
Abstract
A recent genetic association study1 identified a gene cluster on chromosome 3 as a risk locus for respiratory failure after infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A separate study (COVID-19 Host Genetics Initiative)2 comprising 3,199 hospitalized patients with coronavirus disease 2019 (COVID-19) and control individuals showed that this cluster is the major genetic risk factor for severe symptoms after SARS-CoV-2 infection and hospitalization. Here we show that the risk is conferred by a genomic segment of around 50 kilobases in size that is inherited from Neanderthals and is carried by around 50% of people in south Asia and around 16% of people in Europe.
Similar content being viewed by others
Main
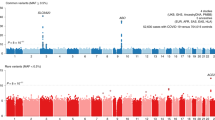
The COVID-19 pandemic has caused considerable morbidity and mortality, and has resulted in the death of over a million people to date3. The clinical manifestations of the disease caused by the virus, SARS-CoV-2, vary widely in severity, ranging from no or mild symptoms to rapid progression to respiratory failure4. Early in the pandemic, it became clear that advanced age is a major risk factor, as well as being male and some co-morbidities5. These risk factors, however, do not fully explain why some people have no or mild symptoms whereas others have severe symptoms. Thus, genetic risk factors may have a role in disease progression. A previous study1 identified two genomic regions that are associated with severe COVID-19: one region on chromosome 3, which contains six genes, and one region on chromosome 9 that determines ABO blood groups. Recently, a dataset was released by the COVID-19 Host Genetics Initiative in which the region on chromosome 3 is the only region that is significantly associated with severe COVID-19 at the genome-wide level (Fig. 1a). The risk variant in this region confers an odds ratio for requiring hospitalization of 1.6 (95% confidence interval, 1.42–1.79) (Extended Data Fig. 1).

a, Manhattan plot of a genome-wide association study of 3,199 hospitalized patients with COVID-19 and 897,488 population controls. The dashed line indicates genome-wide significance (P = 5 × 10−8). Data were modified from the COVID-19 Host Genetics Initiative2 (https://www.covid19hg.org/). b, Linkage disequilibrium between the index risk variant (rs35044562) and genetic variants in the 1000 Genomes Project. Red circles indicate genetic variants for which the alleles are correlated to the risk variant (r2 > 0.1) and the risk alleles match the Vindija 33.19 Neanderthal genome. The core Neanderthal haplotype (r2 > 0.98) is indicated by a black bar. Some individuals carry longer Neanderthal-like haplotypes. The location of the genes in the region are indicated below using standard gene symbols. The x axis shows hg19 coordinates.
The genetic variants that are most associated with severe COVID-19 on chromosome 3 (45,859,651–45,909,024 (hg19)) are all in high linkage disequilibrium (LD)—that is, they are all strongly associated with each other in the population (r2 > 0.98)—and span 49.4 thousand bases (kb) (Fig. 1b). This ‘core’ haplotype is furthermore in weaker linkage disequilibrium with longer haplotypes of up to 333.8 kb (r2 > 0.32) (Extended Data Fig. 2). Some such long haplotypes have entered the human population by gene flow from Neanderthals or Denisovans, extinct hominins that contributed genetic variants to the ancestors of present-day humans around 40,000–60,000 years ago6,7. We therefore investigated whether the haplotype may have come from Neanderthals or Denisovans.
The index variants of the two studies1,2 are in high linkage disequilibrium (r2 > 0.98) in non-African populations (Extended Data Fig. 3). We found that the risk alleles of both of these variants are present in a homozygous form in the genome of the Vindija 33.19 Neanderthal, an approximately 50,000-year-old Neanderthal from Croatia in southern Europe8. Of the 13 single nucleotides polymorphisms constituting the core haplotype, 11 occur in a homozygous form in the Vindija 33.19 Neanderthal (Fig. 1b). Three of these variants occur in the Altai9 and Chagyrskaya 810 Neanderthals, both of whom come from the Altai Mountains in southern Siberia and are around 120,000 and about 60,000 years old, respectively (Extended Data Table 1), whereas none of the variants occurs in the Denisovan genome11. In the 333.8-kb haplotype, the alleles associated with risk of severe COVID-19 similarly match alleles in the genome of the Vindija 33.19 Neanderthal (Fig. 1b). Thus, the risk haplotype is similar to the corresponding genomic region in the Neanderthal from Croatia and less similar to the Neanderthals from Siberia.
