
Abstract
A safe and effective vaccine against COVID-19 is urgently needed in quantities that are sufficient to immunize large populations. Here we report the preclinical development of two vaccine candidates (BNT162b1 and BNT162b2) that contain nucleoside-modified messenger RNA that encodes immunogens derived from the spike glycoprotein (S) of SARS-CoV-2, formulated in lipid nanoparticles. BNT162b1 encodes a soluble, secreted trimerized receptor-binding domain (known as the RBD–foldon). BNT162b2 encodes the full-length transmembrane S glycoprotein, locked in its prefusion conformation by the substitution of two residues with proline (S(K986P/V987P); hereafter, S(P2) (also known as P2 S)). The flexibly tethered RBDs of the RBD–foldon bind to human ACE2 with high avidity. Approximately 20% of the S(P2) trimers are in the two-RBD ‘down’, one-RBD ‘up’ state. In mice, one intramuscular dose of either candidate vaccine elicits a dose-dependent antibody response with high virus-entry inhibition titres and strong T-helper-1 CD4+ and IFNγ+CD8+ T cell responses. Prime–boost vaccination of rhesus macaques (Macaca mulatta) with the BNT162b candidates elicits SARS-CoV-2-neutralizing geometric mean titres that are 8.2–18.2× that of a panel of SARS-CoV-2-convalescent human sera. The vaccine candidates protect macaques against challenge with SARS-CoV-2; in particular, BNT162b2 protects the lower respiratory tract against the presence of viral RNA and shows no evidence of disease enhancement. Both candidates are being evaluated in phase I trials in Germany and the USA1,2,3, and BNT162b2 is being evaluated in an ongoing global phase II/III trial (NCT04380701 and NCT04368728).
Similar content being viewed by others

Main
Owing to the effects of the current pandemic of coronavirus disease 2019 (COVID-19) on human health and society, several collaborative research programmes have been launched and have generated insights and progress in vaccine development. Soon after emerging in December 2019, SARS-CoV-2 was identified as a betacoronavirus with high sequence similarity to bat-derived SARS-like coronaviruses4,5. The fast availability of vaccines is critical in the pandemic, and the rapid globalized response is mirrored by the upload of over 212,000 viral genome sequences as of 23 November 2020 to the Global Initiative on Sharing All Influenza Data.
The trimeric S of SARS-CoV-2 is a key target for virus-neutralizing antibodies6 and the prime candidate for vaccine development. S binds its cellular receptor ACE2 through an RBD, which is part of S1 (the N-terminal furin cleavage fragment of S)7,8. On S, the RBDs are in ’up’ positions, in which the receptor-binding sites and their dense cluster of neutralizing epitopes are exposed, or in ‘down’ positions, in which the receptor-binding sites are buried but some S neutralizing epitopes on and off the RBDs remain available9,10,11,12. S rearranges to translocate the virus into cells by membrane fusion9,13. The C-terminal furin cleavage fragment of S (S2) contains the fusion machinery14.
Messenger RNA (mRNA) technology allows the versatile design of vaccine antigens as well as highly scalable and fast manufacturing. With efficient lipid-nanoparticle-formulation processes, RNA vaccines are highly suited to the rapid development and supply needed during a pandemic15. RNA generated from DNA templates by a highly productive, cell-free in vitro transcription process is molecularly well-defined and free of materials of animal origin. Here we report the preclinical development of lipid-nanoparticle-formulated, N1-methyl-pseudouridine (m1Ψ) nucleoside-modified mRNA (modRNA) BNT162b vaccine candidates (BNT162b1 and BNT162b2) that encode immunogens derived from the S of SARS-CoV-2 (Fig. 1a). The m1Ψ modification dampens innate immune sensing and—together with optimized noncoding sequence elements—increases the efficiency of RNA translation in vivo16,17,18. Vaccines based on modRNA have proven to be immunogenic for several viral targets19,20.

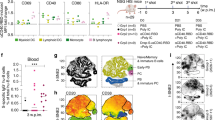
a, Structure of BNT162b1 and BNT162b2 RNA. UTR, untranslated region; SP, signal peptide. The proline subsitutions of S(P2) (K986P and V897P) are indicated. b, Liquid capillary electropherograms of in vitro-transcribed BNT162b1 and BNT162b2 RNA. Peaks represent individual samples merged into one graph. c, Representative 2D class averages from electron microscopy of negatively stained RBD–foldon trimers. Box edge, 37 nm. d, Two-dimensional class average from cryo-EM of the ACE2–B0AT1–RBD–foldon trimer complex. Long box edge, 39.2 nm. Peripheral to the relatively well-defined density of each RBD domain bound to ACE2, there is diffuse density that we attribute to the remainder of the flexibly tethered RBD–foldon trimer. A detergent micelle forms the density at the end of the complex opposite the RBD–foldon. e, Density map of the ACE2–B0AT1–RBD–foldon trimer complex at 3.24 Å, after focused refinement of the ACE2 extracellular domain bound to a RBD monomer. Surface colour-coding is by subunit. The ribbon model refined to the density shows the RBD–ACE2 binding interface.Residues that potentially mediate polar interactions are labelled. f, A 3.29 Å cryo-EM map of S(P2) with fitted and refined atomic model, viewed down the threefold axis towards the membrane (left) and viewed perpendicular to the threefold axis (right). The map is coloured by protomer. g, Mass density map of TwinStrep-tagged S(P2) produced by 3D classification of images extracted from cryo-EM micrographs with no symmetry averaging, showing the class in the one-RBD-up and two-RBD-down position.
Both of the BNT162b vaccines are being evaluated in phase I clinical trials in the USA (NCT04368728) and Germany (NCT04380701, EudraCT: 2020-001038-36), and BNT162b2 is being evaluated in a global phase II/III safety and efficacy study1,2,3.
Construct design and analysis of expressed antigen
BNT162b1 RNA encodes the RBD with the SARS-CoV-2 S signal peptide fused to its N terminus (to enable endoplasmic reticulum translocation and secretion) and with the trimerization domain (foldon) of T4 fibritin21 fused to its C terminus for multimeric display. BNT162b2 RNA encodes full-length S that is stabilized in the prefusion conformation by substitution of residues 986 and 987 to proline (that is, S(P2))10,22,23 (Fig. 1a). The microfluidic capillary electrophoresis profiles of both of the RNAs show single sharp peaks that are consistent with their calculated lengths, indicating high purity and integrity (Fig. 1b). We detected robust expression of RBD–foldon or S(P2) by flow cytometry upon transfection of HEK293T cells with BNT162b1 RNA or BNT162b2 RNA formulated as lipid nanoparticles or mixed with a transfection reagent, respectively (Extended Data Fig. 1a). In transfected cells, the BNT162b1-encoded RBD or BNT162b2-encoded S(P2) localized to the secretory pathway, as shown by immunofluorescence microscopy (Extended Data Fig. 1b). We performed western blot under denaturing and non-denaturing conditions, and detected a main band of RBD-containing protein with an apparent molecular mass of more than 75 kDa (together with lesser quantities of a faster-migrating species) in the medium of cells transfected with BNT162b1 RNA, consistent with the secretion of trimeric RBD–foldon (which has a predicted molecular mass of 88.4 kDa) (Extended Data Fig. 1c).
For further structural characterization, we expressed the RBD–foldon and S(P2) antigens from DNA that corresponds to the RNA coding sequences. We purified the RBD–foldon from the medium of transfected Expi293F cells by affinity capture with the peptidase domain of ACE2 immobilized on agarose beads, which left little residual RBD–foldon uncaptured from the medium. We obtained evidence that the RBD–foldon has three RBDs flexibly tethered to a central hub using electron microscopy, which revealed a variety of conformations (Fig. 1c). The trimerized RBD bound to the peptidase domain of human ACE2 with an apparent KD of less than 5 pM, which is 1,000-fold the reported KD (5 nM) for monomeric RBD and is consistent with the avidity effect of multivalent binding that is enabled by the flexible tethering (Extended Data Fig. 1d). Although the flexibility of the RBD–foldon precluded direct structural analysis at high resolution, one RBD per trimer could be immobilized by binding to a complex of ACE2 and the B0AT1 neutral amino acid transporter (which is chaperoned by ACE2) when that complex was in the previously reported closed conformation8 (Fig. 1d). The size and symmetry of the RBD–foldon–ACE2–B0AT1 ternary complex aided image reconstruction by cryo-electron microscopy (cryo-EM), and we determined the structure of the RBD in the complex to a resolution of 3.24 Å (Fig. 1e, Extended Data Table 1, Supplementary Fig. 2). One copy of the RBD was resolved for each bound trimer. The binding interface between the resolved RBD and the extracellular domain of ACE2 was fitted to a previously reported structure7, and showed good agreement. The high-avidity binding to ACE2 and well-resolved structure in complex with ACE2 demonstrate that the recombinant RBD–foldon authentically presents the ACE2-binding site that is targeted by many SARS-CoV-2-neutralizing antibodies11,24.
We affinity-purified the trimeric S(P2) from detergent-solubilized protein via a C-terminal TwinStrep tag. S(P2) bound the peptidase domain of human ACE2 and a human anti-RBD neutralizing antibody (B38) with high affinity25 (an apparent KD of 1 nM for each) (Extended Data Fig. 1e, f). Our structural analysis by cryo-EM produced a mass density map at a nominal resolution of 3.29 Å, into which we fitted and rebuilt a previously published atomic model10 (Fig. 1f, Extended Data Fig. 2a, b, Extended Data Table 1). The rebuilt model showed good agreement with previously reported structures of prefusion full-length wild-type S and its ectodomain with the P2 mutations9,10. Three-dimensional classification of the dataset showed a class of particles that was in a one-RBD up (accessible for receptor binding), two-RBD down (closed) conformation; this class represented 20.4% of the trimeric molecules (Fig. 1g, Extended Data Fig. 2c). The remainder of the trimeric molecules were in an all-RBD down conformation. The RBD in the up conformation was less well-resolved than the other parts of the structure, which suggests conformational flexibility and a dynamic equilibrium between the RBD up and RBD down states, as has previously been suggested9,26. Our binding and structural analyses indicate that the BNT162b2 RNA sequence encodes a recombinant S(P2) that can authentically present the ACE2-binding site and other epitopes that are targeted by SARS-CoV-2-neutralizing antibodies.
BNT162b-elicted immunogenicity in mice
To study vaccine immunogenicity, we characterized B and T cell responses in a series of experiments in BALB/c mice after a single intramuscular injection of 0.2, 1 or 5 μg of BNT162b1 or BNT162b2, or of a buffer control. A single immunization using either of the candidate vaccines induced high titres of RBD- and S1-binding serum IgG in a dose-level-dependent manner (Fig. 2a, Extended Data Fig. 3a–d); these titres increased more steeply for BNT162b2. On day 28 after injection with 5 μg BNT162b1 or BNT162b2, geometric mean endpoint titres of RBD-binding serum IgG were 752,680 or 434,560, respectively. Polyclonal IgG elicited by either of the candidate vaccines had strong apparent binding affinity for a recombinant RBD target antigen (geometric mean apparent KD of 717 pM for BNT162b1 and 993 pM for BNT162b2), with a low apparent off-rate and a high apparent on-rate (Extended Data Fig. 3e). Serum samples from buffer-immunized control mice had no detectable RBD- or S1-specific IgG (Fig. 2a, b, Extended Data Fig. 3a–d), and neither did serum samples from mice injected up to two times with equivalent modRNA, formulated in lipid nanoparticles, that encoded a SARS-CoV-2 irrelevant antigen (data not shown).

