
Abstract
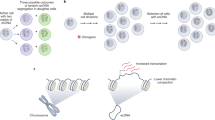
Extrachromosomal DNA (ecDNA) is prevalent in human cancers and mediates high expression of oncogenes through gene amplification and altered gene regulation1. Gene induction typically involves cis-regulatory elements that contact and activate genes on the same chromosome2,3. Here we show that ecDNA hubs—clusters of around 10–100 ecDNAs within the nucleus—enable intermolecular enhancer–gene interactions to promote oncogene overexpression. ecDNAs that encode multiple distinct oncogenes form hubs in diverse cancer cell types and primary tumours. Each ecDNA is more likely to transcribe the oncogene when spatially clustered with additional ecDNAs. ecDNA hubs are tethered by the bromodomain and extraterminal domain (BET) protein BRD4 in a MYC-amplified colorectal cancer cell line. The BET inhibitor JQ1 disperses ecDNA hubs and preferentially inhibits ecDNA-derived-oncogene transcription. The BRD4-bound PVT1 promoter is ectopically fused to MYC and duplicated in ecDNA, receiving promiscuous enhancer input to drive potent expression of MYC. Furthermore, the PVT1 promoter on an exogenous episome suffices to mediate gene activation in trans by ecDNA hubs in a JQ1-sensitive manner. Systematic silencing of ecDNA enhancers by CRISPR interference reveals intermolecular enhancer–gene activation among multiple oncogene loci that are amplified on distinct ecDNAs. Thus, protein-tethered ecDNA hubs enable intermolecular transcriptional regulation and may serve as units of oncogene function and cooperative evolution and as potential targets for cancer therapy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
ChIP–seq, HiChIP, Hi-C, RNA-seq and single-cell multiomics (10x Chromium Single Cell Multiome ATAC + Gene Expression) data generated in this study have been deposited in the GEO and are available under accession number GSE159986. Nanopore sequencing data, WGS data, sgRNA sequencing data and targeted ecDNA sequencing data after CRISPR–Cas9 digestion and PFGE generated in this study have been deposited in the SRA and are available under accession number PRJNA670737. Optical mapping data generated in this study have been deposited in GenBank with BioProject code PRJNA731303. The following publicly available data were also used in this study: TR14 H3K27ac ChIP–seq93 (GEO: GSE90683); COLO320-DM, COLO320-HSR and PC3 WGS1 (SRA: PRJNA506071); SNU16 WGS60 (SRA: PRJNA523380); and HK359 WGS6 (SRA: PRJNA338012). Microscopy image files are available on figshare at https://doi.org/10.6084/m9.figshare.c.5624713.
Code availability
Custom code used in this study is available at https://github.com/ChangLab/ecDNA-hub-code-2021.
References
Wu, S. et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019).
Gorkin, D. U., Leung, D. & Ren, B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14, 762–775 (2014).
Zheng, H. & Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550 (2019).
Bailey, C., Shoura, M. J., Mischel, P. S. & Swanton, C. Extrachromosomal DNA—relieving heredity constraints, accelerating tumour evolution. Ann. Oncol. 31, 884–893 (2020).
Kim, H. et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 52, 891–897 (2020).
Turner, K. M. et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017).
Verhaak, R. G. W., Bafna, V. & Mischel, P. S. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Cancer 19, 283–288 (2019).
Cox, D., Yuncken, C. & Spriggs, A. I. Minute chromatin bodies in malignant tumours of childhood. Lancet 286, 55–58 (1965).
van der Bliek, A. M., Lincke, C. R. & Borst, P. Circular DNA of 3T6R50 double minute chromosomes. Nucleic Acids Res. 16, 4841–4851 (1988).
Hamkalo, B. A., Farnham, P. J., Johnston, R. & Schimke, R. T. Ultrastructural features of minute chromosomes in a methotrexate-resistant mouse 3T3 cell line. Proc. Natl Acad. Sci. 82, 1126–1130 (1985).
Maurer, B. J., Lai, E., Hamkalo, B. A., Hood, L. & Attardi, G. Novel submicroscopic extrachromosomal elements containing amplified genes in human cells. Nature 327, 434–437 (1987).
VanDevanter, D. R., Piaskowski, V. D., Casper, J. T., Douglass, E. C. & Von Hoff, D. D. Ability of circular extrachromosomal DNA molecules to carry amplified MYCN protooncogenes in human neuroblastomas in vivo. J Natl Cancer Inst. 82, 1815–1821 (1990).
Nathanson, D. A. et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76 (2014).
Ståhl, F., Wettergren, Y. & Levan, G. Amplicon structure in multidrug-resistant murine cells: a nonrearranged region of genomic DNA corresponding to large circular DNA. Mol. Cell. Biol. 12, 1179–1187 (1992).
Vicario, R. et al. Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS ONE 10, e0129876 (2015).
Carroll, S. M. et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol. Cell. Biol. 8, 1525–1533 (1988).
Kitajima, K., Haque, M., Nakamura, H., Hirano, T. & Utiyama, H. Loss of irreversibility of granulocytic differentiation induced by dimethyl sulfoxide in HL-60 sublines with a homogeneously staining region. Biochem. Biophys. Res. Commun. 288, 1182–1187 (2001).