We next investigated whether the core 49.4-kb haplotype might be inherited by both Neanderthals and present-day people from the common ancestors of the two groups that lived about 0.5 million years ago9. The longer a present-day human haplotype shared with Neanderthals is, the less likely it is to originate from the common ancestor, because recombination in each generation will tend to break up haplotypes into smaller segments. Assuming a generational time of 29 years12, the local recombination rate13 (0.53 cM per Mb), a split between Neanderthals and modern humans of 550,000 years9 and interbreeding between the two groups around 50,000 years ago, and using a published equation14, we exclude that the Neanderthal-like haplotype derives from the common ancestor (P = 0.0009). For the 333.8-kb-long Neanderthal-like haplotype, the probability of an origin from the common ancestral population is even lower (P = 1.6 × 10−26). The risk haplotype thus entered the modern human population from Neanderthals. This is in agreement with several previous studies, which have identified gene flow from Neanderthals in this chromosomal region15,16,17,18,19,20,21 (Extended Data Table 2). The close relationship of the risk haplotype to the Vindija 33.19 Neanderthal is compatible with this Neanderthal being closer to the majority of the Neanderthals who contributed DNA to present-day people than the other two Neanderthals10.
A Neanderthal haplotype that is found in the genomes of the present human population is expected to be more similar to a Neanderthal genome than to other haplotypes in the current human population. To investigate the relationships of the 49.4-kb haplotype to Neanderthal and other human haplotypes, we analysed all 5,008 haplotypes in the 1000 Genomes Project22 for this genomic region. We included all positions that are called in the Neanderthal genomes and excluded variants found on only one chromosome and haplotypes seen only once in the 1000 Genomes Project data. This resulted in 253 present-day haplotypes that contained 450 variable positions. Figure 2 shows a phylogeny relating the haplotypes that were found more than 10 times (see Extended Data Fig. 4 for all haplotypes). We find that all risk haplotypes associated with severe COVID-19 form a clade with the three high-coverage Neanderthal genomes. Within this clade, they are most closely related to the Vindija 33.19 Neanderthal.

The coloured area highlights the haplotypes that carry the risk allele at rs35044562—that is, the risk haplotypes for severe COVID-19. Arabic numbers indicate bootstrap support (100 replicates). The phylogeny is rooted with the inferred ancestral sequence of present-day humans. The three Neanderthal genomes carry no heterozygous positions in this region. Scale bar, number of substitutions per nucleotide position.
Among the individuals in the 1000 Genomes Project, the Neanderthal-derived haplotypes are almost completely absent from Africa, consistent with the idea that gene flow from Neanderthals into African populations was limited and probably indirect20. The Neanderthal core haplotype occurs in south Asia at an allele frequency of 30%, in Europe at an allele frequency of 8%, among admixed Americans with an allele frequency of 4% and at lower allele frequencies in east Asia23 (Fig. 3). In terms of carrier frequencies, we find that 50% of people in South Asia carry at least one copy of the risk haplotype, whereas 16% of people in Europe and 9% of admixed American individuals carry at least one copy of the risk haplotype. The highest carrier frequency occurs in Bangladesh, where more than half the population (63%) carries at least one copy of the Neanderthal risk haplotype and 13% is homozygous for the haplotype. The Neanderthal haplotype may thus be a substantial contributor to COVID-19 risk in some populations in addition to other risk factors, including advanced age. In apparent agreement with this, individuals of Bangladeshi origin in the UK have an about two times higher risk of dying from COVID-19 than the general population24 (hazard ratio of 2.0, 95% confidence interval, 1.7–2.4).

It is notable that the Neanderthal risk haplotype occurs at a frequency of 30% in south Asia whereas it is almost absent in east Asia (Fig. 3). This extent of difference in allele frequencies between south and east Asia is unusual (P = 0.006, Extended Data Fig. 5) and indicates that it may have been affected by selection in the past. Indeed, previous studies have suggested that the Neanderthal haplotype has been positively selected in Bangladesh25. At this point, we can only speculate about the reason for this—one possibility is protection against other pathogens. It is also possible that the haplotype has decreased in frequency in east Asia owing to negative selection, perhaps because of coronaviruses or other pathogens. In any case, the COVID-19 risk haplotype on chromosome 3 is similar to some other Neanderthal and Denisovan genetic variants that have reached high frequencies in some populations owing to positive selection or drift14,26,27,28, but it is now under negative selection owing to the COVID-19 pandemic.
