
Abstract
In March 2020, the World Health Organization (WHO) declared coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1, a pandemic. With rapidly accumulating numbers of cases and deaths reported globally2, a vaccine is urgently needed. Here we report the available safety, tolerability and immunogenicity data from an ongoing placebo-controlled, observer-blinded dose-escalation study (ClinicalTrials.gov identifier NCT04368728) among 45 healthy adults (18–55 years of age), who were randomized to receive 2 doses—separated by 21 days—of 10 μg, 30 μg or 100 μg of BNT162b1. BNT162b1 is a lipid-nanoparticle-formulated, nucleoside-modified mRNA vaccine that encodes the trimerized receptor-binding domain (RBD) of the spike glycoprotein of SARS-CoV-2. Local reactions and systemic events were dose-dependent, generally mild to moderate, and transient. A second vaccination with 100 μg was not administered because of the increased reactogenicity and a lack of meaningfully increased immunogenicity after a single dose compared with the 30-μg dose. RBD-binding IgG concentrations and SARS-CoV-2 neutralizing titres in sera increased with dose level and after a second dose. Geometric mean neutralizing titres reached 1.9–4.6-fold that of a panel of COVID-19 convalescent human sera, which were obtained at least 14 days after a positive SARS-CoV-2 PCR. These results support further evaluation of this mRNA vaccine candidate.
Similar content being viewed by others


Main
In December 2019, a pneumonia outbreak of unknown cause occurred in Wuhan, China. By January 2020, a new coronavirus was identified as the aetiological agent. Within a month, the genetic sequence of the virus became available (MN908947.3). Infections with SARS-CoV-2 and the resulting disease, COVID-19, have spread globally. On 11 March 2020, the WHO declared the COVID-19 outbreak a pandemic1. So far, the United States has reported the highest number of cases globally2,3. No vaccines are currently available to prevent SARS-CoV-2 infection or COVID-19.
The RNA vaccine platform has enabled rapid vaccine development in response to this pandemic. RNA vaccines provide flexibility in the design and expression of vaccine antigens that can mimic the structure and expression of the antigen during natural infection. RNA is required for protein synthesis, does not integrate into the genome, is transiently expressed, is metabolized and eliminated by the natural mechanisms of the body and is therefore considered safe4,5,6,7. RNA-based prophylactic infectious-disease vaccines and RNA therapeutic agents have been shown to be safe and well-tolerated in clinical trials. In general, vaccination with RNA elicits a robust innate immune response. RNA directs the expression of the vaccine antigen in host cells and has intrinsic adjuvant effects8. A strength of the RNA-vaccine manufacturing platform—irrespective of the encoded pathogen antigen—is the ability to rapidly produce large quantities of vaccine doses against a new pathogen9,10.
Vaccine RNA can be modified by incorporating 1-methyl-pseudouridine, which dampens innate immune sensing and increases mRNA translation in vivo11. The BNT162b1 vaccine candidate that is currently investigated clinically incorporates such nucleoside-modified mRNA and encodes the RBD of the spike protein of SARS-CoV-2, a key target of virus-neutralizing antibodies12,13,14. The RBD antigen expressed by BNT162b1 is modified by the addition of a T4 fibritin-derived foldon trimerization domain to increase its immunogenicity15 by multivalent display16. The proper folding of the RBDs in the resulting protein construct has been confirmed by high resolution structural analysis (A.B.V. et al., manuscript in preparation). The vaccine RNA is formulated in lipid nanoparticles for more-efficient delivery into cells after intramuscular injection17. BNT162b1 is one of several RNA-based SARS-CoV-2 vaccine candidates18 that are studied in parallel for selection to advance to a safety and efficacy trial. Here, we present the available data, up to 14 days after a second dose in adults (18–55 years of age) from an ongoing phase I/II vaccine study with BNT162b1, which is also enrolling adults who are 65–85 years of age (ClinicalTrials.gov identifier, NCT04368728).