Quinn, L. A., Moore, G. E., Morgan, R. T. & Woods, L. K. Cell lines from human colon carcinoma with unusual cell products, double minutes, and homogeneously staining regions. Cancer Res. 39, 4914–4924 (1979).
Storlazzi, C. T. et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 20, 1198–1206 (2010).
Wahl, G. M. The importance of circular DNA in mammalian gene amplification. Cancer Res. 49, 1333–1340 (1989).
Kumar, P. et al. ATAC–seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci. Adv. 6, eaba2489 (2020).
Morton, A. R. et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179, 1330–1341 (2019).
Helmsauer, K. et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 11, 5823 (2020).
Itoh, N. & Shimizu, N. DNA replication-dependent intranuclear relocation of double minute chromatin. J. Cell Sci. 111, 3275–3285 (1998).
Kanda, T., Sullivan, K. F. & Wahl, G. M. Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8, 377–385 (1998).
Oobatake, Y. & Shimizu, N. Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes Chromosomes Cancer 59, 133–143 (2020).
Beliveau, B. J. et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl Acad. Sci. USA 109, 21301–21306 (2012).
Koche, R. P. et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genetics 52, 29–34 (2019).
Parker, S. C. J. et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl Acad. Sci. USA 110, 17921–17926 (2013).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Ren, C. et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc. Natl Acad. Sci. USA 115, 7949–7954 (2018).
Deshpande, V. et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 10, 392 (2019).
Luebeck, J. et al. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat. Commun. 11, 4374 (2020).
Schwab, M., Klempnauer, K. H., Alitalo, K., Varmus, H. & Bishop, M. Rearrangement at the 5′ end of amplified c-myc in human COLO 320 cells is associated with abnormal transcription. Mol. Cell. Biol. 6, 2752–2755 (1986).
L’Abbate, A. et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. 42, 9131–9145 (2014).
Hann, S. R., King, M. W., Bentley, D. L., Anderson, C. W. & Eisenman, R. N. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell 52, 185–195 (1988).
Carramusa, L. et al. The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J. Cell. Physiol. 213, 511–518 (2007).
Cho, S. W. et al. Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell 173, 1398–1412 (2018).
Tolomeo, D., Agostini, A., Visci, G., Traversa, D. & Storlazzi, C. T. PVT1: a long non-coding RNA recurrently involved in neoplasia-associated fusion transcripts. Gene 779, 145497 (2021).
Mumbach, M. R. et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919 (2016).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Park, J. et al. A reciprocal regulatory circuit between CD44 and FGFR2 via c-myc controls gastric cancer cell growth. Oncotarget 7, 28670–28683 (2016).
Furlong, E. E. M. & Levine, M. Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018).
Zhu, Y. et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 39, 694–707 (2021).
Xue, K. S., Hooper, K. A., Ollodart, A. R., Dingens, A. S. & Bloom, J. D. Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. Elife 5, e13974 (2016).
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Henssen, A. et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin. Cancer Res. 22, 2470–2481 (2016).
Xie, L. et al. 3D ATAC-PALM: super-resolution imaging of the accessible genome. Nat. Methods 17,430–436 (2020).
Ambros, P. F. et al. International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br. J. Cancer 100, 1471–1482 (2009).
Balaban-Malenbaum, G. & Gilbert, F. Double minute chromosomes and the homogeneously staining regions in chromosomes of a human neuroblastoma cell line. Science 198, 739–741 (1977).
Marrano, P., Irwin, M. S. & Thorner, P. S. Heterogeneity of MYCN amplification in neuroblastoma at diagnosis, treatment, relapse, and metastasis. Genes Chromosomes Cancer 56, 28–41 (2017).
Villamón, E. et al. Genetic instability and intratumoral heterogeneity in neuroblastoma with MYCN amplification plus 11q deletion. PLoS ONE 8, e53740 (2013).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Rajkumar, U. et al. EcSeg: semantic segmentation of metaphase images containing extrachromosomal DNA. Iscience 21, 428–435 (2019).
Veatch, S. L. et al. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS ONE 7, e31457 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114 (2014).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Normanno, D. et al. Probing the target search of DNA-binding proteins in mammalian cells using TetR as model searcher. Nat. Commun. 6, 7357 (2015).
Mirkin, E. V., Chang, F. S. & Kleckner, N. Protein-mediated chromosome pairing of repetitive arrays. J. Mol. Biol. 426, 550–557 (2014).
Grimm, J. B. et al. A general method to optimize and functionalize red-shifted rhodamine dyes. Nat. Methods 17, 815–821 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Overhauser, J. in Pulsed-Field Gel Electrophoresis, Methods in Molecular Biology Vol. 12 (eds. Burmeister, M. & Ulanovsky, L.) 129–134 (Humana Press, 1992).
Picelli, S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014).
Corces, M. R. et al. An improved ATAC–seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873 (2016).
Raeisi Dehkordi, S., Luebeck, J. & Bafna, V. FaNDOM: fast nested distance-based seeding of optical maps. Patterns 2, 100248 (2021).