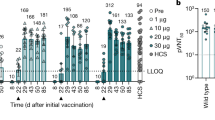
We injected BALB/c mice (n = 8) intramuscularly with a single dose of one of the BNT162b vaccine candidates or a buffer control. Geometric means of each group ± 95% confidence interval are shown. Day-28 P values compared to control (multiple comparison of mixed-effect analysis using Dunnett’s multiple comparisons test) for the single time points and groups are provided in a, b. a, Levels of RBD-specific IgG in sera of mice immunized using 5 μg of BNT162b1 or BNT162b2, determined by enzyme-linked immunosorbent assay (ELISA). For day-0 values, a prescreening of randomly selected mice was performed (n = 4). Extended Data Figure 3a, b shows IgG levels with lower BNT162b doses and sera testing for detection of S1. b, Pseudovirus-based VSV-SARS-CoV-2 50% neutralization titres (pVNT50) in sera of mice immunized using BNT162b1 (left) or BNT162b2 (right). Extended Data Figure 3g–i provides the number of infected cells per well with serum samples drawn 28 d after injection and titre correlation to a SARS-CoV-2 virus neutralization assay. For cellular response analysis in c, d, splenocytes of BALB/c mice (n = 8, unless stated otherwise) injected intramuscularly with BNT162b1 (green) or BNT162b2 (pink) were restimulated ex vivo with full-length S peptide mix or cell culture medium. Symbols represent individual mice. Heights of bars indicate the mean. P values compare immunized groups with the control (parametric, two-tailed paired t-test). c, IFNγ ELISpot of splenocytes 12 d after injection with 5 μg of one of the BNT162b vaccines. d, Cytokine production by splenocytes 28 d after injection with 0.2 μg BNT162b1 or 1 μg BNT162b2, determined by bead-based multiplex analysis (BNT162b2: n = 7 for IL-4, IL-5 and IL-13, one outlier removed by the ROUT method (Q = 1%) for the S peptide stimulated samples).
We measured the inhibition of virus entry by BNT162b-immunized mouse serum in a neutralization assay using vesicular stomatitis virus (VSV)-based SARS-CoV-2 pseudovirus. As with the antigen-specific IgG geometric mean titres (GMTs), 50% pseudovirus-neutralization GMTs increased steadily after injection of 5 μg of either candidate vaccine, and reached 1,056 for BNT162b1 and 296 for BNT162b2 on day 28 after injection (Fig. 2b, Extended Data Fig. 3f, g). We tested a random selection of samples in a SARS-CoV-2 neutralization assay, which demonstrated strong correlation of pseudovirus and SARS-CoV-2 neutralization (Pearson correlation of 0.9479 between the tests) (Extended Data Fig. 3h). In summary, each candidate vaccine induced a high functional antibody response in mice, and BNT162b1 induced higher titres than BNT162b2 after one injection.
Our characterization of antigen-specific responses of splenic T cells in mice at 12 and 28 days after injection with the BNT162b vaccines revealed a high fraction of CD4+ and CD8+ T cells that produced IFNγ and CD8+ cells that produced IL-2, as shown by enzyme-linked immunospot assay (ELISpot) or intracellular-cytokine-staining flow cytometry analysis after ex vivo restimulation with a full-length S peptide pool (Fig. 2c, Extended Data Fig. 4a, b). Total splenocytes collected on day 28 and restimulated with the full-length S peptide pool secreted high levels of the T-helper-1 (TH1) cytokines IL-2 or IFNγ, and minute or undetectable levels of the T-helper-2 (TH2) cytokines IL-4, IL-5 or IL-13, as measured in multiplex immunoassays (Fig. 2d). Overall, the patterns of CD4+ and CD8+ T cell responses were similar for the two vaccine candidates, with a somewhat stronger IFNγ-producing CD8+ T cell response in mice immunized with BNT162b2.
We assessed vaccine-induced effects on the proliferation and dynamics of immune-cell populations in injection-site draining lymph nodes (to evaluate the principal immune-educated compartments for proficient T and B cell priming) as well as in blood and spleen (to evaluate systemic effects of the vaccines). We observed higher numbers of plasma cells, class-switched IgG1+ and IgG2a+ B cells, and germinal-centre B cells in draining lymph nodes, and higher numbers of class-switched IgG1+ and germinal-centre B cells in spleens of mice at 12 days after injection with 5 μg of either vaccine as compared to control (Extended Data Fig. 4c, d). Vaccine-immunized mice had significantly fewer circulating B cells than did control mice as measured in blood at day 7 after injection (Extended Data Fig. 4e), which may imply that B cell homing to lymphoid compartments contributed to augmented B cell counts in the draining lymph nodes and spleen.
The draining lymph nodes from BNT162b1- or BNT162b2-immunized mice also displayed significantly higher counts of CD8+ and CD4+ T cells (as compared to buffer-immunized mice) at 12 days after injection, which were most pronounced for T follicular helper (TFH) cells—including the ICOS+ subsets that are essential for the formation of germinal centres (Extended Data Fig. 4c). Both of the BNT162b vaccines increased TFH cell counts in the spleen and blood, whereas an increase in circulating CD8+ T cells was detected only in BNT162b2-immunized mice (Extended Data Fig. 4d, e). In aggregate, these data indicate a strong induction of SARS-CoV-2-pseudovirus neutralization titres and systemic CD8+ and TH1-driven CD4+ T-cell responses by both of the vaccine candidates, and a somewhat more-pronounced cellular response to BNT162b2.
BNT162b-elicted immunogenicity in macaques
To assess the immunogenicity of BNT162b1 and BNT162b2 in nonhuman primates, we intramuscularly injected groups of six macaques (male, 2–4 years old) with 30 or 100 μg of BNT162b1, BNT162b2 or saline control on day 0 (dose 1) and day 21 (dose 2). RBD-binding IgG was readily detectable by day 14 after dose 1, and levels had increased further 7 days after dose 2 (day 28) (Fig. 3a). On day 28, geometric mean concentrations of RBD-binding IgG were 20,962 units (U) ml−1 (at 30-μg dose level) and 48,575 U ml−1 (at 100-μg dose level) for BNT162b1, and 23,781 U ml−1 (30-μg dose level) and 26,170 U ml−1 (100-μg dose level) for BNT162b2. For comparison, the geometric mean concentration of RBD-binding IgG of a panel of 38 SARS-CoV-2-convalescent human sera was 602 U ml−1, which is lower than the geometric mean concentration of the immunized macaques after one or two doses.

Male macaques (2–4 years old) were injected on day 0 and day 21 (arrows below the x-axes indicate the day of second injection) with 30 μg or 100 μg of BNT162b1 (green) or BNT162b2 (pink) (n = 6 each). Additional macaques received saline (control (C), n = 9)). Human convalescent sera (HCS) were obtained from patients infected with SARS-CoV-2 at least 14 d after PCR-confirmed diagnosis and at a time when acute COVID-19 symptoms had resolved (n = 38). The HCS panel is a benchmark for serology studies in this Article and previous publications1,2,3. a, Concentrations (in arbitrary units) of IgG that binds recombinant SARS-CoV-2 RBD (lower limit of detection (LLOD) = 1.72 U ml−1). b, SARS-CoV-2 50% virus-neutralization titres (VNT50) (LLOD = 20). c, d, Peripheral blood mononuclear cells collected on days 0, 14, 28 and 42 after first injection of BNT162b2 were restimulated ex vivo with full-length S peptide mix. Arrows below the x-axis indicate the days of dose 1 and dose 2. c, IFNγ ELISpot. d, IL-4 ELISpot. Heights of bars indicate the geometric (a, b) or arithmetic (c, d) means for each group, and values are written above the bars (a, b). Whiskers indicate 95% confidence intervals (a, b) or s.e.m. (c, d). Each symbol represents one macaque. Horizontal dashed line marks the LLOD. Values below the LLOD were set to 1/2 the LLOD.
Fifty per cent virus-neutralization GMTs—measured by a SARS-CoV-2-neutralization assay27 (rather than a pseudovirus-neutralization assay)—were detectable in the sera of most BNT162b1-immunized macaques by day 21 after dose 1, and in all BNT162b2-immunized macaques by day 14 after dose 1 (Fig. 3b). There was a strong boosting effect; comparable peak measured GMTs were elicited by BNT162b1 (768 for 30 μg and 1,714 for 100 μg) and BNT162b2 (962 for 30 μg and 1,689 for 100 μg), as measured in sera drawn 7 or 14 days after dose 2. For BNT162b2, sera were available up to day 56 after dose 1 (28 days after dose 2); robust GMTs of 285 for 30-μg and 283 for 100-μg dose levels persisted to this time point. For comparison, the neutralization GMT of the human convalescent sera was 94, which is substantially lower than the GMTs of macaque sera drawn 21 or 35 days after dose 2.
We analysed the S-specific T cell responses of BNT162b2- or saline-injected macaques using peripheral blood mononuclear cells collected before immunization and at the times indicated after doses 1 and 2. ELISpot demonstrated strong IFNγ, but minimal IL-4, responses after dose 2 (Fig. 3c, d, Extended Data Fig. 5a). Intracellular cytokine staining confirmed that BNT162b2 elicited a high frequency of CD4+ T cells that produced IFNγ, IL-2 or TNF, but a low frequency of CD4+ T cells that produced IL-4, which indicates a TH1-biased response (Extended Data Fig. 5b, c). Intracellular cytokine staining also demonstrated that BNT162b2 elicited circulating S-specific CD8+ T cells that produced IFNγ (Extended Data Fig. 5d).
BNT162b-elicted protection in macaques
Forty-one to fifty-five days after dose 2, six of the 2–4-year-old macaques that had been immunized using 100 μg BNT162b1, and six that had been immunized using 100 μg BNT162b2, were challenged with 1.05 × 106 plaque-forming units of SARS-CoV-2 (strain USA-WA1/2020) split equally between the intranasal and intratracheal routes, as previously described28 (Extended Data Fig. 6, Extended Data Table 2). In addition, nine age-matched macaques (controls) that had been mock-immunized with saline received the same SARS-CoV-2 challenge, and six age-matched macaques (sentinels)—three of which had been immunized using 30 μg BNT162b2—were mock-challenged with cell culture medium. We collected nasal, oropharyngeal and rectal swabs, and performed bronchoalveolar lavage at the times indicated (Extended Data Table 2). We then tested samples for SARS-CoV-2 RNA (genomic RNA and subgenomic transcripts) using reverse-transcription quantitative polymerase chain reaction (RT–qPCR). All personnel who performed clinical, radiological, histopathological or RT–qPCR evaluations were blinded to the group assignments of the macaques.
Viral RNA was detected in bronchoalveolar lavage fluid from seven of nine control macaques on day 3; from four of eight control macaques on day 6 after challenge (with one indeterminant result); and from none of the six control macaques that underwent bronchoalveolar lavage at the end of project (EOP; days 7–23 after challenge) (Fig. 4a). Viral RNA was detected in the bronchoalveolar lavage fluid of two of six BNT162b1-immunized macaques on day 3 after challenge, and from none thereafter. Viral RNA was not detected in bronchoalveolar lavage fluid from the BNT162b2-immunized, SARS-CoV-2 challenged macaques at any of the time points we sampled.

Macaques that had been immunized using 100 μg of BNT162b1 or BNT162b2 (n = 6 each) or mock-immunized with saline challenge (control) (n = 9) were challenged with 1.05 × 106 total plaque-forming units of SARS-CoV-2 split equally between the intranasal and intratracheal routes. Additional macaques (sentinel) (n = 6) were mock-challenged with cell culture medium. Macaque assignments to cohorts and schedules of immunization, challenge and sample collection are provided in Extended Data Fig. 6, Extended Data Table 2. Viral RNA levels were detected by RT–qPCR. a, Viral RNA in bronchoalveolar lavage fluid. ‘Pre’, before challenge; 10/EOP, day 10 or end of the project. b, Viral RNA in nasal swabs; 7–23/EOP, days 7–23 or end of project. Symbols represent individual macaques. Ratios above bars indicate the number of viral-RNA-positive macaques among all macaques in a group with evaluable samples. Heights of bars indicate geometric mean viral RNA copies; whiskers indicate geometric s.d. Dotted lines indicate LLOD. Values below the LLOD were set to 1/2 the LLOD. Two-sided statistical significance by a nonparametric test (Friedman’s test) of differences in viral RNA detection after challenge between six BNT162b1-immunized and six mock-immunized macaques (challenge cohorts 1 and 2) was P = 0.0152 for bronchoalveolar lavage fluid and P = 0.0048 for nasal swab; between six BNT162b2-immunized macaques and three mock-immunized macaques (challenge cohort 3), the statistical significance was P = 0.0014 for bronchoalveolar lavage fluid and P = 0.2622 for nasal swabs. Serum samples were assayed for SARS-CoV-2 VNT50. c, BNT162b1-immunized macaques and controls (challenge cohorts 1 and 2). d, BNT162b2-immunized macaques and controls (challenge cohort 3). Symbols represent titres from individual macaques. Horizontal dashed line indicates the lower limit of quantification of 20.