Study design and demographics
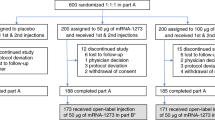
Between 4 May 2020 and 19 June 2020, 76 participants were screened, and 45 participants were randomized and vaccinated. Per dose level (10 μg and 30 μg), 12 participants were vaccinated with BNT162b1 on days 1 and 21, 12 participants received a 100-μg dose on day 1 and 9 participants received placebo (Fig. 1). The study population consisted of healthy male and female participants with a mean age of 35.4 years (range, 19–54 years); 51.1% were male and 48.9% were female. Most participants self-reported as white (82.2%) and non-Hispanic/non-Latinx (93.3%) (Extended Data Table 1).

Participants who were not assigned (n = 20) were screened but not randomized because enrolment had closed.
Safety and tolerability
In the 7 days after vaccination doses 1 and 2, pain at the injection site was the most-frequent solicited local reaction, reported after the first dose by 58.3% (7 out of 12) in the 10-μg BNT162b1 group, 100.0% (12 out of 12 each) in the 30-μg and 100-μg BNT162b1 groups, and 22.2% (2 out of 9) in the placebo group. After the second dose, pain was reported by 83.3% (10 out of 12) and 100.0% of individuals who received 10 μg and 30 μg BNT162b1, respectively, and by 16.7% of individuals who received the placebo. All local reactions were mild or moderate in severity except for one report of severe pain after the first dose of 100 μg BNT162b1 (Fig. 2).

Solicited injection-site (local) reactions were: pain at injection site (mild, does not interfere with activity; moderate, interferes with activity; severe, prevents daily activity; grade 4, emergency room visit or hospitalization) and redness and swelling (mild, 2.0–5.0 cm in diameter; moderate, >5.0–10.0 cm in diameter; severe, >10.0 cm in diameter; grade 4: necrosis or exfoliative dermatitis for redness, and necrosis for swelling). Data were collected with the use of electronic diaries for 7 days after each vaccination.
The most-common systemic events reported in the 7 days after each vaccination in both BNT162b1 and placebo groups were mild to moderate fatigue and headache. Reports of fatigue and headache were more common in the BNT162b1 groups than in the placebo group. In addition, chills, muscle pain and joint pain were reported by individuals who received BNT162b1 but not by individuals who received the placebo. Systemic events increased with dose level and were reported in a greater number of participants after the second dose (10-μg and 30-μg groups). After the first dose, fever (defined as ≥38.0 °C) was reported by 8.3% (1 out of 12) of participants who received 10 μg and 30 μg BNT162b1 and by 50.0% (6 out of 12) of individuals who received 100 μg BNT162b1. After the second dose, 8.3% (1 out of 12) of participants who received 10 μg BNT162b1 and 75.0% (9 out of 12) of participants who received 30 μg BNT162b1 reported fever of ≥38.0 °C. On the basis of the reactogenicity reported after the first dose of 100 μg and the second dose of 30 μg, participants who received an initial 100-μg dose did not receive a second 100-μg dose. Fevers generally resolved within 1 day of onset. No grade 4 systemic events or fever were reported (Fig. 3a, b). Most local reactions and systemic events peaked by day 2 after vaccination and resolved by day 7.

a, Systemic events and medication use reported within 7 days after vaccination 1 for all dose levels. b, Systemic events and medication use reported within 7 days after vaccination 2 for the 10-μg and 30-μg dose levels. Solicited systemic events were: fatigue, headache, chills, new or worsened muscle pain, new or worsened joint pain (mild, does not interfere with activity; moderate, some interference with activity; severe, prevents daily activity), vomiting (mild, 1–2 times in 24 h; moderate, >2 times in 24 h; severe, requires intravenous hydration), diarrhoea (mild, 2–3 loose stools in 24 h; moderate, 4–5 loose stools in 24 h; severe: 6 or more loose stools in 24 h); grade 4 for all events: emergency room visit or hospitalization; and fever (mild, 38.0–38.4 °C; moderate, 38.5–38.9 °C; severe, 39.0–40.0 °C; grade 4, >40.0 °C). Medication indicates the proportion of participants who reported the use of antipyretic or pain medication. Data were collected with the use of electronic diaries for 7 days after each vaccination.