Haas, B. J. et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 20, 213 (2019).
Hahne, F. & Ivanek, R. in Statistical Genomics, Methods in Molecular Biology Vol. 1418 (eds. Mathé, E. & Davis, S.) 335–351 (Humana Press, 2016).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Satpathy, A. T. et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019).
Mumbach, M. R. et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 49, 1602–1612 (2017).
Mumbach, M. R. et al. HiChIRP reveals RNA-associated chromosome conformation. Nat. Methods 16, 489–492 (2019).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Bhattacharyya, S., Chandra, V., Vijayanand, P. & Ay, F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 10, 4221 (2019).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Vidal, E. et al. OneD: increasing reproducibility of Hi-C samples with abnormal karyotypes. Nucleic Acids Res. 46, e49 (2018).
Flynn, R. A. et al. Discovery and functional interrogation of SARS-CoV-2 RNA–host protein interactions. Cell 184, 2394–2411 (2021).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Scheinin, I. et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014).
Hadi, K. et al. Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs. Cell 183, 197–210 (2020).
Blumrich, A. et al. The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum. Mol. Genet. 20, 1488–1501 (2011).
Gogolin, S. et al. CDK4 inhibition restores G1-S arrest in MYCN-amplified neuroblastoma cells in the context of doxorubicin-induced DNA damage. Cell Cycle 12, 1091–1104 (2013).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Knight, P. A. & Ruiz, D. A fast algorithm for matrix balancing. IMA J. Numer. Anal. 33, 1029–1047 (2013).
Boeva, V. et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 49, 1408–1413 (2017).
Acknowledgements
We thank members of the Chang, Liu, Mischel, and Bafna laboratories for discussions; R. Zermeno, M. Weglarz and L. Nichols at the Stanford Shared FACS Facility for assistance with cell sorting experiments; X. Ji, D. Wagh and J. Coller at the Stanford Functional Genomics Facility for assistance with high-throughput sequencing; and A. Pang of Bionano Genomics for assistance with optical mapping. H.Y.C. was supported by NIH R35-CA209919 and RM1-HG007735; K.L.H. was supported by a Stanford Graduate Fellowship; and K.E.Y. was supported by the National Science Foundation Graduate Research Fellowship Program (NSF DGE-1656518), a Stanford Graduate Fellowship and a NCI Predoctoral to Postdoctoral Fellow Transition Award (NIH F99CA253729). Cell sorting for this project was done on instruments in the Stanford Shared FACS Facility. Sequencing was performed by the Stanford Functional Genomics Facility (supported by NIH grants S10OD018220 and 1S10OD021763). Microscopy was performed on instruments in the UCSD Microscopy Core (supported by NINDS NS047101). A.G.H. is supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) (398299703) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 949172). Z.L. is a Janelia Group Leader, and H.Y.C. and R.T. are Investigators of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
K.L.H., K.E.Y. and H.Y.C. conceived the project. K.L.H. performed and analysed CRISPRi and in vitro ecDNA digestion and PFGE experiments, and analysed single-cell multiomics, RNA-seq and ATAC–seq experiments. K.E.Y. performed and analysed metaphase DNA FISH imaging, ChIP–seq, HiChIP, WGS, COLO320-DM nanopore sequencing and JQ1 perturbation experiments. L.X. performed and analysed interphase DNA and RNA FISH imaging, TetO-eGFP cell line generation and live-cell imaging, and PVT1p-nLuc imaging experiments. Q.S. performed and analysed all luciferase reporter experiments, with the exception of PVT1p-nLuc RNA and DNA FISH, and helped with CRISPRi experiments. K.H. and R.S. analysed TR14 Hi-C data and amplicon reconstruction. J.L. and S.R.D. analysed COLO320-DM WGS, nanopore sequencing, optical mapping data and amplicon reconstruction. J.T.L., S.W., C.C. and J.T. performed and analysed DNA FISH imaging. R.C.G. generated Hi-C, DNA FISH, WGS and nanopore sequencing data for TR14. N.E.W. performed and analysed small-molecule inhibitor experiments and DNA FISH imaging after MS645 treatment. M.E.V. performed Hi-C experiments and data analysis for TR14. I.T.-L.W. performed metaphase DNA FISH imaging. C.V.D. performed and analysed ChIP–seq experiments. K.K. performed HiChIP experiments. J.A.B. helped with CRISPRi experimental design and cloning of the sgRNA pool. R.L. performed RNA-seq experiments. U.R. analysed metaphase DNA FISH data. J.F. generated COLO320-DM WGS data. M.R.C. and J.M.G. wrote the HiChIP data processing pipeline. M.R.C., J.C.R., A.B., A.T.S., R.T., S.M., V.B., A.G.H., P.S.M., Z.L. and H.Y.C. guided data analysis and provided feedback on experimental design. K.L.H., K.E.Y. and H.Y.C. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio and Cartography Biosciences, and an advisor of 10x Genomics, Arsenal Biosciences and Spring Discovery. P.S.M. is a co-founder of Boundless Bio. He has equity and chairs the scientific advisory board, for which he is compensated. V.B. is a co-founder and advisor of Boundless Bio. A.T.S. is a founder of Immunai and Cartography Biosciences. K.E.Y. is a consultant for Cartography Biosciences.
