Community Health and Wellbeing Cloud Innovation Centre, powered by AWS.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to this work. A.B. conceived and led the study. A.B. and B.T. designed and implemented the Serratus architecture. A.B. and R.C.E. constructed the virus pangenomes and RdRP query. R.C.E. developed the SerraTax and Summarizer modules. P.B. developed the SerraPlace tree placement and taxonomy prediction code and calculated maximum likelihood trees. T.A. developed the DARTH annotation pipeline and submitted the annotated genomes to ENA. D.M. and A.K. developed the coronaSPAdes assembler. R.C. implemented the assembly pipeline, and deployed the assembly and annotation pipeline. B.B. optimized the DIAMOND algorithm for RdRP search. A.B., V.L. and D.L. designed and developed https://serratus.io and the SQL server. A.B. and G.N. developed the Tantalus R package. A.B., R.C.E., T.A., P.B., D.M., M.d.l.P., A.K. and R.C. analysed the coronavirus, RdRP and delta virus data. B.A.-S. and J.F.B. designed the phage panproteome, assembled phage genomes and conducted phage phylogenetic analyses. All authors contributed to data interpretation and writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Nature thanks C. Titus Brown, Alice McHardy and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Overview of the Serratus infrastructure.
a Schematic and data workflow (b) as described in the methods for sequence alignment. c The align module accepts either a nucleotide or protein sequence query. d A nucleotide alignment completion rate for Serratus shows stable and linear performance to complete 1.29 million SRA accessions in a 24-hour period and the e cost breakdown for this run. Compute costs between modules are an approximate comparison of CPU requirements of each step. The total average cost per completed SRA accession was US$0.0062 for nucleotide search or US$0.0042 for translated-nucleotide search. f Tukey boxplot of biological cross-validation to measure alignment sensitivity for bowtie2 (nucleotide search), DIAMOND14 (translated nucleotide search) or 32-mer for exact search. In brief, two RdRP sequences sharing the nominal amino acid identities form a ”pair”. 100 bp reads were simulated from the coding sequence of one pair and mapped onto the second pair, with the fraction of reads mapped reported. A fraction of 0.5 indicates that half the simulated reads at the given RdRP percent identity are mappable and thus detectable (see Methods). For each of the 12 percent identity categories, n = 10 biologically independent RdRP pairs were analysed.
Extended Data Fig. 2 Analysis of palmprint contigs recovered by Serratus.
a Length distribution of amino acid sequences in the rdrp1 query (upper histogram) and microassembled contigs (lower histogram, length=nucleotides/3). b Distribution of Palmscan confidence scores. c Observations of the 10 most frequent “super-motifs” (six well-conserved residues marked with asterisk) reported by Palmscan. d Kernel distribution and mean (white cross) of coverage vs. abundance (number of runs where a given palmprint is observed), showing that palmprints have similar underlying coverage distributions at all abundances. e Preston plot of distinct palmprints vs. abundance exhibiting similar, approximately log-log-linear relationships to totals at end-of-year 2015 to 2019 and final totals at approx. end of 2020 (all). f Preston plot of number of distinct palmprints observed in a given run vs. number of runs with 95% confidence interval. g Numbers of singletons and second observations (confirmations) at the end of each year showing that the growth in singletons is matched by a comparable growth in confirmations. h Kingdom predicted by Virsorter2 for RdRP+ contigs (by Palmscan) obtained by full assembly of 880 randomly chosen RdRP+ runs. i Number of palmprints in each phylum assigned by taxonomy (known) or predicted (novel). j Number of OTUs as a function of clustering identity.
Extended Data Fig. 3 Distribution of select RNA virus families.
a Histograms of datasets matching select RNA viral family by translated-nucleotide search against RdRP query, binned by the average amino acid identity. Score (gradient colouring) function approximates pangenome/gene coverage (see methods) used for manual inspection and to prioritize assembly. Interactive and queryable versions of these plots for extended virus families are available at https://serratus.io/explorer. b Relationship between the nucleotide pangenome score function and the subsequent assembly success (defined by the presence of an RdRP+ contig) measured from 52,772 libraries with reads aligning to Coronaviridae. c Histogram of all detected sOTUs classified to Riboviria order (>40% amino acid identity to a named species) with unclassified sOTUs not shown. Segmented bars (left) show the fraction of sOTUs with similarity to known sOTU, binned into intervals 90+ (>=90%, ~species), 75+ (75% to 90%, ~genus), 50+ (50% to 75%, ~family), and <50% (~novel family). Complete multiple sequence alignments and tree files for per-order and per-family trees is available at https://serratus.io/trees.
Extended Data Fig. 4 Genome organization of Coronaviridae and neighbours.
a Length distribution for 11,120 assembled contigs classified as CoV-positive, showing a peak around the typical CoV genome length, 4,179 (37.58%) of contigs also contained a match for RdRP. b Phylogram shown in Figure 3 showing the Mesoniviridae, Tobaniviridae, and Roniviridae outgroups. c Triangular matrix showing median RdRP sequence identities between selected Nidovirales and group-E sequences. d Hidden Markov Model (HMM) protein domain matches from the RdRp in exemplar sequences (contigs or GenBank sequences), grouped by genus. Novel sOTUs identified in this analysis indicated by a coloured circle.
Extended Data Fig. 5 Newly characterized delta virus and delta-virus-like genomes.