Adverse events (Extended Data Table 2) were reported by 50.0% (6 out of 12) of participants who received either 10 or 30 μg of BNT162b1, 58.3% (7 out of 12) of participants who received 100 μg of BNT162b1, and 11.1% (1 out of 9) of placebo recipients. Two participants reported a severe adverse event: grade 3 fever 2 days after vaccination in the 30-μg group, and sleep disturbance 1 day after vaccination in the 100-μg group. Related adverse events were reported by 25% (3 out of 12 in the 10-μg group) to 50% (6 out of 12 each in the 30-μg and 100-μg groups) of individuals who received BNT162b1 and by 11.1% (1 out of 9) of participants who received the placebo. No serious adverse events were reported.
No grade 1 or greater change in routine clinical laboratory values or laboratory abnormalities were observed for most participants after either of the BNT162b1 vaccinations. Of those with laboratory changes, the largest changes were decreases in the lymphocyte count after the first dose in 8.3% (1 out of 12), 45.5% (5 out of 11) and 50.0% (6 out of 12) of participants who received 10 μg, 30 μg and 100 μg BNT162b1, respectively. One participant each in the 10-μg (8.3% (1 out of 12)) and 30-μg (9.1% (1 out of 11)) groups and 4 participants in the 100-μg group (33.3% (4 out of 12)) had grade 3 decreases in the lymphocyte count. These decreases in lymphocyte count after the first dose were transient and returned to normal 6–8 days after vaccination (Extended Data Fig. 1). In addition, grade-2 neutropenia was noted 6–8 days after the second dose in 1 participant each in the 10-μg and 30-μg BNT162b1 groups. These two participants continue to be followed in the study, and no adverse events or clinical manifestations of neutropenia have been reported to date. None of the post-vaccination abnormalities observed were associated with clinical findings.
Immunogenicity
RBD-binding IgG concentrations and SARS-CoV-2-neutralizing titres were assessed at baseline, at 7 and 21 days after the first dose, at 7 days (day 28) and 14 days (day 35) after the second dose of BNT162b1. By 21 days after the first dose (for all three dose levels), geometric mean concentrations (GMCs) of RBD-binding IgG ranged from 534 to 1,778 U ml−1 (Fig. 4a). In comparison, a panel of 38 SARS-CoV-2 infection and/or COVID-19 convalescent sera drawn at least 14 days after a PCR-confirmed diagnosis from patients with COVID-19 (18–83 years of age) had an RBD-binding IgG GMC of 602 U ml−1. (Additional information on the convalescent serum panel is included in the Methods.) By 7 days after the second dose (for the 10-μg and 30-μg dose levels), RBD-binding IgG GMCs had increased to 4,813 and to 27,872 U ml−1, respectively. RBD-binding antibody concentrations among participants who received one dose of 100 μg BNT162b1 did not increase further at 21 days after the first vaccination. In the participants who received the 10-μg and 30-μg doses of BNT162b1, highly elevated RBD-binding antibody concentrations persisted to the last time point evaluated (day 35, 14 days after the second dose). These RBD-binding antibody concentrations were 5,880–16,166 U ml−1 compared to 602 U ml−1 in the panel of human convalescent sera.