Additional information
Peer review information Nature thanks Charles Lin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 ecDNA FISH strategies and copy number estimation.
a, WGS tracks with DNA FISH probe locations. For COLO320-DM and PC3, a 1.5 Mb MYC FISH probe (Fig. 1a, b), a 100 kb MYC FISH probe (Fig. 1d–f), or a 1.5 Mb chromosome 8 FISH probe was used. Commercial probes were used in SNU16 and HK359 cells. b, Representative DNA FISH image using chromosomal and 1.5 Mb MYC probes in non-ecDNA amplified HCC1569 showing paired signals as expected from the chromosomal loci. c, ecDNA clustering of individual COLO320-DM cells by autocorrelation g(r). d, Representative FISH images showing ecDNA clustering in primary neuroblastoma tumours (patients 11 and 17). e, ecDNA clustering of individual primary tumour cells from all three patients using autocorrelation g(r). f, Comparison of MYC copy number in COLO320-DM calculated based on WGS (n=7 genomic bins overlapping with DNA FISH probes), metaphase FISH (n=82 cells) and interphase FISH (n=47 cells). P-values determined by two-sided Wilcoxon test. g, Representative images of nascent MYC RNA FISH showing overlap of nascent RNA (intronic) and total RNA (exonic) FISH probes in PC3 cells (independently repeated twice). h, Representative images from combined DNA FISH for MYC ecDNA (100 kb probe) and chromosomal DNA with nascent MYC RNA FISH in COLO320-DM cells (independently repeated four times). i, MYC transcription probability measured by nascent RNA FISH normalized to DNA copy number by FISH comparing singleton ecDNAs to those found in hubs in COLO320-DM (box centre line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). To control for noise in transcriptional probability for small numbers of ecDNAs, we randomly re-sampled RNA FISH data grouped by hub size and calculated transcription probability. The violin plot represents transcriptional probability per ecDNA hub based on the hub size matched sampling. P-value determined by two-sided Wilcoxon test.
Extended Data Fig. 2 Generation of TetR-GFP COLO320-DM cells for ecDNA imaging in live cells.
a, ecDNA imaging based on TetO array knock-in and labelling with TetR-eGFP (left). Representative images of TetR-eGFP signal in TetO-eGFP COLO320-DM cells at indicated timepoints in a time course (right; independently repeated twice). b, GFP signal in ecDNA-TetO COLO320-DM cells. TetR-eGFP and monomeric TetR-eGFP(A206K)-labelled ecDNA hubs appear to be smaller in living cells than in DNA FISH studies of fixed cells, probably because the TetO array is not integrated in all ecDNA molecules and there are potential differences caused by denaturation during DNA FISH and eGFP dimerization. c, ecDNA hub diameter in microns (box centre line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). Tet-eGFP-labelled hubs are slightly smaller than monomeric TetR-eGFP(A206K)-labelled hubs, potentially due to eGFP dimerization effects (Methods). P-value determined by two-sided Wilcoxon test. d, ecDNA hub number per cell. Line represents median. P-value determined by two-sided Wilcoxon test. e, TetR-eGFP signal in chr8-chromosomal-TetO (chr8:116,860,000–118,680,000, left) and ecDNA-TetO (TetO-eGFP COLO320-DM, right) COLO320-DM cells. f, Fluorescence intensity for chr8-chromosomal-TetO and ecDNA-TetO foci. g, h, Inferred ecDNA copy number per foci (g; n = number of foci/cell) and per cell (h; n = number of cells) for ecDNA-TetO labelled cells based on summed fluorescence intensity relative to chr8-chromosomal-TetO foci. Line represents median. i, Representative images of TetR-GFP signal in parental COLO320-DM without TetO array integration which shows minimal TetR-GFP foci. j, Mean fluorescence intensities for ecDNA (TetO-eGFP) and BRD4 (HaloTag) foci across a line drawn across the centre of the largest ecDNA (TetO-eGFP) signal. Data are mean ± SEM for n=5 ecDNA foci. k, Representative image of TetR-eGFP signal in COLO320-DM cells without TetO array integration overlaid with BRD4-HaloTag signal. Dashed line indicates nucleus boundary. We noted cytoplasmic TetR-eGFP signal in a subset of COLO320-DM cells without TetO array integration but it did not colocalize with BRD4-HaloTag. l, MYC RNA measured by RT–qPCR for parental COLO320-DM and BRD4-HaloTag COLO320-DM cells treated with DMSO or 500 nM JQ1 for 6 h which shows similar levels of MYC transcription and sensitivity to JQ1 inhibition following epitope tagging of BRD4. Data are mean ± SD between 3 biological replicates. P values determined by two-sided Student’s t-test.
