
Abstract
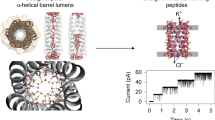
Transmembrane channels and pores have key roles in fundamental biological processes1 and in biotechnological applications such as DNA nanopore sequencing2,3,4, resulting in considerable interest in the design of pore-containing proteins. Synthetic amphiphilic peptides have been found to form ion channels5,6, and there have been recent advances in de novo membrane protein design7,8 and in redesigning naturally occurring channel-containing proteins9,10. However, the de novo design of stable, well-defined transmembrane protein pores that are capable of conducting ions selectively or are large enough to enable the passage of small-molecule fluorophores remains an outstanding challenge11,12. Here we report the computational design of protein pores formed by two concentric rings of α-helices that are stable and monodisperse in both their water-soluble and their transmembrane forms. Crystal structures of the water-soluble forms of a 12-helical pore and a 16-helical pore closely match the computational design models. Patch-clamp electrophysiology experiments show that, when expressed in insect cells, the transmembrane form of the 12-helix pore enables the passage of ions across the membrane with high selectivity for potassium over sodium; ion passage is blocked by specific chemical modification at the pore entrance. When incorporated into liposomes using in vitro protein synthesis, the transmembrane form of the 16-helix pore—but not the 12-helix pore—enables the passage of biotinylated Alexa Fluor 488. A cryo-electron microscopy structure of the 16-helix transmembrane pore closely matches the design model. The ability to produce structurally and functionally well-defined transmembrane pores opens the door to the creation of designer channels and pores for a wide variety of applications.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
Coordinates and structure files have been deposited to the Protein Data Bank with accession codes 6TJ1 (WSHC6, P21221), 6TMS (WSHC6, P1) and 6O35 (WSHC8). An electron microscopy map of TMH4C4 and the associated structure model have been deposited in the Electron Microscopy Data Bank and Protein Data Bank with accession codes EMD-30126 and 6M6Z, respectively. Source data are provided with this paper.
Code availability
All program code is in Rosetta distribution (https://www.rosettacommons.org). Example design protocols are provided in the Supplementary Information.
References
Gilbert, R. J. C., Bayley, H. & Anderluh, G. Membrane pores: from structure and assembly, to medicine and technology. Phil. Trans. R. Soc. Lond. B 372, 20160208 (2017).
Eisenstein, M. An ace in the hole for DNA sequencing. Nature 550, 285–288 (2017).
Kasianowicz, J. J., Brandin, E., Branton, D. & Deamer, D. W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl Acad. Sci. USA 93, 13770–13773 (1996).
Clarke, J. et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 4, 265–270 (2009).
Lear, J. D., Wasserman, Z. R. & DeGrado, W. F. Synthetic amphiphilic peptide models for protein ion channels. Science 240, 1177–1181 (1988).
Akerfeldt, K. S., Lear, J. D., Wasserman, Z. R., Chung, L. A. & DeGrado, W. F. Synthetic peptides as models for ion channel proteins. Acc. Chem. Res. 26, 191–197 (1993).
Joh, N. H. et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346, 1520–1524 (2014).
Lu, P. et al. Accurate computational design of multipass transmembrane proteins. Science 359, 1042–1046 (2018).
Mahendran, K. R. et al. A monodisperse transmembrane α-helical peptide barrel. Nat. Chem. 9, 411–419 (2017).
Mravic, M. et al. Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 363, 1418–1423 (2019).
Joh, N. H., Grigoryan, G., Wu, Y. & DeGrado, W. F. Design of self-assembling transmembrane helical bundles to elucidate principles required for membrane protein folding and ion transport. Phil. Trans. R. Soc. Lond. B 372, 20160214 (2017).
Niitsu, A., Heal, J. W., Fauland, K., Thomson, A. R. & Woolfson, D. N. Membrane-spanning α-helical barrels as tractable protein-design targets. Phil. Trans. R. Soc. Lond. B 372, 20160213 (2017).
Thomson, A. R. et al. Computational design of water-soluble α-helical barrels. Science 346, 485–488 (2014).
Rhys, G. G. et al. Maintaining and breaking symmetry in homomeric coiled-coil assemblies. Nat. Commun. 9, 4132 (2018).
Crick, F. H. C. The Fourier transform of a coiled-coil. Acta Crystallogr. 6, 685–689 (1953).
Grigoryan, G. & Degrado, W. F. Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol. 405, 1079–1100 (2011).
Huang, P. S. et al. High thermodynamic stability of parametrically designed helical bundles. Science 346, 481–485 (2014).
Boyken, S. E. et al. De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science 352, 680–687 (2016).
Das, R. et al. Simultaneous prediction of protein folding and docking at high resolution. Proc. Natl Acad. Sci. USA 106, 18978–18983 (2009).
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. P. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360 (1996).