Participants in groups of 15 were vaccinated with the indicated dose levels of BNT162b1 (n = 12) or with placebo (n = 3) on days 1 (all dose levels and placebo) and 21 (10-μg and 30-μg dose levels and placebo). Reponses in individuals who received the placebo for each of the dosing groups are combined. The 28- and 35-day blood samples were obtained 7 and 14 days after the second vaccination. Sera were obtained before vaccination (day 1), and 7, 21, 28 and 35 days after the first vaccination. Human COVID-19 convalescent sera (HCS, n = 38) were obtained at least 14 days after PCR-confirmed diagnosis and at a time when the donors were asymptomatic. a, GMCs of recombinant RBD-binding IgG. Because the measured antibody concentrations using the Luminex assay are obtained in arbitrary units, they cannot be directly translated into concentrations on a molar or mass basis. The lower limit of quantitation is 1.15. b, The 50% SARS-CoV-2-neutralizing GMTs. Each data point represents a serum sample, and each vertical bar represents a geometric mean with 95% confidence interval. The number above the bars are either the GMC (a) or GMT (b) for the group. Arrows indicate the timing of vaccination (blood was obtained before vaccination on the vaccination days).
For all doses, small increases in SARS-CoV-2-neutralizing geometric mean titres (GMTs) were observed 21 days after the first dose (Fig. 4b). Substantially greater serum neutralizing GMTs were achieved 7 days after the second 10-μg and 30-μg dose, reaching 168–267. Neutralizing GMTs further increased by 14 days after the second dose to 180 (10-μg dose level) and 437 (30-μg dose level), compared to 94 for the panel of human convalescent sera. The kinetics and durability of the neutralizing titres are being monitored.
Discussion
The RNA-based SARS-CoV-2 vaccine candidate BNT162b1, which was administered as 10-μg, 30-μg or 100-μg doses in healthy adults (18–55 years of age), exhibited a tolerability and safety profile consistent with those previously observed for mRNA-based vaccines5. A clear dose-level response in elicited neutralizing titres was observed after doses 1 and 2 in participants with a particularly steep dose response between the 10 μg and 30 μg dose levels.
On the basis of the tolerability profile of the first dose at 100 μg and the second dose at 30 μg, participants randomized to the 100-μg group did not receive a second vaccination. Reactogenicity was generally greater after the second dose in the other two dosing levels; however, symptoms were transient and resolved within a few days. Transient decreases in lymphocyte counts (grades 1–3) were observed within a few days after vaccination, and returned to baseline within 6–8 days in all participants. These laboratory abnormalities were not associated with clinical findings. RNA vaccines are known to induce type-I interferon, which has been associated with transient migration of lymphocytes into tissues19,20,21,22.
Robust immunogenicity was observed after vaccination with BNT162b1. RBD-binding IgG concentrations were detected at 21 days after the first dose, and these were substantially increased 7 days after the second dose given at day 21. After the first dose, the RBD-binding IgG GMCs (10-μg dose) were similar to those observed in a panel of 38 convalescent human serum samples, obtained at least 14 days after a PCR-confirmed diagnosis of SARS-CoV-2 infection and/or COVID-19. After the first dose, GMCs were similar in the 30-μg and 100-μg groups and higher than those in the panel of human convalescent sera. After the second dose, with 10 μg or 30 μg BNT162b1, the RBD-binding IgG GMCs were around 8.0–50-fold that of the GMC of the convalescent serum panel.
The higher RBD-binding IgG GMC elicited by the vaccine relative to the GMC of the human convalescent serum panel may be attributed, in part, to antibodies that bind to epitopes that are exposed on the RNA-expressed RBD immunogen and the recombinant RBD target antigen of the binding assay but are buried and inaccessible to antibodies on the RBDs that are incorporated into the spikes of SARS-CoV-2 virions. Neutralization provides a measure of the vaccine-elicited antibody response that is more relevant to potential protection. Neutralization titres were measurable after a single vaccination at day 21 for all dose levels. At day 28 (7 days after the second dose), substantial SARS-CoV-2 neutralization titres were observed. The virus-neutralizing GMTs after the second dose of 10 μg and 30 μg were, respectively, 1.8-fold and 2.8-fold the GMT of the convalescent serum panel. By day 35 (14 days after the second dose)—despite a decrease in RBD-binding IgG titres since day 28—neutralizing GMTs continued to increase, to 1.9-fold and 4.6-fold the GMT of the convalescent panel for the 10 μg and 30 μg doses, respectively, which is consistent with affinity maturation.