In nasal swabs obtained on the day after challenge, viral RNA was detected from control-immunized macaques (4 of 9) and BNT162b2-immunized macaques (5 of 6), but not from BNT162b1-immunized macaques (Fig. 4b). In subsequent nasal swabs, viral RNA was detected from some of the control-immunized macaques at each sampling time point (5 of 9 on day 3, 4 of 9 on day 6 and 2 of 9 on days 7–23), from some BNT162b1-immunized macaques at only one sampling time point (2 of 6 on day 6) and from none of the BNT162b2-immunized macaques at any sampling time point. Similar patterns were seen in oropharyngeal and rectal swabs: viral RNA was more often detected in control-immunized macaques than in BNT162b1- or BNT162b2-immunized macaques, and there was more persistence of viral RNA in rectal swabs than in oropharyngeal swabs (Extended Data Fig. 7a, b).
At the time of challenge, SARS-CoV-2-neutralizing titres ranged from 208 to 1,185 in the BNT162b1-immunized macaques and from 260 to 1,004 in the BNT162b2-immunized macaques. Neutralizing titres were below the limit of detection in the control macaques (Fig. 4c, d). The control macaques responded to challenge with infectious virus with an increase in SARS-CoV-2-neutralizing titres, consistent with an immune response to viral infection. However, there was no trend towards increasing SARS-CoV-2-neutralizing titres in response to viral challenge in the BNT162b1-immunized or BNT162b2-immunized macaques, consistent with their immunization suppressing SARS-CoV-2 infection. The maximum SARS-CoV-2-neutralizing titre elicited by virus challenge of control macaques remained below 150 through to the time of necropsy, whereas all immunized macaques maintained neutralizing titres greater than 150 throughout the challenge experiment.
None of the challenged macaques—whether immunized or not—showed clinical signs of illness (Extended Data Fig. 7c–f). Radiographic abnormalities were generally minimal or mild, and were not consistently associated with viral challenge (Extended Data Fig. 8a, b). The histopathology of necropsy specimens obtained 7–8 days after challenge revealed localized areas of pulmonary inflammation that were limited in extent even in the control macaques challenged after mock immunization with saline (Extended Data Fig. 8c). We conclude that the 2–4-year-old male-macaque challenge model is primarily a model of SARS-CoV-2 infection rather a model than of COVID-19 diseae.
Discussion
We demonstrate that the candidate vaccines BNT162b1 or BNT162b2—lipid-nanoparticle-formulated, m1Ψ nucleoside-modified mRNAs that encode secreted, trimerized SARS-CoV-2 RBD or prefusion-stabilized S, respectively—induce strong antigen-specific immune responses in mice and macaques. The RBD–foldon coding sequence directs the expression and secretion of a flexible, trimeric protein that binds to ACE2 with high affinity and has structurally intact ACE2 receptor-binding sites. We confirmed that protein expressed from DNA with the BNT162b2-encoded S(P2) amino acid sequence was in the prefusion conformation using cryo-EM. This analysis showed that the antigenically important RBD can assume the up conformation, in which the receptor-binding site that is rich in neutralizing epitopes is accessible in a proportion of the molecules24. The alternative states observed probably reflect a dynamic equilibrium between RBD up and down positions10,26. The binding of expressed and purified S(P2) to ACE2 and a neutralizing monoclonal antibody further demonstrates the conformational and antigenic integrity of this prefusion-stabilized S.
In mice, a single sub-microgram immunization using either of the BNT162b candidates rapidly induced high antibody titres that inhibited pseudovirus entry in the range of—or above—recently reported neutralizing titres that are elicited by other candicate vaccines against SARS-CoV-229,30. The candidate vaccines discussed in this Article also induced strong TFH and TH1-type CD4+ T cell responses, the latter of which are thought to be a more general effect of lipid-nanoparticle-formulated modRNA vaccines against SARS-CoV-230. Both CD4+ T cell types are known to support antigen-specific antibody generation and maturation. In some animal models of respiratory virus infection, a TH2-type CD4+ T cell response has previously been associated with vaccine-associated enhanced respiratory disease31,32. Therefore, a TH1-type response to immunization is preferred as it may reduce the theoretical risk of enhanced pulmonary disease during subsequent viral infection. Immunization with the vaccine candidates triggered redistribution of B cells from the blood to lymphoid tissues, where antigen presentation occurs. In humans, TFH cells in the circulation after vaccination with a VSV-vectored Ebola vaccine candidate have previously been correlated with a high frequency of antigen-specific antibodies33. After vaccination of mice with BNT162b1 or BNT162b2, high numbers of TFH cells were present in both the blood and lymph nodes, a potential correlate for the generation of a strong adaptive B cell response in germinal centres. In addition to eliciting favourable CD4+ T cell responses, BNT162b1 and BNT162b2 both elicit CD8+ T cell responses in mice, and BNT162b2 appears to be somewhat more efficient at eliciting antigen-specific cytotoxic IFNγ CD8+ T cells.
BNT162b1 and BNT162b2 elicit immune profiles in macaques similar to those observed in mice. Seven days after dose 2 (of 100 μg of the candidate) was administered to macaques (during the expansion phase of the antibody response), the neutralizing GMTs elicited by either candidate reached approximately 18× the GMT of a panel of SARS-CoV-2-convalescent human sera. Neutralizing GMTs declined by day 56 (35 days after dose 2), consistent with the contraction phase; however, they remained well above the GMT of the human sera panel. The duration of the study was not long enough to assess the rate of decline during the plateau phase of the antibody response. As in mice, BNT162b2 elicted a strongly TH1-biased CD4+ T cell response and IFNγ+ CD8+ T cell response in macaques.
Limitation and clearance of virus infection is promoted by the interplay between neutralizing antibodies that eliminate infectious particles and CD8+ T cells that target intracellular reservoirs of virus. CD8+ T cells may also reduce the influx of monocytes into infected lung tissue, which can be associated with undesirable IL-6 and TNF production and impaired antigen presentation34,35. The responses elicited by the vaccine candidates reflect a pattern that is favourable for vaccine safety and efficacy, which provides added reassurance for clinical translation36. The contributions of the individual immune effector systems to human protection from SARS-CoV-2 are not yet understood. Therefore, it appears prudent to develop COVID-19 vaccines that enlist concomitant cognate B cell, CD4+ T cell and CD8+ T cell responses.
Both candidates protected 2–4-year-old macaques from challenge with infectious SARS-CoV-2, and there was reduced detection of viral RNA in immunized macaques as compared to those that received saline. Immunization with BNT162b2 provided particularly strong RT–qPCR evidence for protection of the lower respiratory tract, as demonstrated by the absence of detectable SARS-CoV-2 RNA in serial bronchoalveolar lavage samples that were obtained starting 3 days after challenge. The lack of serological response to SARS-CoV-2 challenge in BNT162b1- or BNT162b2-immunized macaques—despite a neutralizing response to challenge in control-immunized macaques—suggests suppression of infection by the vaccine candidates. Clinical signs of disease were absent, and radiological and pathological abnormalities were generally mild after challenge. As in other published reports of immunization and SARS-CoV-2 challenge of nonhuman primates, there was no evidence of vaccine-mediated enhancement of viral replication, disease or pathology37,38. The interpretation of vaccine-mediated protection in nonhuman primates is limited by the small number of animals and the inherent limitations of animal models. Nevertheless, these preclinical results provided key support for the immunization of large numbers of clinical-trial participants with BNT162b2.
The selection of BNT162b2 over BNT162b1 for further clinical testing was largely driven by the greater tolerability of BNT162b2 with comparable immunogenicity in clinical trials3, and the broader range and MHC diversity of T cell epitopes on the much larger full-length S. A global phase III safety and efficacy study of immunization with BNT162b2 (NCT04368728) is ongoing, and may answer open questions that cannot be addressed by preclinical models.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment, except for the performance of serological assays of nonhuman primates and RT–PCR-based viral load measurements and the intepretation of radiographs, computed tomography scans, and histopathology specimens.
Ethics statement
All mouse studies were performed at BioNTech SE, and protocols were approved by the local authorities (local welfare committee) and conducted according to Federation of European Laboratory Animal Science Associations recommendations. Study execution and housing were in compliance with the German Animal Welfare Act and Directive 2010/63/EU. Mice were kept in individually ventilated cages with a 12-h light/dark cycle, controlled environmental conditions (22 ± 2 °C, 45% to 65% relative humidity) and under specific-pathogen-free conditions. Food and water was available ad libitum. Only mice with an unobjectionable health status were selected for testing procedures.
Immunizations for the nonhuman primate study were performed at the University of Louisiana at Lafayette-New Iberia Research Centre (NIRC), which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) (animal assurance no. 000452). The work was in accordance with United States Department of Agriculture Animal Welfare Act and Regulations and the NIH Guidelines for Research Involving Recombinant DNA Molecules, and Biosafety in Microbiological and Biomedical Laboratories. All procedures performed on these macaques were in accordance with regulations and established guidelines, and were reviewed and approved by an Institutional Animal Care and Use Committee or through an ethical review process. Challenge of nonhuman primates with infectious SARS-CoV-2 after immunization was performed at the Southwest National Primate Research Centre (SNPRC), Texas Biomedical Research Institute (San Antonio), which is also accredited by the AAALAC (animal assurance no. 000246). Animal husbandry followed standards recommended by AAALAC International and the NIH Guide for the Care of Use of Laboratory Animals. This study was approved by the Texas Biomedical Research Institute Animal Care and Use Committee.
Protein and peptide reagents
Purified recombinant SARS-CoV-2 RBD (Sino Biological) or trimeric S (Acro Biosystems) was used as a target for western blot, and the RBD tagged with a human Fc (Sino Biological) was used in ELISA to detect SARS-CoV-2 S-specific IgG. A recombinant SARS-CoV-2 RBD containing a C-terminal Avitag (Acro Biosystems) was used as a target antigen in Luminex immunoassays. Purified recombinant SARS-CoV-2 S1 including a histidine tag (Sino Biological) was used in ELISA to detect SARS-CoV-2 S-specific IgG in mice. Purified recombinant SARS-CoV-2 S1 and RBD with histidine tags (both Sino Biological) were used for surface plasmon resonance spectroscopy. A peptide pool of 15-mer peptides overlapping by 11 amino acids covering the full-length S was used for restimulation in ELISpot, cytokine profiling and intracellular cytokine staining followed by flow cytometry. An irrelevant peptide (SPSYVYHQF, derived from gp70 AH-139) or a cytomegalovirus (CMV) peptide pool was used as control for ELISpot assays. All peptides were obtained from JPT Peptide Technologies.
Panel of SARS-CoV-2-convalescent human sera
A previously described1,2,3 panel of SARS-CoV-2-convalescent human sera was used as a benchmark for nonhuman primate serology. The sera (n = 38) were drawn from donors 18–83 years of age, at least 14 days after PCR-confirmed diagnosis and at a time when the participants were asymptomatic. Most serum donors had outpatient (35/38) or inpatient (1/38) COVID-19; 2 of 38 had asymptomatic SARS-CoV-2 infections. Sera were obtained from Sanguine Biosciences, the MT Group and Pfizer Occupational Health and Wellness.
Cell culture
HEK293T and Vero 76 cells (both from ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX (Gibco) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich). Cell lines were tested for mycoplasma contamination after receipt, before expansion and cryopreservation. For studies including nonhuman primate samples, Vero 76 and Vero CCL81 cells (both from ATCC) were cultured in DMEM (Gibco) containing 2% HyClone fetal bovine and 100 U ml−1 penicillium–streptomycin (Gibco). Expi293F cells were grown in Expi293 medium and transiently transfected using ExpiFectamine293 (all from Thermo Fisher Scientific).