Hou, X., Pedi, L., Diver, M. M. & Long, S. B. Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313 (2012).
Hou, X., Burstein, S. R. & Long, S. B. Structures reveal opening of the store-operated calcium channel Orai. eLife 7, e36758 (2018).
Dynes, J. L., Amcheslavsky, A. & Cahalan, M. D. Genetically targeted single-channel optical recording reveals multiple Orai1 gating states and oscillations in calcium influx. Proc. Natl Acad. Sci. USA 113, 440–445 (2016).
Jiang, Y. et al. X-ray structure of a voltage-dependent K+ channel. Nature 423, 33–41 (2003).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Tang, L. et al. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505, 56–61 (2014).
Pan, X. et al. Structure of the human voltage-gated sodium channel NaV1.4 in complex with β1. Science 362, eaau2486 (2018).
Fujii, S. et al. Liposome display for in vitro selection and evolution of membrane proteins. Nat. Protoc. 9, 1578–1591 (2014).
Fujii, S., Matsuura, T., Sunami, T., Kazuta, Y. & Yomo, T. In vitro evolution of α-hemolysin using a liposome display. Proc. Natl Acad. Sci. USA 110, 16796–16801 (2013).
Dwidar, M. et al. Programmable artificial cells using histamine-responsive synthetic riboswitch. J. Am. Chem. Soc. 141, 11103–11114 (2019).
Sim, A. Y. L., Lipfert, J., Herschlag, D. & Doniach, S. Salt dependence of the radius of gyration and flexibility of single-stranded DNA in solution probed by small-angle X-ray scattering. Phys. Rev. E 86, 021901 (2012).
Huang, P.-S., Boyken, S. E. & Baker, D. The coming of age of de novo protein design. Nature 537, 320–327 (2016).
Song, L. et al. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1865 (1996).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank A. Kang for assistance with crystallization; B. Sankaran of the Berkeley Center for Structural Biology at the Advanced Light Source for help with X-ray diffraction data collection; J. Sumida for AUC support; L. Carter for assistance with SEC–MALS; U. Nattermann, Q. Zhou and H. Shen for assistance with EM; SIBYLS mail-in SAXS program at the Advanced Light Source (ALS) for SAXS data collection; L. Jan, T. Cheng and R. Zhou from UCSF for sharing yeast strain SGY1528; and Rosetta@Home volunteers for contributing computing resources. The cryo-EM work was performed at the Cryo-EM Facility of Westlake University. We thank the Arnold and Mabel Beckman Cryo-EM Center at the University of Washington for access to electron microscopes, the Information Technology Center of Westlake University for providing computation support, and the Mass Spectrometry and Metabolomics Core Facility of Westlake University for mass spectrometry analysis. This work was supported by the Howard Hughes Medical Institute, the Air Force Office of Scientific Research (FA9550-18-1-0297) and NSF grant CHE 1629214. W.A.C. was supported by NIH research grant R35 NS111573. P.L. was supported by NSFC Project 31901054, Tencent Foundation and the foundation of Westlake University. SAXS data collection at SIBYLS is funded through DOE BER Integrated Diffraction Analysis Technologies (IDAT) program and NIGMS grant P30 GM124169-01, ALS-ENABLE. T.M. was in part supported by KAKENHI grants 17H00888 from JSPS. B.F.L. and X.Y.P. are supported by a Wellcome Trust Investigator award (200873/Z/16/Z). The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169-01 and the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under contract number DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
C.X., P.L., T.M.G., L.S., T.M., W.A.C. and D.B. planned the research and designed experiments. C.X., P.L. and D.B. designed the proteins. C.X., P.L., T.M.G., J.D., T.M., W.A.C. and D.B. wrote the manuscript. C.X. and P.L. carried out biophysical characterizations. T.M.G. and W.A.C. performed patch–clamp experiments and analysed data. A.U. and T.M. performed liposome permeability assays and analysed data. X.Y.P., M.J.B. and B.F.L. solved crystal structures. P.L., Q.X., M.C.J. and J.M.K. determined the cryo-EM structure. G.R. and F.D. refined the cryo-EM structure. J.D. carried out the yeast growth assay. D.J. and D.M. prepared samples for patch–clamp experiments. E.M.L. took negatively stained electron micrographs. H.B., Y.H., T.B., S.E.B. and P.-S.H. helped with design calculations. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The University of Washington has submitted a US provisional patent application (63/017,810) that covers the computational design of transmembrane pores described in this paper, listing C.X., P.L., T.M.G., W.A.C. and D.B. as inventors.