Assuming that the neutralization titres that are induced by natural infection provide protection from COVID-19, comparing vaccine-induced SARS-CoV-2 neutralization titres to those from sera of convalescent humans provides a benchmark for the magnitude of the vaccine-elicited response and the potential of the vaccine to provide protection. Because the titre at which human neutralizing antibodies are protective remains unknown, these findings are not proof of vaccine efficacy. Efficacy will be determined in a pivotal phase III trial. Because the cohort that received the 100 μg dose level did not receive the booster dose, no data for immunogenicity after a second vaccination at this dose level are available; however, there were no substantial differences in immunogenicity between the 30-μg and 100-μg dose levels after the first dose. This observation suggests that a well-tolerated and immunogenic dose level may be between 10 μg and 30 μg for this vaccine candidate.
Our study had several limitations. Although we used convalescent sera as a comparator, the kind of immunity (T cells versus B cells or both) and level of immunity needed to protect from COVID-19 are unknown. Furthermore, this analysis of available data did not assess immune responses or safety beyond 2 weeks after the second dose of vaccine. Both are important to inform the public health use of this vaccine. Follow-up will continue for all participants and will include collection of serious adverse events for 6 months and COVID-19 infection and multiple additional immunogenicity measurements for up to 2 years. Although our population of healthy adults up to 55 years of age is appropriate for a phase I/II study, it does not accurately reflect the population at highest risk for COVID-19. Adults who are 65 years of age and over have already been enrolled in this study and results will be reported as they become available. Later phases of this study will prioritize enrolment of more diverse populations, including those with chronic underlying health conditions and from racial and ethnic groups that are adversely affected by COVID-1923.
The clinical testing of BNT162b1 described here has taken place in the context of a broader, ongoing COVID-19-vaccine-development program. That program includes the clinical testing of three additional vaccine candidates, including candidates that encode the full-length spike protein, and a parallel trial in Germany, in which additional immune responses, including neutralizing responses against variant strains and cell-mediated responses, are being assessed24. The resulting comparative data will allow us to address whether a full-length spike immunogen, which presents additional epitopes, is better able to elicit high virus-neutralizing titres that are robust to potential antigenic drift of SARS-CoV-2 than the relatively small RBD immunogen that is encoded by BNT162b1. The clinical findings for the BNT162b1 RNA-based vaccine candidate are encouraging and strongly support accelerated clinical development, including efficacy testing, and at-risk manufacturing to maximize the opportunity for the rapid production of a SARS-CoV-2 vaccine to prevent COVID-19.
Methods
Study design
This study was conducted in healthy men and women (who were not pregnant) who were 18–55 years of age to assess the safety, tolerability and immunogenicity of ascending dose levels of various BNT162 mRNA vaccine candidates. In the part of the study reported here, assessment of three dose levels (10 μg, 30 μg or 100 μg) of the BNT162b1 candidate was conducted at two sites in the USA. This study used a sentinel cohort design with progression and dose escalation taking place after review of data from the sentinel cohort at each dose level. The study is registered at ClinicalTrials.gov (NCT04368728). The phase I portion of this study was observer-blinded at the site level. Investigators were blinded to participant-level study intervention assignment; but investigators were not blinded to group-level assignment for the dataset included in this Article.
Eligibility
Key exclusion criteria included individuals with known infection with human immunodeficiency virus, hepatitis C virus or hepatitis B virus; immunocompromised individuals and those with a history of autoimmune disease; and those with increased risk for severe COVID-19, previous clinical or microbiological diagnosis of COVID-19, receipt of medications intended to prevent COVID-19, previous vaccination with any coronavirus vaccine, a positive serological test for SARS-CoV-2 IgM and/or IgG at the screening visit, and a SARS-CoV-2 nucleic acid amplification test-positive nasal swab within 24 h before study vaccination.