In vitro transcription and purification of RNA
Antigens encoded by BNT162b vaccine candidates were designed on a background of S sequences from SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank MN908947.3). The DNA template for the BNT162b1 RNA is a DNA fragment encoding a fusion protein of the SARS-CoV-2 S signal peptide (SP) (amino acids 1–16), the SARS-CoV-2 S RBD and the T4 bacteriophage fibritin trimerization motif21 (foldon). The template for the BNT162b2 RNA is a DNA fragment encoding SARS-CoV-2 S with K986P and V987P substitutions. BNT162b1 and BNT162b2 DNA templates were cloned into a plasmid vector with backbone sequence elements (T7 promoter, 5′ and 3′ UTR, 100 nucleotide poly(A) tail) interrupted by a linker (A30LA70, 10 nucleotides) for improved RNA stability and translational efficiency17,40. The DNA was purified, spectrophotometrically quantified and in vitro-transcribed by T7 RNA polymerase in the presence of a trinucleotide cap1 analogue ((m27,3′-O)Gppp(m2’-O)ApG) (TriLink) and with N1-methylpseudouridine-5′-triphosphate (m1ΨTP) (Thermo Fisher Scientific) replacing uridine-5′-triphosphate (UTP)41. RNA was purified using magnetic particles42. RNA integrity was assessed by microfluidic capillary electrophoresis (Agilent Fragment Analyzer), and the concentration, pH, osmolality, endotoxin level and bioburden of the solution were determined.
Lipid nanoparticle formulation of the RNA
Purified RNA was formulated into lipid nanoparticles using an ethanolic lipid mixture of ionizable cationic lipid and transferred into an aqueous buffer system via diafiltration to yield a lipid nanoparticle composition similar to one previously described43. The lipid nanoparticle contains RNA, an ionizable lipid, ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate)), a PEGylated lipid, 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide and two structural lipids (1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC])and cholesterol). The vaccine candidates were stored at −70 to −80 °C at a concentration of 0.5 mg ml−1.
Transfection of HEK cells
HEK293T cells were transfected with 1 μg RiboJuice transfection reagent-mixed BNT162b1 RNA or BNT162b2 RNA, or with the vaccine candidates BNT162b1 (lipid-nanoparticle-formulated BNT162b1 RNA) or BNT162b2 (lipid-nanoparticle-formulated BNT162b2 RNA) by incubation for 18 h. Non-lipid-nanoparticle-formulated mRNA was diluted in Opti-MEM medium (Thermo Fisher Scientific) and mixed with the transfection reagent according to the manufacturer’s instructions (RiboJuice, Merck Millipore).
Western blot analysis of size fractions of the medium of BNT162b1-RNA-transfected cells
Medium from cultured HEK293T cells was collected. After 13-fold concentration via Vivaspin 20 centrifugal concentrators with a molecular weight cut off of 10 kDa, supernatants were applied to a preparative HiLoad 16/600 Superdex 200 pg column (both Sigma Aldrich). The column was run at 29.8 cm h−1 in phosphate buffered saline (PBS), and 500-μl fractions were collected (Supplementary Fig. 1). The gel filtration column was calibrated with well-defined protein standards separated under identical conditions in a second run. Size-fractioned FBS-free medium from BNT162b1-RNA-transfected HEK293T cells was analysed by denaturing (95 °C) and non-denaturating (no heating) PAGE using 4–15% Criterion TGX Stain-Free Gel (Bio-Rad) and western blot. Transfer to a nitrocellulose membrane (Bio-Rad) was performed using a semi-dry transfer system (Trans-Blot Turbo Transfer System, Bio-Rad). Blotted proteins were detected with a monoclonal antibody that recognizes SARS-CoV-2 S1 (SinoBiological) and a secondary anti-rabbit horse radish peroxidase (HRP)-conjugated antibody (Sigma Aldrich). Blots were developed with Clarity Western ECL Substrate (Bio-Rad) and imaged with a Fusion FX Imager (Vilber) using the Image Lab software version 6.0.
Vaccine antigen detection by flow cytometry
Transfected HEK293T cells were stained with Fixable Viability Dye (eBioscience). After fixation (Fixation Buffer, Biolegend), cells were permeabilized (Perm Buffer, eBioscience) and stained with a monoclonal antibody that recognizes SARS-CoV-2 S1 (SinoBiological). Cells were acquired on a FACSCanto II flow cytometer (BD Biosciences) using BD FACSDiva software version 8.0.1 and analysed by FlowJo software version 10.6.2 (FlowJo, BD Biosciences).
Localization of expressed vaccine antigens by immunofluorescence
Transfected HEK293T cells were fixed in 4% paraformaldehyde (PFA) and permeabilized in PBS/0.2% Triton X-100. Free binding sites were blocked and cells incubated with a rabbit monoclonal antibody that recognizes the SARS-CoV-2 S1 subunit (SinoBiological), an anti-rabbit IgG secondary antibody (Jackson ImmunoResearch), labelled lectin HPA (Thermo Fisher Scientific) and concanavalin A (Fisher Scientific). DNA was stained with Hoechst (Life Technologies). Images were acquired with a Leica SP8 confocal microscope and Application Suite LAS-X Version 3.1.5.
SARS-CoV-2 RBD–foldon and S(P2) expression and purification
To express the RBD–foldon encoded by BNT162b1 for ACE2-binding analysis and cryo-EM, DNA corresponding to the RNA coding sequence was cloned into the pMCG1309 vector. A plasmid encoding amino acids 1–615 of human ACE2 with C-terminal His-10 and Avi tags was generated for transient expression of the peptidase domain of ACE2 (ACE2 PD) in Expi293F cells. The ACE2–B0AT1 complex was produced by co-expression of two plasmids in Expi293F cells, one of them encoding ACE2 amino acids 1–17 followed by haemagglutinin and Strep II tags and ACE2 amino acids 18–805, and the other containing a methionine followed by a Flag tag and amino acids 2–634 of human B0AT1. Secreted ACE2 PD was isolated from conditioned cell culture medium using Nickel Excel resin (GE Healthcare) followed by gel filtration chromatography on a Superdex200 10/30 column (GE Healthcare) in PBS. Approximately 5 mg of purified ACE2 PD was covalently attached per 1 ml of 4% beaded agarose by amine coupling using AminoLink Plus resin (Thermo Fisher Scientific).
The RBD trimer was purified from conditioned medium by affinity capture with the ACE2 PD crosslinked agarose and was eluted from the resin with 3 M MgCl2. Following dialysis, the protein was concentrated and purified by gel filtration using a Superdex200 10/300 column in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline (HBS) with 10% glycerol. Purification of the ACE2–B0AT1 complex was based on a previously described procedure8. To form the ACE2–B0AT1–RBD-trimer complex, ACE2–B0AT1 aliquots were combined with purified RBD–foldon diluted in size-exclusion chromatography buffer (25 mM Tris pH 8.0, 150 mM NaCl, 0.02% glyco diosgenin) for a 3:1 molar ratio of RBD trimers to ACE2 protomers. After incubation at 4 °C for 30 min, the sample was concentrated and resolved on a Superose 6 Increase 10/300 GL column. Peak fractions containing the complex were pooled and concentrated.
To express SARS-CoV-2 S(P2) encoded by BNT162b2 for characterization by size-exclusion chromatography, ACE2-PD binding, monoclonal antibody binding and cryo-EM, a gene encoding the full length of SARS-CoV-2 (GenBank MN908947) with two prolines substituted at residues 986 and 987 (K986P and V987P) followed with a C-terminal HRV3C protease site and a TwinStrep tag was cloned into a modified pcDNA3.1(+) vector with the CAG promoter. The TwinStrep-tagged S(P2) was expressed in Expi293F cells.
Purification of the recombinant protein was based on a previously described procedure, with minor modifications9. Upon cell lysis, S(P2) was solubilized in 1% NP-40 detergent. The TwinStrep-tagged protein was then captured with StrepTactin Sepharose HP resin in 0.5% NP-40. S(P2) was further purified by size-exclusion chromatography and eluted as three distinct peaks in 0.02% NP-40, as previously reported9 (chromatogram not shown). A peak that consists of intact S(P2) migrating at around 150 kDa, as well as dissociated S1 and S2 subunits (which co-migrate at just above 75 kDa), was used in the structural characterization. Spontaneous dissociation of the S1 and S2 subunits occurs throughout the course of protein purification, starting at the point of detergent-mediated protein extraction, so that S(P2) preparations also contain dissociated S1 and S2.
Binding kinetics of the RBD–foldon trimer and S(P2) to immobilized human ACE2 and a neutralizing monoclonal antibody by biolayer interferometry
Binding of purified RBD–foldon to ACE2 PD and of NP-40 solubilized, purified S(P2) to ACE2 PD and human neutralizing monoclonal antibody B3825 was measured by biolayer interferometry at 25 °C on an Octet RED384 (FortéBio). RBD–foldon binding was measured in 10 mM HEPES pH 7.5, 150 mM NaCl and 1 mM EDTA (EDTA). S(P2) binding was measured in 25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA and 0.02% NP-40. Avi-tagged human ACE2 PD was immobilized on streptavidin-coated sensors; B38 antibody was immobilized on protein G-coated sensors. For an RBD–foldon concentration series, binding data were collected for 600 s of association and 900 s of dissociation. For an S(P2) concentration series, after initial baseline equilibration of 120 s, the sensors were dipped in a 10 μg ml−1 solution of Avi-tagged ACE2 PD or B38 monoclonal antibody for 300 s to achieve capture levels of 1 nM using the threshold function. Then, after another 120 s of baseline, binding data were collected for 300 s of association and 600 s of dissociation.
Biolayer interferometry data were collected with Octet Data Acquisition software version 10.0.0.87 and processed using ForteBio Data Analysis software version 10.0. Data were reference-subtracted and fit to a 1:1 binding model with R2 value greater than 0.96 for the RBD and 0.95 for S(P2). Potential avidity effects for the RBD–foldon and potential ongoing dissociation of S1 from S(P2) could make the actual binding events more complicated than represented by 1:1 binding model. Therefore, we report apparent kinetics and affinity (S(P2)) or avidity (RBD–foldon) of binding as calculated using Octet Data Analysis Software v.10.0 (FortéBio). For the RBD–foldon, the dissociation rate of interaction (kd) with ACE2 PD was slower than the limit of measurement of the instrument, and the apparent minimum binding avidity (KD) was estimated using an assumed dissociation rate kd of 1 × 10−6 s−1.
Electron microscopy of negatively stained RBD–foldon trimers
Purified RBD–foldon in 4 μl was applied to a glow-discharged copper grid overlaid with formvar and amorphous carbon (Ted Pella). Negative staining was performed with Nano-W organotungstate stain (Nanoprobes) according to the manufacturer’s protocol. The sample imaged using an FEI TF-20 microscope operating at 200 kV, with a magnification of 62,000× and defocus of −2.5 μm. Micrographs were contrast transfer function (CTF)-corrected in RELION using CTFFIND-4.144. A small manually picked dataset was used to generate 2D references for autopicking. The resulting particle set was subjected to 2D classification in RELION 3.0.645.
Cryo-EM of the ACE2–B0AT1–RBD-trimer complex
Cryo-EM was performed using a Titan Krios operating at 300 keV equipped with a Gatan K2 Summit direct electron detector in super-resolution mode at a magnification of 165,000×, for a magnified pixel size of 0.435 Å at the specimen level.
Purified ACE2–B0AT1–RBD-trimer complex at 6 mg ml−1 in 4 μl was applied to gold Quantifoil R1.2/1.3 200 mesh grids glow-discharged in residual air for 30 s at 20 mA using a Pelco Easiglow. The sample was blotted using a Vitrobot Mark IV for 5 s with a force of −3 before being plunged into liquid ethane cooled by liquid nitrogen. In total, 7,455 micrographs were collected from a single grid. Data were collected over a defocus range of −1.2 to −3.4 μm with a total electron dose of 52.06 e− per Å2 fractionated into 40 frames over a 6-s exposure for 1.30 e− per Å2per frame. Initial motion correction was performed in Warp46, during which super-resolution data were binned to give a pixel size of 0.87 Å. Corrected micrographs were imported into RELION 3.1-beta45 for CTF estimation with CTFFIND-4.144.