Structure and organization of selected examples from the 14 delta virus-, 39 epsilon virus- and 311 zeta virus-like genomes identified in our study. a Similar to human delta virus (HDV), delta virus-like genomes from vertebrates (PmacDV SRR7910143; MmonDV SRR2136906; TgutDV SRR5001850; IchiDV SRR8954566 and BglaDV SRR8242383) and environmental datasets (SRR7286070 and SRR6943136) share similar predicted stable rod-like folding, a predicted ORF coding for the delta antigen (δAg) and a delta ribozyme (dvrbz) on each polarity. Folding of the circular DNA virus Porcine Circovirus 2 (PCV2) and a shuffled MmonDV sequence are shown as negative controls. b Epsilon virus-like genomes detected in invertebrates (SulaEV SRR8739608; GsulEV SRR7170939 and BaerEV SRR12300397) and environmental datasets (SRR8840728 and SRR6943136) show similar structure and organization to delta viruses, with one or two predicted ORFs (epsilon antigen or Ag) and two hammerhead ribozymes (hhrbz) in equivalent genomic regions. c Zeta virus-like genomes detected in invertebrate (Ocassitermes sp. ZVs SRR8924823) and environmental datasets (SRR7286070, SRR6943136, SRR8840728, SRR6201737, SRR5864109 and SRR12063536) are smaller than delta and epsilon agents. Up to 90% of the zeta genomes have sizes multiple of 3 and predicted ORFs without stop codons, capable to encode endless tandem-repeated zeta antigens in both polarities (ζAg+ and ζAg– shown as yellow and red arrows, respectively). Both genomic zeta polarities keep hhrbzs (shown as arrows overlapping the ORFs) similar to the epsilon ribozymes (Extended Fig 6). Larger zeta virus-like genomes (>651 nt) were less abundant (7% of all zeta genomes) and frequently show stop codons, or their sizes are not multiple of 3.
Extended Data Fig. 6 Evolutionary history of delta-virus-like agents.
a Consensus structures (weighted nucleotide conservation threshold of 90%) of delta virus ribozymes, including the 14 genomes described in this work. b Consensus structures of the two hammerhead ribozyme families (type III and extended-type III39) detected in epsilon and zeta agents. Most positions of epsilon and zeta motifs are sequence conserved for each ribozyme family. c MSA of the predicted antigen (N-term domain) from delta and epsilon agents (genomes detected in this study are indicated with a red asterisk). The antiparallel coiled-coil of the HDV is delimited with a grey box, and conserved residues involved in hydrophobic interactions are shown at the bottom40, supporting a highly divergent connection between delta and epsilon genomes. d Human HDV delta virus is known to contain a viroid-like domain related to the Pospiviroidae family of plant viroids. Both families of agents conserve a tertiary structure reminiscent of the E-loop 5S rRNA (nucleotides in green) and are replicated by the RNA Pol II of the host41. Pospiviroids, despite lacking hhrbzs, share with zeta genomes a small rod structure, and in some cases, the presence of predicted endless tandem-repeat ORFs, most notably in both polarities of numerous variants of the Hop Stunt Viroid (HSVd). Whereas viroids have been historically regarded as non-protein-coding RNAs, our reported observations warrant further investigation.
Extended Data Fig. 7 Huge phage and Lak phage detail.
Expanded view of maximum likelihood terminase large subunit protein phylogenetic trees for (a) the expansion of the Kabirphage clade by newly recovered sequences from different animal types (coloured dots). Red branches are public data recovered by Serratus, black branches indicate the previously reported genomes from42. b Publicly available Lak phage genomes98 with sequences of two newly reconstructed complete Lak megaphage genomes. These are the first reported Lak megaphages from dogs (assembled from faecal sample metagenome reads from Allaway et al.99). The genomes have identical terminase sequences (at the nucleotide level) although the dogs were in different housing areas and were sampled at different times (D. Allaway, personal communication).
Supplementary information
Supplementary Table 1
SRA run queries and CoV assembly table: Queries and accessions from this study. a SRA queries to retrieve collections of datasets. b Run accessions, assembly statistics and select metadata for the 11,120 runs for which Coronaviridae, or Coronaviridae-like sequences were assembled. c Assignment of assembled runs to operational taxonomic units (OTUs) based on 97% nucleotide identity of the RNA dependent RNA polymerase (RdRP) domain. d Assignment of GenBank records to RdRP OTUs. e Sub-palmprint RdRP fragments which are possibly novel Coronaviruses (50-90% amino acid identity to a known CoV). Non-overlapping fragments from the same library, or identical overlapping fragments from different libraries are estimated to be the same virus. f Assignment of expected viral host for GenBank records based on Sequence Read Archive and JGI GOLD metadata1,30. g Taxonomic source for RdRP containing assemblies.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Edgar, R.C., Taylor, B., Lin, V. et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 602, 142–147 (2022). https://doi.org/10.1038/s41586-021-04332-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04332-2
This article is cited by
-
Mixed infection of ITPase-encoding potyvirid and secovirid in Mercurialis perennis: evidences for a convergent euphorbia-specific viral counterstrike
Virology Journal (2024)
-
Benchmarking bioinformatic virus identification tools using real-world metagenomic data across biomes
Genome Biology (2024)
-
Uncovered diversity of infectious circular RNAs: A new paradigm for the minimal parasites?
npj Viruses (2024)
-
Indexing and real-time user-friendly queries in terabyte-sized complex genomic datasets with kmindex and ORA
Nature Computational Science (2024)
-
Diverse and abundant phages exploit conjugative plasmids
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