The final protocol and informed consent document were approved by institutional review boards for each of the participating investigational centres. This study was conducted in compliance with all International Council for Harmonisation good clinical practice guidelines and the ethical principles of the Declaration of Helsinki. A signed and dated informed consent form was required before any study-specific activity was performed.
End points
In this report, results from the following study primary end points are presented: the proportion of participants who reported solicited local reactions, systemic events and use of antipyretic and/or pain medication within 7 days after vaccination, adverse events and serious adverse events (available up to around 45 days after dose 1), and the proportion of participants with clinical laboratory abnormalities 1 and 7 days after vaccination and grading shifts in laboratory assessments between baseline and 1 and 7 days after dose 1, and between dose 2 and 7 days after dose 2. Secondary end points included: SARS-CoV-2-neutralizing GMTs and SARS-CoV-2 RBD-binding IgG GMCs 7 and 21 days after dose 1, and 7 and 14 days after dose 2.
Procedures
Study participants were randomly assigned to a vaccine group using an interactive web-based response technology system with each group comprising 15 participants (12 active vaccine recipients and 3 placebo recipients). Participants received two 0.5-ml doses of either BNT162b1 or placebo, administered by intramuscular injection into the deltoid muscle.
BNT162b1 incorporates a good manufacturing practice-grade mRNA drug substance that encodes the trimerized SARS-CoV-2 spike glycoprotein RBD antigen. The coding sequence for the antigen has been deposited with GenBank (accession number, MN908947.3). The mRNA is formulated with lipids as the mRNA–lipid nanoparticle drug product. The vaccine was supplied as a buffered-liquid solution for intramuscular injection and was stored at −80 °C. The placebo was a sterile saline solution for injection (0.9% sodium chloride injection, in a 0.5-ml dose).
Safety assessments
Safety assessments included a 4-h observation after vaccination (for the first 5 participants vaccinated in each group), or a 30-min observation (for the remainder of participants) for immediate adverse events. The safety assessments also included self-reporting of solicited local reactions (redness, swelling and pain at the injection site), systemic events (fever, fatigue, headache, chills, vomiting, diarrhoea, muscle pain and joint pain), the use of antipyretic and/or pain medication in an electronic diary for 7 days after vaccination, and the reporting of unsolicited adverse events and serious adverse events after vaccination. Haematology and chemistry assessments were conducted at screening, 1 and 7 days after the first dose, and 7 days after the second dose.
There were protocol-specified safety stopping rules for all sentinel cohort participants. Both an internal review committee and an external data monitoring committee reviewed all safety data. No stopping rules were met before the publication of this report.
Human convalescent serum panel
The 38 human SARS-CoV-2 infection and/or COVID-19 convalescent sera were drawn from participants, who were 18–83 years of age, at least 14 days after PCR-confirmed diagnosis, and at a time when participants were asymptomatic. The mean age of the donors was 45 years of age. Neutralizing GMTs in subgroups of the donors were as follows: ≤55 years of age, 82 (n = 29); >55 years of age, 142 (n = 9); symptomatic infections, 90 (n = 35); asymptomatic infections, 156 (n = 3). The antibody titre for the one individual who was hospitalized was 618. The sera were obtained from Sanguine Biosciences, the MT Group and Pfizer Occupational Health and Wellness.