Extended Data Fig. 3 BET inhibition leads to ecDNA hub dispersal.
a, Representative metaphase FISH images and schematic showing ecDNA in COLO320-DM and chromosomal HSRs in COLO320-HSR (independently repeated twice for COLO320-DM and not repeated for COLO320-HSR). b, Ranked BRD4 ChIP–seq signal. Peaks in ecDNA or HSR amplifications are highlighted and labelled with nearest gene. c, ATAC–seq, BRD4 ChIP–seq, H3K27ac ChIP–seq and WGS at amplified MYC locus. d, Number of ecDNA locations (including ecDNA hubs with >1 ecDNA and singleton ecDNAs) from interphase FISH imaging for individual COLO320-DM cells after treatment with DMSO or 500 nM JQ1 for 6 h. N = number of cells quantified per condition. P-value determined by two-sided Wilcoxon test. e, ecDNA copies in each ecDNA location from interphase FISH imaging in COLO320-DM after treatment with DMSO or 500 nM JQ1 for 6 h (box centre line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). N = number of ecDNA locations quantified per condition. P-value determined by two-sided Wilcoxon test. f, Representative live images of TetR-eGFP-labelled ecDNA after treatment with DMSO or 500 nM JQ1 at indicated timepoints in a time course (top; independently repeated twice) and ecDNA hub zoom-ins (bottom). g, Representative image from combined DNA/RNA FISH in COLO320-DM cells treated with DMSO, 500 nM JQ1, or 1% 1,6-hexanediol for 6 h. h, MYC transcription probability measured by dual DNA/RNA FISH after treatment with DMSO, 1% 1,6-hexanediol, or 100 µg/mL alpha-amanitin for 6 h (box centre line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range; n = number of cells). P-values determined by two-sided Wilcoxon test. i, Representative DNA FISH images for MYC ecDNA in interphase COLO320-DM treated with either 1% 1,6-hexanediol or 100 µg/mL alpha-amanitin for 6 h. j, ecDNA clustering in interphase cells by autocorrelation g(r) for COLO320-DM treated with DMSO, 1% 1,6-hexanediol, or 100 µg/mL alpha-amanitin for 6 h. Data are mean ± SEM (n = 10 cells quantified per condition). k, Averaged BRD4 ChIP–seq signal and heat map over all BRD4 peaks for cells treated with DMSO or 500 nM JQ1 for 6 h. l, Cell viability measured by ATP levels (CellTiterGlo) after treatment with different JQ1 concentrations for 48 h normalized to DMSO-treated cells. Data are mean ± SD between 3 biological replicates. P values determined by two-sided Student’s t-test. m, Cell proliferation after treatment with different JQ1 concentrations over 72 h. Data are mean ± SD between 3 biological replicates. n, Cell doubling times after treatment with different JQ1 concentrations over 72 h in hours (top) or after normalization to DMSO-treated cells (bottom). Data are mean ± SD between 3 biological replicates. P values determined by two-sided Student’s t-test. o, MYC RNA measured by RT–qPCR after treatment with indicated inhibitors for 6 h (top; each point represents a biological replicate, n=6 for DMSO and JQ1 treatments, n=3 for all other drug treatments). Data are mean ± SD. P values determined by two-sided Student’s t-test. Details of inhibitor panel, protein target, significance of effect on MYC transcription, and comparison of effect on ecDNA and HSR transcription (bottom). p, q, Representative DNA FISH images (p) and clustering by autocorrelation g(r) (q) for MYC ecDNAs in COLO320-DM treated with DMSO or 500 nM MS645 for 6 h. Data are mean ± SEM. P-value determined by two-sided Wilcoxon test at radius = 0.
Extended Data Fig. 4 Reconstruction of COLO320-DM ecDNA amplicon structure.
a, Structural variant (SV) view of AmpliconArchitect (AA) reconstruction of the MYC amplicon in COLO320-DM cells. b, Nanopore sequencing of COLO320-DM cells (left) and distribution of read lengths. c, WGS for COLO320-DM with junctions detected by WGS and nanopore sequencing. d, Molecule lengths used for optical mapping and statistics. e, Reconstructed COLO320-DM ecDNA after integrating WGS, optical mapping, and in-vitro ecDNA digestion. Chromosomes of origin and corresponding coordinates (hg19) are labelled. Three inner circular tracks (light tan, slate and brown in colour; guides A, B and C, respectively) representing expected fragments as a result of Cas9 cleavage using three distinct sgRNAs and their expected sizes. Guide sequences are in Supplementary Table 2 (PFGE_guide_A-C). f, In-vitro Cas9 digestion of COLO320-DM ecDNA followed by PFGE (left). Fragment sizes were determined based on H. wingei and S. cerevisiae ladders. Uncropped gel image is in Supplementary Fig. 1. Middle panel shows short-read sequencing of the MYC ecDNA amplicon for all isolated fragments, ordered by fragment size. Right panel shows concordance of expected fragment sizes by optical mapping reconstruction, and observed fragment sizes by in-vitro Cas9 digestion (discordant fragments circled). Each sgRNA digestion was performed in one independent experiment. g, Metaphase FISH images showing colocalization of MYC, PCAT1 and PLUT as predicted by optical mapping and in-vitro digestion. N = 20 cells and 1,270 ecDNAs quantified for MYC/PCAT1 DNA FISH and n = 15 cells and 678 ecDNAs for MYC/PLUT DNA FISH from one experiment. h, RNA expression measured by RT–qPCR for indicated transcripts in COLO320-DM cells stably expressing dCas9-KRAB and indicated sgRNAs (n=2 biological replicates). Canonical MYC was amplified with primers MYC_exon1_fw and MYC_exon2_rv; fusion PVT1-MYC was amplified with PVT1_exon1_fw and MYC_exon2_rv; total MYC was amplified with total_MYC_exon2_fw and total_MYC_exon2_rv. All primer sequences are in Supplementary Table 1 and guide sequences are in Supplementary Table 2. i, Alignment of junction reads at the PVT1-MYC breakpoint.