Additional information
Peer review information Nature thanks Eric Gouaux and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Design and characterization of water-soluble pores.
a, f, Design models (insets) and energy versus r.m.s.d. plots generated from Rosetta ‘fold-and-dock’ structure prediction calculations. The predicted structures converge on the design models with r.m.s.d. values less than 2.0 Å. Structures in the alternative energy minima at large r.m.s.d. positions also recapitulate the design models but with chain identities in the r.m.s.d. calculations reversed. b, g, Wavelength-scan and temperature-scan (insets) circular dichroism spectra. WSHC6 does not melt up to 95 °C, while WSHC8 has a melting temperature of 85 °C. The overlap of the pre- and post-annealing circular dichroism spectra indicates that the thermal denaturation is reversible. c, h, Representative analytical ultracentrifugation sedimentation-equilibrium curves at three different rotor speeds for WSHC6 and WSHC8, 0.2 OD230 and 0.3 OD230 in PBS (pH 7.4), respectively. The determined oligomeric states match those of the design models. d, i, Small-angle X-ray scattering (SAXS) characterization. The experimental scattering profiles (black) are similar to scattering profiles computed from the design models (red). e, The chain of water molecules in the pore of WSHC6 crystal structure (red spheres) is verified by processing the data and refining the structure in the P1 space group. j, Overlay of the crystal structure (blue) and the design model (grey) of WSHC8. Helices are more tilted in the crystal structure than in the design model.
Extended Data Fig. 2 Representative SEC and SDS–PAGE of the designed proteins.
a, b, WSHC6 and WSHC8. Molecular masses are determined by coupling SEC with MALS. c, TMHC6 and the E44F mutant. d, TMHC6(E44C) treated or untreated with MTSES. e, TMHC8. f, TMH4C4. These experiments were repeated twice with similar results.
Extended Data Fig. 3 Comparisons between WSHC6 and TMHC6 and additional characterizations of TMHC6 and mutants.
a, Sequence alignment of TMHC6 with WSHC6. b, Pore-lining residues in WSHC6 and TMHC6. Top row, overlay of the crystal structure (colours) and the design model (grey) of WSHC6. The pore is lined with alternating leucine (red layer) and isoleucine (blue layer) residues. Bottom row, the TMHC6 pore is lined with E44 ring (red layer) and K65 ring (blue layer) at the extracellular and intracellular sides, respectively. c, Negative stain EM for TMHC6 in amphipols. Protein particles on the EM grid showed round shape and size consistent with the design model (scale bar at the bottom left, 100 nm). Inset, close-up view of representative particles; each side of the particle frames represent 12.8 nm. d, Disrupting mutation in the TMHC6 pore entrance reduces the current. The E44F single mutant reduced the K+ current to half that of TMHC6. TMHC2, a previously designed transmembrane protein without a pore, does not conduct ions across the membrane. Three cells were measured for each protein; data are mean ± s.e.m. e, The covalent modification of TMHC6(E44C) by MTSES. Mass spectrometry analysis that there is a 140 Da increase in molecular mass for the mutant after MTSES treatment, in agreement with the predicted value. f, Expression of TMHC6 and mutants in insect cells for the whole-cell patch-clamp experiments. The same amount of cells were loaded into the gel and the expression levels for two variants were examined by western blot. The E44F mutant had a similar, if not higher, expression level to TMHC6. The E44C mutant was expressed at a slightly lower level compared to TMHC6. These experiments were repeated three times with similar results.
Extended Data Fig. 4 The expression of TMHC6 complements a yeast strain defective in K+ uptake.
a, SGY1528 with an empty vector MCS grew slowly when K+ concentration was lower than 5 mM. The observed growth rate showed dependency on extracellular K+ concentration. b, SGY1528 supplemented with the TMHC6 gene rescued the yeast growth at lower extracellular K+ concentrations (1 mM–5 mM) and showed increased growth rates at higher extracellular K+ concentrations (7.5 mM–100 mM). c, d, With increasing concentrations of extracellular Na+, TMHC6 yeast showed decreased growth rate (d) in comparison with the insensitive growth rates of MCS yeast (c). These results suggest that TMHC6 conducts K+ and complements the defective K+ uptake in strain SGY1528; this rescuing effect is sensitive to extracellular Na+ concentrations indicating an increased Na+ permeability. Detailed methods are described in the Supplementary Information. The minimal medium and the seeding process are carefully designed to not contain or bring in potassium. The background K+ concentration should be low, which is suggested by the sharp difference between curves for 0 and 1 mM K+ in the case of TMHC6.