Immunogenicity assessments
For immunogenicity assessments, 50 ml of blood was collected before each study vaccination, at 7 and 21 days after the first dose, and at 7 and 14 days after the second dose. In the RBD-binding IgG assay, a recombinant SARS-CoV-2 RBD containing a C-terminal Avitag (Acro Biosystems, SPD-C82E9) and no foldon domain was bound to streptavidin-coated Luminex microspheres. In brief, 1.25 × 107 microspheres/ml were coated with streptavidin by 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride reaction. Recombinant RBD Avitag was coupled to streptavidin beads by incubating for 90 min at room temperature with shaking (35 rpm). Beads were blocked in 1% BSA buffer for 30 min at room temperature. Heat-inactivated serum from participants was diluted 1:500, 1:5,000 and 1:50,000 in assay buffer (PBS with 0.5% BSA, 0.05% Tween-20 and 0.02% sodium azide). Following a 16–20-h incubation at 2–8 °C with shaking (300 rpm), plates were washed three times in a solution containing 0.05% Tween-20. An R-phycoerythrin-conjugated goat anti-human polyclonal antibody (Jackson Labs) was then added to plates for 90 min at room temperature with shaking (300 RPM). Plates were then washed a final time in a solution containing 0.05% Tween-20. Data were captured as median fluorescent intensities using a Luminex reader and converted to U/ml antibody concentrations using a reference standard curve with arbitrary assigned concentrations of 100 U/ml and accounting for the serum dilution factor. The reference standard was composed of a pool of five COVID-19 convalescent serum samples (>14 days after PCR diagnosis). Three dilutions are used to increase the likelihood that at least one result for any sample will fall within the usable range of the standard curve. Assay results were reported in U/ml of IgG. The final assay results are expressed as the GMC of all sample dilutions that produced a valid assay result within the assay range.
The SARS-CoV-2 neutralization assay used a previously described strain of SARS-CoV-2 (USA_WA1/2020) that had been rescued by reverse genetics and engineered by the insertion of an mNeonGreen gene into open-reading frame 7 of the viral genome25. This reporter virus generates similar plaque morphologies and indistinguishable growth curves from the wild-type virus. Viral master stocks (2 × 107 plaque-forming units per ml) used for the neutralization assay were grown in Vero E6 cells as previously described25. When testing patient convalescent serum specimens, the fluorescent neutralization assay produced comparable results as the conventional plaque reduction neutralization assay26. In brief, serial dilutions of heat-inactivated sera from participants were incubated with the reporter virus to yield an infection rate of approximately 10–30% of the Vero monolayer) for 1 h at 37 °C before inoculating Vero CCL81 cell monolayers (targeted to have 8,000–15,000 cells per well) in 96-well plates to enable the accurate quantification of infected cells. Total cell counts per well were enumerated by nuclear stain (Hoechst 33342) and fluorescent virally infected foci were detected 16–24 h after inoculation with a Cytation 7 Cell Imaging Multi-Mode Reader (BioTek) with Gen5 Image Prime v.3.09. Titres were calculated in GraphPad Prism v.8.4.2 by generating a four-parameter logistical fit of the percentage neutralization at each serial serum dilution. The 50% neutralization titre was reported as the interpolated reciprocal of the dilution that yielded a 50% reduction in fluorescent viral foci.
Statistical analysis
The sample size for the reported part of the study was not based on statistical hypothesis testing. The primary safety objective was evaluated by descriptive summary statistics for local reactions, systemic events, abnormal haematology and chemistry laboratory parameters, adverse events and serious adverse events after each vaccine dose for each vaccine group. The secondary immunogenicity objectives were descriptively summarized at the various time points. All participants with data available were included in the safety and immunogenicity analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual anonymized participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information. These data are interim data from an ongoing study for which the database is not locked. Data have not yet been source-verified or subjected to standard quality check procedures that would occur at the time of database lock and may therefore be subject to change.
Change history
19 January 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-03098-3.
References
World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---7-september-2020 (2020).
Coronavirus Resource Center. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). https://coronavirus.jhu.edu/map.html (Johns Hopkins University & Medicine, 2020).
World Health Organization. Coronavirus Disease 2019 (COVID-19) Situation Report 154. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200622-covid-19-sitrep-154.pdf?sfvrsn=d0249d8d_2 (2020).
Alberer, M. et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390, 1511–1520 (2017).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Şahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Petsch, B. et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 30, 1210–1216 (2012).