Extended Data Fig. 5 Single-cell multiomic analysis reveals combinatorial and heterogeneous ecDNA regulatory element activities associated with MYC expression.
a, Joint single-cell RNA and ATAC–seq for simultaneously assaying gene expression and chromatin accessibility and identifying regulatory elements associated with MYC expression. b, Unique ATAC–seq fragments and RNA features for cells passing filter (both log2-transformed). c, Correlation between MYC accessibility score and normalized RNA expression. d, UMAP from the RNA or the ATAC–seq data (left). Log-normalized and scaled MYC RNA expression (top right) and MYC accessibility scores (bottom right) were visualized on the ATAC–seq UMAP, showing cell-level heterogeneity in MYC RNA-seq and ATAC-seq signals in ecDNA-containing COLO320-DM. e, Gene expression scores (calculated using Seurat in R) of MYC-upregulated genes (Gene Set M6506, Molecular Signatures Database; MSigDB) across all MYC RNA quantile bins. Horizontal line marks median. Population variances for all individual cells are shown (top). P-value determined by two-sided F-test. f, MYC expression levels of top and bottom bins (left). Normalized ATAC–seq coverages are shown (right). g, Number of variable elements identified on COLO320-DM ecDNAs compared to chromosomal HSRs in COLO320-HSR (left). 45 variable elements were uniquely observed on ecDNA. All variable elements on ecDNA are shown on the right (y-axis shows -log10(FDR) and dot size represents log2 fold change. Five most significantly variable elements are highlighted and named based on relative position in kb to the MYC TSS (negative, 5′; positive, 3′). h, Correlation between estimated MYC copy numbers and normalized log2-transformed MYC expression of all individual cells showing a high level of copy number variability associated with increased expression, in particular for COLO320-DM. i, Estimated MYC amplicon copy number of all cell bins separated by MYC RNA expression. j, Zoom-ins of the ATAC–seq coverage of each of the five most significantly variable elements identified in g (marked by dashed boxes). k, Similar distributions of TSS enrichment in the high and low cell bins, indicating differences in accessibility at variable elements are not an artifact of differences in data quality. l, Mean copy number regressed, log-normalized, scaled ATAC–seq coverage of the differential peaks against mean MYC RNA (log-normalized, mean-centred, scaled) for each cell bin in orange. Same number of random non-differential peaks from the same amplicon interval and shown in grey. Error bands show 95% confidence intervals for the linear models. m, Cumulative probability of MYC amplicon copy number distributions (mean-centred, scaled) of single-cell ATAC–seq data and DNA FISH data, suggesting that copy number estimates from single cell ATAC-seq data reflect heterogeneity in ecDNA copy number as measured by DNA FISH. P-values determined by Kolmogorov-Smirnov test (1,000 bootstrap simulations).
Extended Data Fig. 6 Endogenous enhancer connectome of COLO320-DM MYC ecDNA amplicon and effect of promoter sequence, cis enhancers, and BET inhibition on episomal reporter activation.
a, Top to bottom: COLO320-DM H3K27ac HiChIP contact map (KR-normalized read counts, 10-kb resolution), reconstructed COLO320-DM amplicon, H3K27ac ChIP–seq signal, BRD4 ChIP–seq signal, WGS coverage, interaction profile of PVT1 (top, dark pink) and MYC (bottom, light pink) promoters at 10-kb resolution with FitHiChIP loops shown below, coloured by adjusted p-value. Active elements identified by scATAC and overlapping H3K27ac HiChIP contacts named by genomic distance to MYC start site: −1132E, −1087E, −679E, −655E, −401E, −328E, −85E. b, Comparison of HiChIP matrix normalization methods for COLO320-DM H3K27ac HiChIP at 10-kb resolution. HiChIP signal is robust to different normalization methods. c, Quantification of NanoLuc luciferase signal for plasmids with PVT1p-, minp-, or MYCp-driven NanoLuc reporter expression. Luciferase signal was calculated by normalizing NanoLuc readings to Firefly readings. Bar plot shows mean ± SEM. P values were calculated using a two-sided Student’s t-test (n=3 biological replicates). d, Violin plots showing mean fluorescence intensities and signal sizes of the NanoLuc reporter RNA in PVT1p-reporter and minp-reporter transfected cells. P-values were calculated using a two-sided Wilcoxon test. e, Schematic of PVT1 promoter-driven luciferase reporter plasmid with a cis-enhancer. Details of cis-enhancer are in Methods. f, Bar plot showing luciferase signal driven by PVT1p, MYCp or the constitutive TKp with or without a cis-enhancer (mean ± SEM). All values are normalized to the corresponding promoter-only construct without a cis-enhancer. P values were calculated using a two-sided Student’s t-test (n=3 biological replicates). g, Dot plots showing fold change in luciferase signal (Firefly-normalized NanoLuc signal) in JQ1-treated over DMSO-treated COLO320-DM and COLO320-HSR cells after transfection with the PVT1p or the MYCp plasmid with or without a cis-enhancer. P values were calculated using a two-sided Student’s t-test (n=3 biological replicates).