Particles were picked using the LaPlacian-of-Gaussian particle-picking algorithm as implemented in RELION, and extracted with a box size of 450 pixels. References obtained by 2D classification were used for a second round of reference-based autopicking, yielding a dataset of 715,356 particles. Two of the three RBDs of each particle (the two not constrained by binding to ACE2–B0AT1) exhibited diffuse density in 2D classification that reflected high particle flexibility, consistent with the conformational flexibility of RBD trimers observed by negative-stain electron microscopy (Fig. 1c, d). This flexibility precluded the inclusion of all three RBDs in the final structural solution. Particle heterogeneity was filtered out with 2D and 3D classification with a mask size of 280 Å to filter out the diffuse density of the two non-ACE2-bound RBD copies in each RBD trimer, yielding a set of 87,487 particles that refined to 3.73 Å with C2 symmetry. Refinement after subtraction of micelle and B0AT1 density from the particles yielded an improved map of 3.24 Å. The atomic model from Protein Data Bank code (PDB) 6M178 was rigid-body-fitted into the 3.24 Å density and then flexibly fitted to the density using real-space refinement in Phenix47 alternating with manual building in Coot48. The microscope was operated for image acquisition using SerialEM software version 3.8.0 beta49. Validation of this model is shown in Supplementary Fig. 2. Data collection, 3D reconstruction and model refinement statistics are listed in Extended Data Table 1.
Cryo-EM of S(P2)
For TwinStrep-tagged S(P2), 4 μl purified protein at 0.5 mg ml−1 were applied to gold Quantifoil R1.2/1.3 300 mesh grids freshly overlaid with graphene oxide. The sample was blotted using a Vitrobot Mark IV for 4 s with a force of −2 before being plunged into liquid ethane cooled by liquid nitrogen. We collected 27,701 micrographs from 2 identically prepared grids. Data were collected from each grid over a defocus range of −1.2 to −3.4 μm with a total electron dose of 50.32 and 50.12 e− per Å2, respectively, fractionated into 40 frames over a 6-s exposure for 1.26 and 1.25 e− per Å2 per frame. On-the-fly motion correction, CTF estimation, and particle-picking and extraction with a box size of 450 pixels were performed in Warp46, during which super-resolution data were binned to give a pixel size of 0.87 Å. A total of 1,119,906 particles were extracted. All subsequent processing was performed in RELION 3.1-beta45. Particle heterogeneity was filtered out with 2D and 3D classification, yielding a set of 73,393 particles that refined to 3.6 Å with C3 symmetry. Three-dimensional classification of this dataset without particle alignment separated out one class with a single RBD up, representing 15,098 particles. The remaining 58,295 particles, in the three-RBD-down conformation, were refined to give a final model at 3.29 Å. The atomic model from PDB 6XR89 was rigid-body fitted into the map density, then flexibly fitted to the density using real-space refinement in Phenix47 alternating with manual building in Coot48. The cryo-EM model validation is provided in Extended Data Fig. 2, and the full cryo-EM data processing workflow and the model refinement statistics in provided in Extended Data Table 1.
Immunization
Mice
Female BALB/c mice (Janvier) (8–12 weeks old) were randomly allocated to groups. BNT162b1 and BNT162b2 diluted in PBS with 300 mM sucrose (Fig. 2a–c, Extended Data Figs. 3 for both BNT162 vaccine candidates; Fig. 2e, Extended Data Fig. 4a for BNT162b2) or 0.9% NaCl (Fig. 2d, Extended Data Fig. 4b–e for both BNT162 vaccine candidates; Fig. 2e, Extended Data Fig. 4a for BNT162b1) were injected into the gastrocnemius muscle at a volume of 20 μl under isoflurane anaesthesia. PBS with 300 mM sucrose or 0.9% NaCl served as buffer controls, respectively.
Macaques
Male macaques (2–4 years old) were randomly assigned to receive BNT162b1 or BNT162b2 on days 0 and 21 or saline control on days 0 and 21 or 35. Vaccine was administered in 0.5 ml by intramuscular injection in the left quadriceps muscle. Macaques were anaesthetized with ketamine HCl (10 mg kg−1; intramuscular) during immunization and were monitored for adequate sedation.
Phlebotomy and tissue preparation
Mice
Peripheral blood was collected from the retro-orbital venous plexus under isoflurane anaesthesia or the vena facialis without anaesthesia. For flow cytometry, blood was heparinized. For serum generation, blood was centrifuged for 5 min at 16,000g and the serum was immediately used for downstream assays or stored at −20 °C. Spleen single-cell suspensions were prepared in PBS by mashing tissue against the surface of a 70-μm cell strainer (BD Falcon). Erythrocytes were removed by hypotonic lysis. Popliteal, inguinal and iliac lymph nodes were pooled, cut into pieces, digested with collagenase D (1 mg ml−1) (Roche) and passed through cell strainers.
Macaques
Serum was obtained before, 6 h after and 1, 14, 21, 28, 35 and 42 days after injection with BNT162b1, BNT162b2 or saline (Extended Data Table 2). For BNT162b2 and challenge cohort 3 controls, serum was also obtained on day 56, and peripheral blood mononuclear cells (PBMCs) were obtained before immunization and on days 7, 28, and 42 (except that PBMCs were not obtained from the challenge cohort 3 control macaques on day 28). Blood for serum and PBMCs was collected in compliance with animal protocol 2017-8725-023, approved by the NIRC Institutional Animal Care and Use Committee. Macaques were anaesthetized with ketamine HCl (10 mg kg−1; intramuscular) during blood collection and were monitored for adequate sedation.
Analysis of S1- and RBD-specific serum IgG
Mice
MaxiSorp plates (Thermo Fisher Scientific) were coated with recombinant S1 or RBD (1 μg ml−1) in sodium carbonate buffer, and serum-derived bound IgG was detected using an HRP-conjugated secondary antibody and tetramethylbenzidine substrate (Biotrend). Data collection was performed using a BioTek Epoch reader and Gen5 software version 3.0.9. For concentration analysis, an IgG mouse isotype control was used in parallel in a serial dilution, and the sample signals were correlated to a standard curve of the isotype control.
Macaques and humans
Recombinant SARS-CoV-2 S1 containing a C-terminal Avitag (Acro Biosystems) was bound to streptavidin-coated Luminex microspheres. Bound macaque or human anti-S1 antibodies present in the serum were detected with a fluorescently labelled goat anti-human polyclonal secondary antibody (Jackson ImmunoResearch). Data were captured as median fluorescent intensities using a Bioplex200 system (Bio-Rad) and converted to U ml−1 antibody concentrations using a reference standard consisting of 5 pooled SARS-CoV-2-convalescent human serum samples (obtained >14 days after PCR diagnosis, from the panel described in ‘Panel of SARS-CoV-2-convalescent human sera’), diluted in antibody-depleted human serum with arbitrary assigned concentrations of 100 U ml−1 and accounting for the serum dilution factor.
Surface plasmon resonance spectroscopy of polyclonal mouse immune sera
Binding kinetics of mouse S1- and RBD-specific serum IgG to recombinant S1 and RBD was determined using a Biacore T200 device (Cytiva) with 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% v/v surfactant P20 (HBS-EP running buffer, BR100669, Cytiva) at 25 °C. Carboxyl groups on the CM5 sensor chip matrix were activated with a mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimidehydrochloride and N-hydroxysuccinimide to form active esters for the reaction with amine groups. Anti-mouse IgG Fc-antibody (Jackson ImmunoResearch) was diluted in 10 mM sodium acetate buffer pH 5 (30 μg ml−1) for covalent coupling to immobilization level of about 10,000 response units. Free N-hydroxysuccinimide esters on the sensor surface were deactivated with ethanolamine.
Mouse serum was diluted 1:50 in HBS-EP buffer and applied at 10 μl min−1 for 30 s to the active flow cell for capture by immobilized antibody, and the reference flow cell was treated with buffer. Binding analysis of captured mouse IgG antibodies to S1–His or RBD–His (Sino Biological) was performed using a multicycle kinetic method with concentrations ranging from 25 to 400 nM or 1.56 to 50 nM, respectively. An association period of 180 s was followed by a dissociation period of 600 s with a constant flow rate of 40 μl min−1 and a final regeneration step. Apparent binding kinetics for the captured polyclonal IgG were calculated using a global kinetic fit model (1:1 Langmuir, Biacore T200 Evaluation Software Version 3.1, Cytiva).
VSV-SARS-CoV-2 S pseudovirus entry-inhibition assay by serum IgG in mice
A recombinant replication-deficient VSV vector that encodes green fluorescent protein (GFP) instead of VSV-G (VSV(ΔG–GFP)) was pseudotyped with SARS-CoV-2 S according to published pseudotyping protocols50,51. In brief, HEK293T/17 monolayers transfected to express SARS-CoV-2 S truncated of the C-terminal cytoplasmic 19 amino acids (SARS-CoV-2-S(CΔ19)) were inoculated with VSVΔG-GFP vector (rescued from pVSVΔG-GFP plasmid expression vector; Kerafast). After incubation for 1 h at 37 °C, the inoculum was removed, and cells were washed with PBS before medium supplemented with anti-VSV-G antibody (clone 8G5F11, Kerafast) was added to neutralize residual input virus. VSV-SARS-CoV-2 pseudovirus-containing medium was collected 20 h after inoculation, 0.2-μm-filtered and stored at −80 °C.
Vero-76 cells were seeded in 96-well plates. Serial dilutions of mouse serum samples were prepared and pre-incubated for 10 min at room temperature with VSV-SARS-CoV-2 pseudovirus suspension (4.8 × 103 infectious units per ml) before transferring the mix to Vero-76 cells. Inoculated Vero-76 cells were incubated for 20 h at 37 °C. Plates were placed in an IncuCyte Live Cell Analysis system (Sartorius) and incubated for 30 min before the analysis (IncuCyte 2019B Rev2 software). Whole-well scanning for bright-field and GFP fluorescence was performed using a 4× objective. The pVNT50 is reported as the reciprocal of the highest dilution of serum that still yielded a 50% reduction in GFP-positive infected cell number per well, compared to the mean of the no-serum pseudovirus positive control. Each serum sample dilution was tested in duplicate.
IFNγ and IL-4 ELISpot
Mice
ELISpot assays were performed with mouse IFNγ ELISpotPLUS kits according to the manufacturer’s instructions (Mabtech). A total of 5 × 105 splenocytes was ex vivo restimulated with the full-length S peptide mix (0.1 μg ml−1 final concentration per peptide) or controls (gp70-AH1 (SPSYVYHQF)39, 4 μg ml−1; concanavalin A, 2 μg ml−1 (Sigma)). Streptavidin–alkaline phosphatase and 5-bromo-4-chloro-3′-indolyl phosphate/nitro blue tetrazolium-plus substrate were added, and spots counted using an ELISpot plate reader (ImmunoSpot S6 Core Analyzer (CTL)). Spot numbers were evaluated using ImmunoCapture Image Acquisition Software v.7.0 and ImmunoSpot 7.0.17.0 Professional. Spot counts denoted too numerous to count by the software were set to 1,500. For T cell subtyping, CD8+ T cells and CD4+ T cells were isolated from splenocyte suspensions using MACS MicroBeads (CD8a (Ly-2) and CD4 (L3T4) (Miltenyi Biotec)) according to the manufacturer’s instructions. CD8+ or CD4+ T cells (1 × 105) were subsequently restimulated with 5 × 104 syngeneic bone-marrow-derived dendritic cells loaded with full-length S peptide mix (0.1 μg ml−1 final concentration per peptide), or cell culture medium as control. The purity of isolated T cell subsets was determined by flow cytometry to calculate spot counts per 1 × 105 CD8+ or CD4+ T cells.