It is currently not known what feature in the Neanderthal-derived region confers risk for severe COVID-19 and whether the effects of any such feature are specific to SARS-CoV-2, to other coronaviruses or to other pathogens. Once the functional feature is elucidated, it may be possible to speculate about the susceptibility of Neanderthals to relevant pathogens. However, with respect to the current pandemic, it is clear that gene flow from Neanderthals has tragic consequences.
Methods
Linkage disequilibrium was calculated using LDlink 4.129 and alleles were compared to the archaic genomes8,9,10,11 using tabix30 (HTSlib 1.10). Haplotypes were constructed from the phase 3 release of the 1000 Genomes Project22 as described. Phylogenies were estimated with phyML 3.331 using the Hasegawa–Kishino–Yano-8532 substitution model with a gamma shape parameter and the proportion of invariant sites estimated from the data. The probability of observing a haplotype of a particular length or longer owing to incomplete lineage sorting was calculated as previously described14. The inferred ancestral states at variable positions among present-day humans were taken from Ensembl33. The distribution of frequency differences of Neanderthal haplotypes between east and south Asia was computed by filtering diagnostic Neanderthal variants (fixed positions in the three high-coverage Neanderthal genomes and the Neanderthal allele missing in 108 Yoruba individuals) using a published introgression map20, followed by pruning using PLINK1.9034 (r2 cut-off of 0.5 in a sliding window of 100 variants) and allele frequency assessment in the 1000 Genomes Project. Maps displaying allele frequencies and linkage disequilibrium in different populations were made using Mathematica 11.0 (Wolfram Research) and OpenStreetMap data.
For the meta-analysis carried out by the COVID-19 Host Genetics Initiative2, participants consented and ethical approvals were obtained (https://www.covid19hg.org/partners/). The following eight studies contributed to the meta-analysis of hospitalization versus population controls: Genetic modifiers for COVID-19-related disease ‘BelCovid’ (Université Libre de Bruxelles, Belgium), Genetic determinants of COVID-19 complications in the Brazilian population ‘BRACOVID’ (University of Sao Paulo, Brazil), deCODE (deCODE Genetics, Iceland), FinnGen (Institute for Molecular Medicine Finland, Finland), GEN-COVID (University of Siena, Italy), Genes & Health (Queen Mary University of London, UK), COVID-19-Host(age) (Kiel University and University Hospitals of Oslo and Schleswig-Holstein, Germany and Norway) and the UK Biobank (UK).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The summary statistics of the genome-wide association study that support the finding of this study are available from the COVID-19 Host Genetics Initiative (round 3, ANA_B2_V2: hospitalized patients with COVID-19 compared with population controls; https://www.covid19hg.org/). The genomes used are available from the 1000 Genomes Project (phase 3 release, https://www.internationalgenome.org/) and the Max Planck Institute for Evolutionary Anthropology (Chagyrskaya, Altai and Vindija 33.19, http://cdna.eva.mpg.de/neandertal/). The ancestral alleles are available at Ensembl (release 100, https://www.ensembl.org/). Map data are from OpenStreetMap and available from https://www.openstreetmap.org.
References
Ellinghaus, D. et al. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2020283 (2020).
COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
WHO. Coronavirus disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update: Weekly Epidemiological Update 14 September 2020 https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (2020).
Vetter, P. et al. Clinical features of COVID-19. Br. Med. J. 369, m1470 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Sankararaman, S., Patterson, N., Li, H., Pääbo, S. & Reich, D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 8, e1002947 (2012).
Prüfer, K. et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017).
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014).
Mafessoni, F. et al. A high-coverage Neandertal genome from Chagyrskaya Cave. Proc. Natl Acad. Sci. USA 117, 15132–15136 (2020).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Langergraber, K. E. et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl Acad. Sci. USA 109, 15716–15721 (2012).
Kong, A. et al. A high-resolution recombination map of the human genome. Nat. Genet. 31, 241–247 (2002).
Huerta-Sánchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
Sankararaman, S. et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014).
Vernot, B. & Akey, J. M. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343, 1017–1021 (2014).
Vernot, B. et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 352, 235–239 (2016).
Steinrücken, M., Spence, J. P., Kamm, J. A., Wieczorek, E. & Song, Y. S. Model-based detection and analysis of introgressed Neanderthal ancestry in modern humans. Mol. Ecol. 27, 3873–3888 (2018).
Gittelman, R. M. et al. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol. 26, 3375–3382 (2016).
Chen, L., Wolf, A. B., Fu, W., Li, L. & Akey, J. M. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell 180, 677–687 (2020).
Skov, L. et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature 582, 78–83 (2020).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
OpenStreetMap. Planet OSM. https://planet.osm.org/ (2017).