Extended Data Fig. 7 Generation of monoclonal SNU16-dCas9-KRAB with reduced ecDNA fusions.
a, Representative DNA FISH images showing extrachromosomal single-positive MYC and FGFR2 amplifications (top left and top middle) and double-positive MYC and FGFR2 amplifications in metaphase spreads in parental SNU16 cells (top right) with zoom in (top right). N = 42 cells and 8,222 ecDNAs. Representative DNA FISH images showing distinct extrachromosomal MYC and FGFR2 amplifications in metaphase spreads in SNU16-dCas9-KRAB cells (bottom). N = 29 cells and 3,893 ecDNAs. b, Ranked plot showing number of junction reads supporting each breakpoint in AmpliconArchitect. Breakpoints are coloured based on whether they span regions from the same amplicon (MYC/FGFR2) or regions from two distinct amplicons. c, HiChIP contact matrices at 10-kb resolution with KR normalization for parental SNU16 cell line (left) and SNU16-dCas9-KRAB cell line (right). Contact matrix for parental cells contains regions of increased cis-contact frequency between chr8 and chr10 as indicated, as compared to SNU16-dCas9-KRAB cells with highly reduced contact frequency between chr8 and chr10. Regions of increased focal interaction overlapping low frequency structural rearrangements between chr8 and chr10 described in b indicated with boxes.
Extended Data Fig. 8 Perturbations of ecDNA enhancers by CRISPRi reveal functional intermolecular enhancer–gene interactions.
a, CRISPRi experiments perturbing candidate enhancers in SNU16-dCas9-KRAB cells. Single-guide RNAs (sgRNAs) were designed to target candidate enhancers on FGFR2 and MYC ecDNAs based on chromatin accessibility. b, Experimental workflow for pooled CRISPRi repression of putative enhancers. Stable SNU16-dCas9-KRAB cells were generated from a single cell clone. Cells were transduced with a lentiviral pool of sgRNAs, selected with antibiotics and oncogene RNA was assessed by flowFISH. Cells were sorted into six bins by fluorescence-activated cell sorting (FACS) based on oncogene expression. sgRNAs were quantified for cells in each bin. c, FACS gating strategy. d, Log2 fold changes of sgRNAs for each candidate enhancer element compared to unsorted cells for CRISPRi libraries targeting either MYC or FGFR2 ecDNAs, followed by cell sorting based on expression levels of MYC or FGFR2. Each dot represents the mean log2 fold change of 20 sgRNAs targeting a candidate element. Elements negatively correlated with oncogene expression as compared to the negative control sgRNA distributions in the same pools are marked in red. e, Bar plot showing significance of CRISPRi repression of candidate enhancer elements as in Fig. 4e (top). Significant in-trans and in-cis enhancers are coloured as indicated. SNU16-dCas9-KRAB H3K27ac HiChIP 1D signal track and interaction profiles of FGFR2 and MYC promoters at 10-kb resolution with cis FitHiChIP loops shown below. Interaction profiles in cis shown in purple and in trans shown in orange. f, Spearman correlations of individual sgRNAs that target MYC TSS across fluorescence bins corresponding to MYC and FGFR2 expression. P values using the lower-tailed t-test comparing target sgRNAs with negative control sgRNAs (negcontrols) are shown. Each dot represents an independent sgRNA.
Extended Data Fig. 9 Intermolecular enhancers and MYC are located on distinct molecules for the vast majority of ecDNAs.
a, Top: two-colour DNA FISH on metaphase spreads for quantifying the frequency of colocalization of the MYC gene and intermolecular enhancers shown in Fig. 4e. Above-random colocalization would indicate fusion events. Bottom: representative DNA FISH images. DNA FISH probes target the following hg19 genomic coordinates: E1, chr10:122,635,712–122,782,544 (RP11-95I16; n = 11 cells); E2, chr10:122,973,293–123,129,601 (RP11-57H2; n = 12 cells); E3/E4/E5, chr10:123,300,005–123,474,433 (RP11-1024G22; n = 10 cells). b, Top: numbers of distinct and colocalized FISH signals. To estimate random colocalization, 100 simulated images were generated with matched numbers of signals and mean simulated frequencies were compared with observed colocalization. P values determined by two-sided t-test (Bonferroni-adjusted). Bottom: number of colocalized signals significantly above random chance. Colocalization above simulated random distributions is the sum of colocalized molecules in excess of random means in all FISH images in which total colocalization was above the random mean plus 95% confidence interval (100 simulated images per FISH image). c, In vitro Cas9 digestion of MYC-containing ecDNA in SNU16-dCas9-KRAB followed by PFGE (one independent experiment). Fragment sizes were determined based on H. wingei and S. cerevisiae ladders. Uncropped gel image is in Supplementary Fig. 1. MYC CDS guide corresponds to guide B in Supplementary Table 2. d, Enrichment of enhancer DNA sequences in isolated MYC ecDNAs bands from c over background (DNA isolated from a separate PFGE lane in the corresponding size range resulting from undigested genomic DNA) based on normalized reads in 5kb windows. Each dot represents DNA from a distinct gel band. Red indicates fold change above 4. e, Sequencing track for a gel-purified MYC ecDNA showing enrichment of the MYC amplicon and depletion of the FGFR2 amplicon containing enhancers E1-E5.