Macaques
Macaque PBMCs were tested with commercially available nonhuman primate IFNγ and IL-4 ELISpot assay kits (Mabtech). Cryopreserved macaque PBMCs were thawed in prewarmed AIM-V medium (Thermo Fisher Scientific) with benzonase (EMD Millipore). For IFNγ ELISpot, 1.0 × 105 PBMCs, and for IL-4 ELISpot, 2.5 × 105 PBMCs, were stimulated ex vivo with 1 μg ml−1 of the full-length S overlapping peptide mix. Tests were performed in triplicate wells and medium containing dimethyl sulfoxide (medium–DMSO), a CMV peptide pool and phytohemagglutinin (Sigma) were included as controls. After 24 h for IFNγ and 48 h for IL-4, streptavidin–HRP and 3-amino-9-ethylcarbazole substrate (BD Bioscience) were added and spots counted using a CTL ImmunoSpot S6 Universal Analyzer (CTL). Results shown are background (medium–DMSO) subtracted and normalized to spot-forming cells per 106 PBMCs.
Cell-mediated immunity by flow cytometry
Mice
For T cell analysis in peripheral blood, erythrocytes from 50 μl freshly drawn blood were lysed (ammonium–chloride–potassium lysing buffer (Gibco)), and cells were stained with Fixable Viability Dye (eBioscience) and primary antibodies in the presence of Fc block in flow buffer (Dulbecco’s phosphate-buffered saline (Gibco) supplemented with 2% fetal calf serum (FCS), 2 mM EDTA (both Sigma) and 0.01% sodium azide (Morphisto)). After staining with secondary biotin-coupled antibodies in flow buffer, cells were stained extracellularly against surface markers with directly labelled antibodies and streptavidin in Brilliant Stain Buffer Plus (BD Bioscience) diluted in flow buffer. Cells were washed with 2% RotiHistofix (Carl Roth), fixed (Fix/Perm Buffer, FoxP3/Transcription Factor Staining Buffer Set (eBioscience)) and permeabilized (Perm Buffer, FoxP3/Transcription Factor Staining Buffer Set (eBioscience)) overnight. Permeabilized cells were intracellularly treated with Fc block and stained with antibodies against transcription factors in Perm Buffer.
For T cell analysis in lymphoid tissues, 1 × 106 lymph node cells (for BNT162b1) or 1.5 × 106 lymph node cells (for BNT162b2) and 4 × 106 spleen cells were stained for viability and extracellular antigens with directly labelled antibodies. Fixation, permeabilization and intracellular staining was performed as described for blood T cell staining.
For B cell subtyping in lymphoid tissues, 2.5 × 105 lymph node and 1 × 106 spleen cells were treated with Fc block, stained for viability and extracellular antigens as described for blood T cell staining, and fixed with 2% RotiHistofix overnight.
For intracellular cytokine staining of T cells from BNT162b1-immunized mice, 1 × 106 lymph node and 4 × 106 spleen cells were ex vivo restimulated with 0.2 μg ml−1 final concentration per peptide of full-length S peptide mix. For intracellular cytokine staining of T cells from mice immunized using BNT162b2, 4 × 106 spleen cells were ex vivo restimulated with 0.5 μg ml−1 final concentration per peptide of full-length S peptide mix or cell culture medium (no peptide) as control. The cells were restimulated for 5 h in the presence of GolgiStop and GolgiPlug (both BD Bioscience) for 5 h. Cells were stained for viability and extracellular antigens as described for lymphoid T cell staining. Cells were fixed with 2% RotiHistofix and permeabilized overnight. Intracellular staining was performed as described for blood T cell staining.
Mouse cells were acquired on a BD Symphony A3 or BD Celesta (B cell subtyping) flow cytometer (BD Bioscience) using BD FACSDiva software version 9.1 or 8.0.1.1, respectively, and analysed with FlowJo 10.6 (FlowJo, BD Biosciences).
Macaques
For intracellular cytokine staining in T cells, 1.5 × 106 PBMCs were stimulated with the full-length S peptide mix at 1 μg ml−1 (concentration of all peptides, combined), Staphyloccocus enterotoxin B (2 μg ml−1) as positive control, or 0.2% DMSO as negative control. GolgiStop and GolgiPlug (both BD Bioscience) were added. Following 37-°C incubation for 12 to 16 h, cells were stained for viability and extracellular antigens after blocking Fc binding sites with directly labelled antibodies. Cells were fixed, permeabilized with BDCytoFix/CytoPerm solution (BD Bioscience), and intracellular staining was performed in the permeabilization buffer for 30 min at room temperature. Cells were washed, resuspended in 2% FBS/PBS buffer and acquired on an LSR Fortessa. Data were analysed by FlowJo 10.4.1 (FlowJo, BD Biosciences). Results shown are background (medium–DMSO) subtracted.
Cytokine profiling in mice by bead-based immunoassay
Mouse splenocytes were restimulated for 48 h with full-length S peptide mix (0.1 μg ml−1 final concentration per peptide) or cell culture medium (no peptide) as control. Concentrations of IFNγ, IL-2, IL-4, IL-5 and (for splenocytes from BNT162b2-immunized mice) IL-13 in supernatants were determined using a bead-based, 11-plex TH1/TH2 mouse ProcartaPlex multiplex immunoassay (Thermo Fisher Scientific) according to the manufacturer’s instructions. Fluorescence was measured with a Bioplex200 system (Bio-Rad) and analysed with ProcartaPlex Analyst 1.0 software (Thermo Fisher Scientific). Values below the lower limit of quantification were set to zero.
SARS-CoV-2 neutralization by macaque sera
The SARS-CoV-2 neutralization assay used a previously described strain of SARS-CoV-2 (USA_WA1/2020) that had been rescued by reverse genetics and engineered by the insertion of an mNeonGreen gene into open reading frame 7 of the viral genome27. This reporter virus generates similar plaque morphologies and indistinguishable growth curves from wild-type virus. Viral master stocks were grown in Vero E6 cells as previously described52. When testing human convalescent serum specimens, the fluorescent neutralization assay produced comparable results to the conventional plaque reduction neutralization assay. Serial dilutions of heat-inactivated sera were incubated with the reporter virus (2 × 104 plaque forming units (PFU) per well) to yield an approximately 10–30% infection rate of the Vero CCL81 monolayer for 1 h at 37 °C before inoculating Vero CCL81 cell monolayers (targeted to have 8,000 to 15,000 cells in the central field of each well at the time of seeding, one day before infection) in 96-well plates to allow accurate quantification of infected cells. Cell counts were enumerated by nuclear stain (Hoechst 33342), and fluorescent virus-infected foci were detected 16–24 h after inoculation with a Cytation 7 Cell Imaging Multi-Mode Reader (BioTek) with Gen5 Image Prime version 3.09. Titres were calculated in GraphPad Prism version 8.4.2 by generating a 4-parameter logistical fit of the per cent neutralization at each serial serum dilution. The VNT50 is reported as the interpolated reciprocal of the dilution yielding a 50% reduction in fluorescent viral foci.
SARS-CoV-2 challenge of macaques
The SARS-CoV-2 inoculum was obtained from a stock of 2.1 × 106 PFU ml−1 previously prepared at Texas Biomedical Research Institute, aliquoted into single-use vials and stored at −70 °C. The working virus stock was generated from two passages of the SARS-CoV-2 USA-WA1/2020 isolate (a fourth passage seed stock purchased from BEI Resources; NR-52281) in Vero E6 cells. The virus was confirmed to be SARS-CoV-2 by deep sequencing that demonstrated identity to a published SARS-CoV-2 sequence (GenBank accession number MN985325.1).
BNT162b1-immunized (n = 6) and BNT162b2-immunized (n = 6) male macaques, and age-matched male macaques mock-immunized with saline (n = 9) (control), were challenged with 1.05 × 106 PFU of SARS-CoV-2 USA-WA1/2020 isolate, split equally between the intranasal (0.25 ml) and intratracheal (0.25 ml) routes, as previously described28. Sentinel age- and sex-matched macaques (n = 6) were mock-challenged with DMEM supplemented with 10% FCS intranasally (0.25 ml) and intratracheally (0.25 ml). The macaques were challenged or mock-challenged at the times relative to immunization indicated in Extended Data Fig. 6, Extended Data Table 2.
Twelve to nineteen days before challenge, macaques were moved from the NIRC where they had been immunized to the animal biosafety level 3 facility at SNPRC. Macaques were monitored regularly by a board-certified veterinary clinician for rectal body temperature, weight and physical examination. Specimen collection was performed under tiletamine zolazepam (Telazol) anaesthesia as previously described28. Bronchoalveolar lavage, and nasal, oropharyngeal and rectal swab collection, X-ray and CT examinations and necropsy were performed at the times indicated in Extended Data Fig. 6, Extended Data Table 2. The three control macaques in challenge cohort 3 and three sentinel macaques were not necropsied to allow their subsequent rechallenge (control) or challenge (sentinel). Bronchoalveolar lavage was performed by instilling 20 ml of saline 4 times. These washings were pooled, aliquoted and stored frozen at −70 °C.
SARS-CoV-2 viral RNA quantification by RT–qPCR
To detect and quantify SARS-CoV-2 in nonhuman primates, viral RNA was extracted from bronchoalveolar lavage fluid and from nasal, oropharyngeal and rectal swabs as previously described53,54,55, and tested by RT–qPCR as previously described28. In brief, 10 μg yeast tRNA and 1 × 103 PFU of MS2 phage (Escherichia coli bacteriophage MS2) (ATCC) were added to each thawed sample, and RNA extraction performed using the NucleoMag Pathogen kit (Macherey-Nagel). The SARS-CoV-2 RT–qPCR was performed on extracted RNA using a 2019-nCoV N1 assay developed by the United States Centers for Disease Control and Prevention, on a QuantStudio 3 instrument (Applied Biosystems). The cut-off for positivity (limit of detection) was established at 10 gene equivalents per reaction (800 gene equivalents per ml). Samples were tested in duplicate. One bronchoalveolar lavage specimen from the challenge cohort 2 control group obtained on day 6 after challenge, and one nasal swab from the BNT162b1-immunized group obtained on day 1 after challenge, had—on repeated measurements—viral RNA levels on either side of the LLOD. These specimens were categorized as indeterminate and excluded from the graphs and the analysis.
Radiology
Thoracic radiographs and computed tomography scans were performed under anaesthesia, as previously described28. For radiographic imaging, three-view thoracic radiographs (ventrodorsal, right and left lateral) were obtained at the times relative to challenge indicated in Extended Data Table 2. The macaques were anaesthetized using telazol (2–6 mg kg−1) and maintained by inhaled isoflurane delivered through a Hallowell 2002 ventilator anaesthesia system (Hallowell). Macaques were intubated to perform end inspiratory breath-hold using a remote breath-hold switch. Lung field computed tomography images were acquired using Multiscan LFER150 PET/CT (MEDISO) scanner. Image analysis was performed using 3D region-of-interest tools available in Vivoquant (Invicro). Images were interpreted by a board-certified veterinary radiologist blinded to treatment groups. Scores were assigned to a total of 7 lung regions on a severity scale of 0–3 per region, with a maximum severity score of 21. Pulmonary lesions evident before challenge, or those which could not be unequivocally attributed to the viral challenge (such as atelectasis secondary to recumbency and anaesthesia), received a score of 0.
Histopathology
Lung histopathology is reported on necropsies performed on 2–4-year-old male macaques at the times after challenge indicated in Extended Data Fig. 6, Extended Data Table 2. Necropsy, tissue processing and histology were performed by SNPRC. Samples were fixed in 10% neutral buffered formalin and processed routinely into paraffin blocks. Tissue blocks were sectioned to 5 μm and stained with haematoxylin and eosin. Microscopic evaluation of 7 lung tissue sections per macaque (1 sample of each lobe in the left and right lungs) was performed blindly by SNPRC and Pfizer pathologists. Lungs were evaluated using a semiquantitative scoring system with inclusion of cell types and/or distribution as appropriate. Inflammation score was based on area of tissue in section involved: 0 = normal; 1 ≤ 10%; 2 = 11–30%; 3 = 30–60%; 4 = 60–80%; 5 ≥ 80%. Each lobe received an individual score, and the final score for each macaque was reported as the mean of the individual scores. The pathologists were unblinded to the group assignments after agreement on diagnoses. As indicated in Extended Data Fig. 6, Extended Data Table 2, the BNT162b1-immunized and control macaques were challenged and necropsied in parallel (challenge cohorts 1 and 2), and the BNT162b2-immunized macaques were immunized and challenged subsequently (challenge cohort 3).