Extended Data Fig. 5 Design and additional characterizations of the designed 16-helix pores.
a, b, Design models from the first and second rounds of water-soluble designs. The monomers of the first round designs (a, 70 amino acids) are considerably smaller than those of the second round (b, 100 amino acids). c, Sequence alignment of TMHC8 with WSHC8. d, Pore-lining residues in WSHC8 and TMH4C4. The crystal and cryo-EM structures are in colours. The design models are in grey. Top row, the lumen of WSHC8 pore is freely water-accessible, so the residues inside the pore are all polar. Shown in the figure are three representative layers of the pore-lining residues in the crystal structure, Glu69 ring (red), Lys80 ring (blue), and Glu87 (orange). The missing heavy atoms of these flexible residues are built using Rosetta with backbone constraints. Bottom row, three pore-lining layers in the cryo-EM structure of TMH4C4 corresponding to the three layers in the top row. Glu69 and Glu87 are replaced with glutamine and leucine, respectively. e, f, Circular dichroism spectra and thermal stability of 16-helix transmembrane pores. An unfolding transition is observed at around 75 °C for TMHC8 (e). TMH4C4 (f) is thermally stable up until 95 °C. g, h, Representative AUC sedimentation-equilibrium curves of 16-helix transmembrane nanopores. By fitting the data globally as a single ideal species in solution, TMHC8 is shown to form complexes with a molecular mass of 98.9 kDa, which is in between the masses of a heptamer and an octamer. The molecular mass of TMH4C4 is determined to be 98.1 kDa, very close to that of a tetramer. ‘MW (D)’ refers to the molecular mass of the oligomer design and ‘MW (E)’ refers to the molecular mass determined in the experiment.
Extended Data Fig. 6 In vitro protein synthesis and characterizations of TMHC6 and TMH4C4.
a, SEC analyses of TMHC6 and TMH4C4 purified from E. coli (top) and synthesized in vitro (middle and bottom). b, The in vitro synthesized products were analysed by SDS–PAGE and autoradiography. The means of three independent experiments are shown. The error bars indicate s.e.m. c, EmrE, one of the E. coli-derived membrane proteins, showed strong interaction with LUV, whereas GusA, a soluble enzyme, did not. For TMHC6 and TMH4C4, the fraction interacting with LUV was found to be 25.9% and 17.6%, respectively, among synthesized, indicating that the fraction associated with the membrane is similar between them. The mean of four independent experiments are shown. The error bars indicate s.e.m. Student’s paired t-test with a two-sided distribution was used to calculate the P values (*P < 0.05, **P < 0.01; from left to right P = 1.65 × 10−6, 0.0357, 0.0040, 0.0024). d, The narrowest dimension of the head group of Alexa Fluor 488–biocytin is approximately 12 Å. The van der Waals radius of nitrogen atoms is 1.55 Å. e, Representative original data for Fig. 3f. Data of approximately 15,000 to 20,000 particles are shown. Similar results were obtained with 7 independent experiments. f, Flow cytometry data of the liposomes with pores made of α-haemolysin (AH). Time courses of the median values of the histogram of AF488/OA647 fluorescence, which represents the concentration of the Alexa Fluor 488 inside the liposome, are shown. The means of three independent experiments are plotted with the error bars indicating the s.e.m. g, Representative original data of f. Similar results were obtained with 3 independent experiments.
Extended Data Fig. 7 Cryo-EM resolution estimation and data processing.
a, Exemplary cryo-EM micrograph of purified TMH4C4 after drift correction and dose-weighting. All the micrographs showed similar results. b, Class averages after the final round of 2D classification sorted in descending order by the number of particles in each class. The white scale bar in the bottom right panel indicates 10 nm. c, Angular distribution plot for the final reconstruction from two different views. d, The gold-standard Fourier shell correlation (FSC) curves for the 3D reconstruction. Deriving map resolution from FSC = 0.143 is indicated. e, Processing of 2,166 EM micrographs resulted in a total number of 2,146,524 TMH4C4 particles. After a 2D sub-classification, 3D classifications and refinement, a final data set containing 64,739 particles was used for 3D auto-refinement within RELION 3.0. Local resolution of TMH4C4 was determined within RELION 3.0. Coloured full views (lower lane) from two different orientations illustrate the resolution of different regions in the protein. The low resolution ‘belt’ in the right panel indicates the density for detergents. f, EM density from 213,654 particles. An EM map (grey) from the second round of 3D classification from 213,654 particles (e) is shown in three perpendicular views. Superposition of the cryo-EM structure of TMH4C4 (cyan) to the map shows a good fit.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary References and Appendices (A-C).
Rights and permissions
About this article

Cite this article
Xu, C., Lu, P., Gamal El-Din, T.M. et al. Computational design of transmembrane pores. Nature 585, 129–134 (2020). https://doi.org/10.1038/s41586-020-2646-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2646-5
This article is cited by
-
Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics
Nature Chemistry (2024)
-
Design of complicated all-α protein structures
Nature Structural & Molecular Biology (2024)
-
Hydrophobic mismatch drives self-organization of designer proteins into synthetic membranes
Nature Communications (2024)
-
Machine learning for functional protein design
Nature Biotechnology (2024)
-
Transient water wires mediate selective proton transport in designed channel proteins
Nature Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