Extended Data Fig. 10 Reconstruction of four distinct amplicons in TR14 neuroblastoma cell line and intermolecular amplicon interaction patterns associated with H3K27ac marks.
a, Top to bottom: long read-based reconstruction of four different amplicons; genome graph with long read-based structural variants of >10kb size and >20 supporting reads indicated by red edges; copy number variation and coverage from short-read whole-genome sequencing, positions of the selected genes. b, A representative DNA FISH image of MYCN ecDNAs in interphase TR14 cells (top) and ecDNA clustering compared to DAPI control in the same cells assessed by autocorrelation g(r) (bottom). Data are mean ± SEM (n = 14 cells). c, Custom Hi-C map of reconstructed TR14 amplicons. The MYCN/CDK4 amplicon and the MYCN ecDNA share sequences, which prevented an unambiguous short-read mapping in these regions and appear as white areas. Trans interactions appear locally elevated between MYCN ecDNA and ODC1 amplicon (indicated by arrows). Cis- and trans-contact frequencies are coloured as indicated. d, Read support for structural variants identified by long read sequencing linking amplicons. Only one structural variant between distinct amplicons (MYCN and MDM2 amplicons) was identified with 3 supporting reads. e, Variant allele frequency for structural variants linking amplicons. f, Trans-interaction pattern between enhancers on a MYCN amplicon fragment (vertical) and an ODC1 amplicon fragment (horizontal). Short-read WGS coverage (grey), H3K27ac ChIP–seq track showing mean fold change over input in 1kb bins (yellow) and Hi-C contact map showing (KR-normalized counts in 5kb bins). g, Top to bottom: three amplicon reconstructions, virtual 4C interaction profile of the enhancer-rich HPCAL1 locus on the ODC1 amplicon with loci on other amplicons (red), and H3K27ac ChIP–seq (fold change over input; yellow). h, Trans interaction between different amplicons (KR-normalized counts in 5kb bins) depending on H3K27ac signal of the interaction loci (left; box centre line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). Trans interaction (KR-normalized counts in 5kb bins) separated by amplicon pair (right). H3K27ac High vs. Low denotes at least vs. less than 3-fold mean enrichment over input in 5kb bins. N = 114,636 H3K27ac Low + Low pairs, n = 11,990 H3K27ac High + Low pairs, n = 296 H3K27ac High + High pairs.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1 and 2 and accompanying legends for Supplementary Tables 1–3.
Supplementary Figure 1
Raw images of agarose gels. Related to Extended Data Figs. 4f, 9c.
Supplementary Table 3
See Supplementary Information for Supplementary Table 3 legend.
Supplementary Video 1
Live-cell imaging with untreated TetO–eGFP COLO320-DM cells. Snapshots of an untreated cell are shown over the course of 30 minutes. GFP labels TetO-knock-in MYC ecDNAs.
Supplementary Video 2
Live-cell imaging with DMSO-treated TetO–eGFP COLO320-DM cells. A control cell treated with DMSO was tracked over the course of 1 hour. GFP labels TetO-knock-in MYC ecDNAs.
Supplementary Video 3
Live-cell imaging with TetO-GFP COLO320-DM cells after JQ1 treatment. A cell treated with 500 nM JQ1 was tracked over the course of 1 hour. GFP labels TetO-knock-in MYC ecDNAs.
Rights and permissions
About this article

Cite this article
Hung, K.L., Yost, K.E., Xie, L. et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600, 731–736 (2021). https://doi.org/10.1038/s41586-021-04116-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04116-8
This article is cited by
-
MUC20 regulated by extrachromosomal circular DNA attenuates proteasome inhibitor resistance of multiple myeloma by modulating cuproptosis
Journal of Experimental & Clinical Cancer Research (2024)
-
Aneuploidy and complex genomic rearrangements in cancer evolution
Nature Cancer (2024)
-
Machine learning-based extrachromosomal DNA identification in large-scale cohorts reveals its clinical implications in cancer
Nature Communications (2024)
-
scCircle-seq unveils the diversity and complexity of extrachromosomal circular DNAs in single cells
Nature Communications (2024)
-
Extrachromosomal DNA in cancer
Nature Reviews Cancer (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