Statistics and reproducibility
No statistical methods were used to predetermine group and samples sizes (n). All experiments were performed once. P values reported for RT–qPCR analysis were determined by nonparametric analysis (Friedman’s test) based on the ranking of viral RNA shedding data within each day. PROC RANK and PROC GLM from SAS 9.4 were used to calculate the P values. All available post-challenge bronchoalveolar lavage fluid and nasal, oropharyngeal and rectal swab samples from the necropsied macaques and all available post-challenge samples through day 10 from the macaques that were not necropsied were included in the analysis. Indeterminate results were excluded from this analysis. All remaining analyses were two-tailed and carried out using GraphPad Prism 8.4.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank MN908947.3) is the genetic background of the BNT162b antigens. The cryo-EM maps and atomic coordinates have been deposited to the Electron Microscopy Data Bank (EMDB) and PDB with accession numbers EMD-23211 and 7L7F, respectively, for the ACE2–B0AT1–RBD–foldon complex and EMD-23215 and 7L7K, respectively, for S(P2). The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Sahin, U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586, 594–599 (2020).
Mulligan, M. J. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020).
Walsh, E. E. et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N. Engl. J. Med. 383, 2439–2450 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
He, Y. et al. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 324, 773–781 (2004).
Yi, C. et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 17, 621–630 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655 (2020).
Ou, X. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620 (2020).
Fan, X., Cao, D., Kong, L. & Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 11, 3618 (2020).
Rauch, S., Jasny, E., Schmidt, K. E. & Petsch, B. New vaccine technologies to combat outbreak situations. Front. Immunol. 9, 1963 (2018).
Pardi, N. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 217, 345–351 (2015).
Orlandini von Niessen, A. G. et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol. Ther. 27, 824–836 (2019).
Karikó, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Pardi, N. et al. Characterization of HIV-1 nucleoside-modified mRNA vaccines in rabbits and rhesus macaques. Mol. Ther. Nucleic Acids 15, 36–47 (2019).
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248–251 (2017).
Meier, S., Güthe, S., Kiefhaber, T. & Grzesiek, S. Foldon, the natural trimerization domain of T4 fibritin, dissociates into a monomeric A-state form containing a stable β-hairpin: atomic details of trimer dissociation and local β-hairpin stability from residual dipolar couplings. J. Mol. Biol. 344, 1051–1069 (2004).
Pallesen, J. et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl Acad. Sci. USA 114, E7348–E7357 (2017).
Kirchdoerfer, R. N. et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 8, 15701 (2018).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. 26, 1422–1427 (2020).
Wu, Y. et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020).
Henderson, R. et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 27, 925–933 (2020).
Muruato, A. E. et al. A high-throughput neutralizing antibody assay for COVID-19 diagnosis and vaccine evaluation. Nat. Commun. 11, 4059 (2020).
Singh, D. K. et al. Responses to acute infection with SARS-CoV-2 in the lungs of rhesus macaques, baboons and marmosets. Nat. Microbiol. 6, 73–86 (2021).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Laczkó, D. et al. A single immunization with nucleoside-modified mRNA vaccines elicits strong cellular and humoral immune responses against SARS-CoV-2 in mice. Immunity 53, 724–732.e7 (2020).
Tseng, C.-T. et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 7, e35421 (2012).
Graham, B. S. Rapid COVID-19 vaccine development. Science 368, 945–946 (2020).
Farooq, F. et al. Circulating follicular T helper cells and cytokine profile in humans following vaccination with the rVSV-ZEBOV Ebola vaccine. Sci. Rep. 6, 27944 (2016).
Jafarzadeh, A., Chauhan, P., Saha, B., Jafarzadeh, S. & Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 257, 118102 (2020).
Yang, D. et al. Attenuated interferon and proinflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J. Infect. Dis. 222, 734–745 (2020).
Lambert, P.-H. et al. Consensus summary report for CEPI/BC March 12-13, 2020 meeting: assessment of risk of disease enhancement with COVID-19 vaccines. Vaccine 38, 4783–4791 (2020).
Rauch, S. et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. Preprint at https://doi.org/10.1101/2020.12.23.424138 (2020).
Corbett, K. S. et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med. 383, 1544–1555 (2020).
Slansky, J. E. et al. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC–peptide–TCR complex. Immunity 13, 529–538 (2000).
Holtkamp, S. et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108, 4009–4017 (2006).
Grudzien-Nogalska, E. et al. Synthetic mRNAs with superior translation and stability properties. Methods Mol. Biol. 969, 55–72 (2013).
Berensmeier, S. Magnetic particles for the separation and purification of nucleic acids. Appl. Microbiol. Biotechnol. 73, 495–504 (2006).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Berger Rentsch, M. & Zimmer, G. A vesicular stomatitis virus replicon-based bioassay for the rapid and sensitive determination of multi-species type I interferon. PLoS ONE 6, e25858 (2011).
Lester, S. et al. Middle East respiratory coronavirus (MERS-CoV) spike (S) protein vesicular stomatitis virus pseudoparticle neutralization assays offer a reliable alternative to the conventional neutralization assay in human seroepidemiological studies. Access Microbiol. 1, e000057 (2019).
Xie, X. et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 27, 841–848.e3 (2020).
Joosten, S. A. et al. Mycobacterium tuberculosis peptides presented by HLA-E molecules are targets for human CD8 T-cells with cytotoxic as well as regulatory activity. PLoS Pathog. 6, e1000782 (2010).
Mehra, S. et al. Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. J. Infect. Dis. 207, 1115–1127 (2013).
Gautam, U. S. et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 115, E62–E71 (2018).
Acknowledgements
We thank T. Garretson and D. Cooper for advice on, and M. Cutler for coordination of, serology studies in nonhuman primates; R. F. Sommese and K. F. Fennell for technical assistance with molecular cloning and cell-based binding; M. Dvorak, M. Drude, F. Zehner, T. Lapin, B. Ludloff, S. Hinz, F. Bayer, J. Scholz, A.L. Ernst, T. Sticker and S. Wittig for their valuable support and assistance, which resulted in a rapid availability of oligonucleotides and DNA templates; E. Boehm, K. Goebel, R. Frieling, C. Berger, S. Koch, T. Wachtel, J. Leilich, M. Mechler, R. Wysocki, M. Le Gall, A. Czech and S. Klenk for carrying out RNA production and analysis, without which this vaccine candidate could not have been transferred to nonclinical studies with such speed; B. Weber, J. Vogt, S. Krapp, K. Zwadlo, J. Mottl, J. Mühl and P. Windecker for supporting the mouse studies and serological analysis with excellent technical assistance; A. K. Voges and E. Clemmons for interpreting radiographs for the nonhuman primate study; S. Ganatra for radiology services; E. Romero for veterinary services; K. A. Soileau and the staff of the New Iberia Research Centre for care of the nonhuman primates; E. J. Dick for veterinary pathology; Polymun Scientific for excellent formulation services; Acuitas Therapeutics for fruitful discussions; and S. Wigge and C. Lindemann for scientific writing support and manuscript review. BioNTech is the sponsor of the study, and Pfizer is its agent. BioNTech and Pfizer are responsible for the design, data collection, data analysis, data interpretation and writing of the report. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit the data for publication. This study was not supported by any external funding at the time of submission.
Author information
Authors and Affiliations
Contributions
U.S. conceived and conceptualized the work and strategy. S.Hein, S.C.D., A.A.H.S., C.K., R.d.l.C.G.G., and M.C.G. designed primers, performed oligosynthesis, cloned constructs and performed protein expression experiments. T.Z., S.F., J.S. and A.N.K. developed, planned, performed and supervised RNA synthesis and analysis. E.H.M. purified S(P2). N.L.N. purified RBD trimer and ACE2 peptidase domain. J.A.L. developed ACE2–B0AT1–RBD trimer formation and purified the complex. P.V.S. developed and performed biolayer interferometry experiments. J.A.L. and S. Han performed electron microscopy and solved the structure of the complex. Y.C. supervised the structural and biophysical characterization and analysed the structures. A.M. and B.G.L. performed surface plasmon resonance spectroscopy. A.G., S.A.K., S.S., T.H., L.F. and F.V. planned, performed and analysed in vitro studies. F.B., T.K. and C.R. managed the formulation strategy. A.B.V., M.V., L.M.K. and K.C.W. designed mouse studies, and analysed and interpreted data. A.P., S.E., D.P. and G.S. performed and analysed the S1- and RBD-binding IgG assays. M.G. designed and optimized MS2 SARS-nCoV-2 N1 RT–qPCR assay. M.G., R.C. Jr and K.J.A. performed and analysed viral RT-qPCR data. A.M., B.S. and A.K.-W. performed and analysed pVNT assays. C.F.-G. and P.-Y.S. performed and analysed VNT assays. D.E., D.S., B.J., Y.F. and H.J. performed in vivo studies and ELISpot assays. A.B.V., K.C.W., J.L., M.S.M., A.O.-S. and M.V. planned, analysed and interpreted ELISpot assays. L.M.K., J.L., D.E., Y.F., H.J., A.P.H., M.S.M. and P.A.-Q. planned, performed and analysed flow cytometry assays. A.B.V., L.M.K., Y.F. and H.J. planned, performed, analysed and interpreted cytokine release assays. M.R.G. read and interpreted radiographs and computed tomography scans. O.G. and S.C. read and interpreted histopathology specimens. R.S.S. and S.C. interpreted histopathology data. I.K., K.A.S., K.T., C.Y.T., M.G., D.K. and P.R.D. designed nonhuman primate studies, and analysed and interpreted data. K.T., M.P., I.L.S. and W.V.K. oversaw immunogenicity and serology testing of nonhuman primates. S.H.-U. and K.B. provided veterinary services for nonhuman primates. J.A.F., J.C., T.C. and J.O. managed the nonhuman primate colony. U.S., Ö.T., P.R.D, L.M.K., A.M. and M.V. contributed to synthesis and integrated interpretation of obtained data. A.B.V., I.K., Y.C., A.M., M.V., L.M.K., C.T., K.A.S., Ö.T., P.R.D, K.U.J. and U.S. wrote the manuscript. All authors supported the review of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that U.S. and Ö.T. are management board members and employees at BioNTech SE (Mainz, Germany); K.C.W., B.G.L., D.S., B.J., T.H., T.K. and C.R. are employees at BioNTech SE; A.B.V., A.M., M.V., L.M.K., S. Hein, A.G., T.Z., F.B., A.P., D.E., S.C.D., S.F., S.E., F.B., B.S., A.K.-W., Y.F., H.J., S.A.K., S.S., A.P.H., P.A.-Q., J.S., A.A.H.S., C.K., R.d.l.C.G.G., L.F. and A.N.K. are employees at BioNTech RNA Pharmaceuticals GmbH (Mainz, Germany); A.B.V., A.M., K.C.W., A.G., S.F., A.N.K and U.S. are inventors on patents and patent applications related to RNA technology and COVID-19 vaccines; A.B.V., A.M., M.V., L.M.K., K.C.W., S. Hein, B.G.L., A.P., D.E., S.C.D., S.F., S.E., D.S., B.J., B.S., A.P.H., P.A.-Q., J.S., A.A.H.S., T.H., L.F., C.K., T.K., C.R., A.N.K., Ö.T. and U.S. have securities from BioNTech SE; I.K., Y.C., K.A.S. J.A.L. M.S.M., K.T., A,O.-S., J.A.F., M.C.G., S. Han, J.A.L., E.H.M., N.L.N., P.V.S., C.Y.T., D.P., W.V.K., J.O., R.S.S., S.C., T.C., I.L.S., M.W.P., G.S., and P.R.D., K.U.J. are employees of Pfizer and may hold stock options; C.F.-G. and P.-Y.S. received compensation from Pfizer to perform neutralization assays; M.R.G. received compensation from Pfizer to read and interpret radiographs and computed tomography scans. J.C., S.H.-U., K.B., R.C., Jr., K.J.A. O.G. and D.K., are employees of Southwest National Primate Research Center, which received compensation from Pfizer to conduct the animal challenge work; M.G. is an employee of Texas Biomedical Research Institute, which received compensation from Pfizer to conduct the RT–qPCR quantification of viral load. The authors declare no other relationships or activities that could appear to have influenced the submitted work.