Public Health England. COVID-19: Review of Disparities in Risks and Outcomes. https://www.gov.uk/government/publications/covid-19-review-of-disparities-in-risks-and-outcomes (2020).
Browning, S. R., Browning, B. L., Zhou, Y., Tucci, S. & Akey, J. M. Analysis of human sequence data reveals two pulses of archaic Denisovan admixture. Cell 173, 53–61 (2018).
Dannemann, M., Andrés, A. M. & Kelso, J. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 98, 22–33 (2016).
Zeberg, H., Kelso, J. & Pääbo, S. The Neandertal progesterone receptor. Mol. Biol. Evol. 37, 2655–2660 (2020).
Zeberg, H. et al. A Neanderthal sodium channel increases pain sensitivity in present-day humans. Curr. Biol. 30, 3465–3469 (2020).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Li, H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 27, 718–719 (2011).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Hasegawa, M., Kishino, H. & Yano, T. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Yates, A. D. et al. Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Acknowledgements
We thank the COVID-19 Host Genetics Initiative for making the data from the genome-wide association study available, and the Max Planck Society and the NOMIS Foundation for funding.
Author information
Authors and Affiliations
Contributions
H.Z. performed the haplotype analysis. H.Z. and S.P. jointly wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Tobias Lenz, Yang Luo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Odds ratios for hospitalization owing to COVID-19 for cohorts contributing to the meta-analysis (round 3) of the COVID-19 Host Genetics Initiative (rs35044562).
The odds ratio and the P value for the summary effect are odds ratio = 1.60 (95% confidence interval, 1.42–1.79) and P = 3.1 × 10−15 (two-sided z-test, n = 3,199 patients with COVID-19 and 897,488 controls over 8 independent studies). Data are the odds ratios and 95% confidence intervals. HOST(age), UK Biobank European (EUR), GENCOVID, deCODE and BelCovid use European population controls. BRACOVID, Genes & Health and FinnGen use American, south Asian and Finnish population controls, respectively.
Extended Data Fig. 2 Pairwise linkage disequilibrium between diagnostic Neanderthal variants.
Heat map of linkage disequilibrium between genetic variants in which one allele is shared with three Neanderthal genomes and missing in 108 Yoruba individuals. The black box highlights a haplotype of 333.8 kb between rs17763537 and rs13068572 (chromosome 3: 45,843,315–46,177,096). Red, r2 correlation; blue, D′ correlation.
Extended Data Fig. 3 Linkage disequilibrium between index variant rs11385942 and the index variant of the COVID-19 Host Genetics Initiative (rs35044562).
Shades of red indicate the extent of linkage disequilibrium (r2) in the populations included in the 1000 Genomes Project. Populations labelled ‘n/a’ are monomorphic for the protective allele of rs35044562. The previously described index variant (rs11385942)1 does not have any genetic variants in linkage disequilibrium (r2 > 0.8) in populations from Africa. Map source data from OpenStreetMap23.
Extended Data Fig. 4 Phylogeny of haplotypes in individuals included in the 1000 Genomes Project and Neanderthals covering the genomic region of the core risk haplotype.
The shaded area highlights a monophyletic group that contains all present-day haplotypes carrying the risk allele at rs35044562 and the haplotypes of the three high-coverage Neanderthals. Arabic numbers show bootstrap support (100 replicates). The tree is rooted with the inferred ancestral human sequence. Scale bar, number of substitutions per nucleotide position.
Extended Data Fig. 5 Frequency differences between south and east Asia for haplotypes introgressed from Neanderthals.
The dashed line indicates the frequency difference for the Neanderthal haplotype that confers risk of severe COVID-19.
Supplementary information
Rights and permissions
About this article

Cite this article
Zeberg, H., Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020). https://doi.org/10.1038/s41586-020-2818-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2818-3
This article is cited by
-
Altered expression of serum lncRNA CASC2 and miRNA-21-5p in COVID-19 patients
Human Genomics (2024)
-
Evolutionary origin of germline pathogenic variants in human DNA mismatch repair genes
Human Genomics (2024)
-
Dissecting the low morbidity and mortality during the COVID-19 pandemic in Africa: a critical review of the facts and fallacies
Advances in Traditional Medicine (2024)
-
Role of the Neanderthal Genome in Genetic Susceptibility to COVID-19: 3p21.31 Locus in the Spotlight
Biochemical Genetics (2024)
-
The Human Genome Organisation (HUGO) and a vision for Ecogenomics: the Ecological Genome Project
Human Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