Additional information
Peer review information Nature thanks Wolfgang Baumgärtner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Antigen expression and receptor affinity of vaccines.
a, Detection of BNT162b1-encoded RBD–foldon and BNT162b2-encoded S(P2) in HEK293T cells by S1-specific antibody staining and flow cytometry. HEK293T cells analysed by flow cytometry were incubated with: no RNA (control), BNT162b RNA formulated as lipid nanoparticles (BNT162b1 and BNT162b2) or BNT162b RNAs mixed with a transfection reagent (BNT162b1 RNA and BNT162b2 RNA). Heights of bars indicate the means of technical triplicates. b, Localization of BNT162b1 RNA-encoded RBD–foldon or BNT162b2 RNA-encoded S(P2) in HEK293T cells transfected as in a, determined by immunofluorescence staining. Endoplasmic reticulum and Golgi (ER/Golgi) (red), S1 (green) and DNA (blue). Scale bar, 10 μm. c, Western blot of denatured and non-denatured samples of size-exclusion chromatography fractions (chromatogram in Supplementary Fig. 1) of concentrated medium from HEK293T cells transfected with BNT162b1 RNA. The RBD–foldon was detected with a rabbit monoclonal antibody against the S1 fragment of SARS-CoV-2 S. Protein controls (ctrl): purified, recombinant RBD and S. d, Biolayer interferometry (BLI) sensorgram demonstrating the binding kinetics of the purified RBD–foldon trimer, expressed from DNA, to immobilized human ACE2 PD. e, f, Biolayer inferometry sensorgrams showing binding of a DNA-expressed S(P2) preparation from a size-exclusion chromatography peak (not shown) that contains intact S(P2) and dissociated S1 and S2 to immobilized human ACE2 PD (e) and B38 monoclonal antibody (f). Binding data are in colour; 1:1 binding models fit to the data are in black. Apparent kinetic parameters are provided in the graphs.
Extended Data Fig. 2 Cryo-EM evidence for alternative conformers of S(P2).
a, Representative 2D class averages of TwinStrep-tagged S(P2) particles extracted from cryo-EM micrographs. Box edge, 39.2 nm. b, Fourier shell correlation curve from RELION gold-standard refinement of the S(P2) trimer. c, Flowchart for cryo-EM data processing of the complex, showing 3D class averages.
Extended Data Fig. 3 BNT162b-elicited antibody responses in mice.
a–d, BALB/c mice (n = 8) were injected intramuscularly with a single dose of one of the BNT162b vaccine candidate or buffer (control, n = 8 (a, b) and n = 16 (c, d)). P values of day 28 compared to control (multiple comparison of mixed-effect analysis (a, b) and one-way analysis of variance (c, d), all using Dunnett’s multiple comparisons test) are provided. a, b, RBD- and S1-specific IgG responses (geometric mean of each group ± 95% confidence interval) in sera obtained 7, 14, 21 and 28 days after injection with BNT162b1 (a) or BNT162b2 (b), determined by ELISA. For day-0 values, a prescreening of randomly selected mice was performed (n = 4). c, d, RBD-specific IgG reciprocal serum endpoint titres 14 (c) and 28 days (d) after injection. The horizontal dotted line indicates the LLOD. Each data point represents one mouse, and the height of bar indicates the geometric mean of groups. e, f, Representative surface plasmon resonance sensorgram of the binding kinetics of His-tagged RBD (e) to immobilized mouse IgG from serum drawn 28 days after injection with 5 μg BNT162b candidates. Binding data (in colour) and 1:1 binding model fit to the data (black) are depicted. e, Number of infected cells per well in a VSV-SARS-CoV-2 pVNT50 assay conducted with serial dilutions of mouse serum samples drawn 28 days after injection with BNT162b1 (f) or BNT162b2 (g). Lines represent individual sera measured in triplicate. Horizontal dotted lines indicate geometric mean ± 95% confidence interval (as grey area) of infected cells in the absence of mouse serum (virus-positive control). h, Pearson correlation of VSV-SARS-CoV-2 pVNT50 with live SARS-CoV-2 VNT50 for n = 10 random selected serum samples from mice immunized with BNT162b1 and BNT162b2 each. For several samples, identical pVNT50 and VNT50 values were measured: blue symbols represent individual mice; red symbols represent 2 mice; green symbol represents 3 mice.
Extended Data Fig. 4 T cell response and T cell and B cell phenotyping of BNT162b-immunized mice.
BALB/C mice (n = 8 per group, unless stated otherwise) were injected intramuscularly with one of the BNT162b vaccines or buffer (control). a, b, Separated T cells from spleen or splenocytes of BALB/c mice were ex vivo restimulated with full-length S peptide mix or cell culture medium (medium). Symbols represent individual mice. Height of bars indicate the mean of each group. a, IFNγ ELISpot of separated splenic CD4+ or CD8+ T cells after immunization using 1 μg of one of the BNT162b vaccines (BNT162b1, n = 7 for CD4+ T cells, one outlier removed by Grubbs test, α = 0.05). P values compare immunized groups with the control (parametric, two-tailed paired t-test). b, CD8+ T-cell-specific cytokine release by splenocytes after immunization using 5 μg of one of the BNT162b vaccines or buffer (control), determined by flow cytometry. S-peptide-specific responses are corrected for background (medium). P values compare immunized groups with the control (parametric, two-tailed unpaired t-test assuming equal s.d.). c–e, T cell and B cell phenotype composition after immunization with BNT162b candidates was determined by flow cytometry. P values were determined by a two-tailed, unpaired t-test assuming populations to have the same s.d. c, B cell and T cell numbers in draining lymph nodes (dLN) (popliteal, iliac and inguinal lymph nodes). For B cell subtyping, control, n = 4; BNT162b2, n = 7. #For per cent ICOS+ cells of TFH cells, only BNT162b2 data are available. d, B cell and TFH cell numbers in the spleen. e, B cell and T cell numbers in the blood.
Extended Data Fig. 5 Macaque CD4+ and CD8+ T cell response.
Macaques (n = 6 per group) were injected on day 0 and day 21 with 30 μg or 100 μg BNT162b2, as in Figs. 3, 4. PBMCs collected on days 0, 14, 28 and 42 after BNT162b2 injection were ex vivo restimulated with full-length S peptide mix. a, Scatter plot showing the correlation of IL-4- and IFNγ-secreting cells by ELISpot of day-42 PBMCs. b, IFNγ, IL-2 or TNF release from CD4+ T cells by flow cytometry (LLOD = 0.04). c, IL-4 release from CD4+ T cells by flow cytometry (LLOD = 0.05). d, IFNγ release from CD8+ T cells by flow cytometry (LLOD = 0.03). Heights of bars indicate the arithmetic means for each group. Whiskers indicate s.e.m. (b–d). Each symbol represents one macaque. Horizontal dashed lines mark LLODs. Values below the LLOD were set to 1/2 the LLOD. Arrows below the x axis indicate the days of doses 1 and 2.
Extended Data Fig. 6 Schedule of macaque SARS-CoV-2 challenge and necropsy.
Timing in days from dose 2 of vaccine or saline (numbers to the left of the bars) and of necropsy (numbers inside bars) are presented relative to the day of SARS-CoV-2 or mock-challenge (day 0). Numbers of macaques represented by the bars are indicated by red numbers to the right of the bars. Control, macaques challenged but not immunized with BNT162b. Sentinel, macaques mock-challenged (cell culture medium only). n/a, macaques not necropsied. Additional details, including timing of sample collections and radiographic examinations, are in Extended Data Table 2.
Extended Data Fig. 7 Viral RNA detection in oropharyngeal and rectal swabs, and clinical signs in BNT162b-immunized macaques after challenge with infectious SARS-CoV-2.
Macaques immunized using 100 μg of BNT162b1 or BNT162b2 (n = 6 each) and macaques mock-immunized using saline or not immunized (control, n = 9) (as described in Fig. 4, Extended Data Fig. 6, Extended Data Table 2) were challenged with 1.05 × 106 total PFU of SARS-CoV-2 split equally between the intranasal and intratracheal routes. Additional macaques (sentinel, n = 6) were mock-challenged with cell culture medium. a, b, Viral RNA levels were detected by RT–qPCR in oropharyngeal (a) and rectal (b) swabs. Ratios above data points indicate the number of viral-RNA-positive macaques among all macaques that provided evaluable samples in a group. Heights of bars indicate geometric mean of viral RNA copies; whiskers indicate geometric s.d. Every symbol represents one macaque. Dotted lines indicate the LLODs. Values below the LLOD were set to 1/2 the LLOD. The two-sided statistical significance by Friedman’s nonparametric test of differences in viral RNA detection between 6 BNT162b1-immunized and 6 contemporaneously control-immunized macaques (challenge cohorts 1 and 2) after challenge was P < 0.0001 for oropharyngeal swabs and P = 0.1179 for rectal swabs; between 6 BNT162b2-immunized macaques and 3 contemporaneously control-immunized macaques (challenge cohort 3) after challenge, the statistical significance was P = 0.0007 for oropharyngeal swabs and P = 0.2209 for rectal swabs. c–f, Vital signs were recorded. c, Body weight change. d, Temperature change. e, Heart rate. f, Oxygen saturation. Each data point represents an arithmetic mean. Whiskers indicate s.d.
Extended Data Fig. 8 Radiographic signs and pulmonary histopathology of macaques immunized with BNT162b1 or BNT162b2 and challenged with SARS-CoV-2.
Macaques were immunized using BNT162b1 or BNT162b2 or mock-immunized with saline (control) and challenged with SARS-CoV-2, and a sentinel group was challenged with cell culture medium. The disposition of the macaques for immunization, infectious challenge, imaging and necropsy are described in Figs. 3, 4, Extended Data Fig. 6, Extended Data Table 2. Three-view thoracic radiographs (ventrodorsal, right and left lateral) and lung field computed tomography images were obtained. The macaques were anaesthetized and intubated to perform end inspiratory breath-hold. Images were interpreted by two board-certified veterinary radiologists blinded to treatment groups. Scores were assigned to 7 lung regions on a severity scale of 0–3 per region, with a maximum severity score of 21. Pulmonary lesions evident before challenge, or those which could not be unequivocally attributed to the viral challenge (such as atelectasis secondary to recumbency and anaesthesia), received a score of 0. a, Thoracic radiograph scores. b, Lung field computed tomography scores. Each dot represents the summed radiograph or computed tomography scores for the seven lung lobes of a single macaque. Two veterinary pathologists blindly performed microscopic evaluation of formalin-fixed, haematoxylin and eosin-stained lung tissue sections from each of seven lobes from each macaque that had been necropsied on day 7 or day 8. Inflammation scores were assigned by consensus between the pathologists on a scale of 1–5 on the basis of the area of involvement. c, Pulmonary histopathology scores. Each dot represents the mean inflammation area score from the seven lung lobes of an individual macaque. In a–c, the height of each bar indicates the arithmetic mean of the radiograph, computed tomography or histopathology score, respectively, for the macaques in each group, and whiskers indicate s.d.
Supplementary information
Supplementary Figures
This file contains Supplementary Figs 1-8.
Rights and permissions
About this article

Cite this article
Vogel, A.B., Kanevsky, I., Che, Y. et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 592, 283–289 (2021). https://doi.org/10.1038/s41586-021-03275-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03275-y
This article is cited by
-
Nano-carrier DMSN for effective multi-antigen vaccination against SARS-CoV-2
Journal of Nanobiotechnology (2024)
-
Finely tuned ionizable lipid nanoparticles for CRISPR/Cas9 ribonucleoprotein delivery and gene editing
Journal of Nanobiotechnology (2024)
-
SARS-CoV-2 vaccination may mitigate dysregulation of IL-1/IL-18 and gastrointestinal symptoms of the post-COVID-19 condition
npj Vaccines (2024)
-
Good reasons for structural biology
Nature Structural & Molecular Biology (2024)
-
Branched chemically modified poly(A) tails enhance the translation capacity of mRNA
Nature Biotechnology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