Rauch, S., Jasny, E., Schmidt, K. E. & Petsch, B. New vaccine technologies to combat outbreak situations. Front. Immunol. 9, 1963 (2018).
Şahin, U., Karikó, K. & Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Karikó, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
He, Y. et al. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 324, 773–781 (2004).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. 26, 1422–1427 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020).
Güthe, S. et al. Very fast folding and association of a trimerization domain from bacteriophage T4 fibritin. J. Mol. Biol. 337, 905–915 (2004).
Bachmann, M. F. & Zinkernagel, R. M. Neutralizing antiviral B cell responses. Annu. Rev. Immunol. 15, 235–270 (1997).
Pardi, N. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 217, 345–351 (2015).
Walsh, E. E. RNA-based COVID-19 vaccine BNT162b2 selected for a pivotal efficacy study. Preprint at https://doi.org/10.1101/2020.08.17.20176651 (2020).
Foster, G. R. et al. IFN-α subtypes differentially affect human T cell motility. J. Immunol. 173, 1663–1670 (2004).
Hopkins, R. J. et al. Randomized, double-blind, placebo-controlled, safety and immunogenicity study of 4 formulations of Anthrax Vaccine Adsorbed plus CPG 7909 (AV7909) in healthy adult volunteers. Vaccine 31, 3051–3058 (2013).
Regules, J. A. et al. A recombinant vesicular stomatitis virus Ebola vaccine. N. Engl. J. Med. 376, 330–341 (2017).
Lai, L. et al. Emergency postexposure vaccination with vesicular stomatitis virus-vectored Ebola vaccine after needlestick. J. Am. Med. Assoc. 313, 1249–1255 (2015).
Stokes, E. K. et al. Coronavirus disease 2019 case surveillance — United States, January 22–May 30, 2020. MMWR Morb. Mortal. Wkly. Rep. 69, 759–765 (2020).
Şahin, U. et al. Concurrent human antibody and TH1 type T-cell responses elicited by a COVID-19 RNA vaccine. Preprint at https://doi.org/10.1101/2020.07.17.20140533 (2020).
Xie, X. et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 27, 841–848 (2020).
Muruato, A. E. et al. A high-throughput neutralizing antibody assay for COVID-19 diagnosis and vaccine evaluation. Nat. Commun. 11, 4059 (2020).
Acknowledgements
We thank C. Monahan and D. Gantt for writing and editorial support; H. Ma, J. Trammel and K. Challagali for statistical analysis support in the generation of this manuscript; all of the participants who volunteered for this study; A. Kottkamp, R. Herati, R. Pellet Madan, M. Olson, M. Samanovic-Golden, E. Cohen, A. Cornelius, L. Frye, H. Youn, B. Fran, K. Ballani, N. Veling, J. Erb, M. Ali, L. Zhao, S. Rettig, H. Khan, H. Lambert, K. Hu, J. Hyde, M. McArthur, J. Ortiz, R. Rapaka, L. Wadsworth, G. Cummings, T. Robinson, N. Greenberg, L. Chrisley, W. Somrajit, J. Marron, C. Thomas, K. Brooks, L. Turek, P. Farley, S. Eddington, P. Komninou, M. Reymann, K. Strauss, B. Shrestha, S. Joshi, R. Barnes, R. Sukhavasi, M. Lee, A. Kwon, T. Sharp, E. Pierce, M. Criddle, A. Cline, S. Parker, M. Dickey, K. Buschle, A. Cawein, J. L. Perez, H. Seehra, D. Tresnan, R. Maroko, H. Smith, S. Tweedy, A. Jones, G. Adams, R. Malick, E. Worobetz, E. Weaver, L. Zhang, C. Devlin, D. Boyce, E. Harkins Tull, M. Boaz, M. Cruz, C. Rosenbaum, C. Miculka, A. Kuhn, F. Bates, P. Strecker, A. Kemmer-Brück, and the Vaccines Clinical Assay Team and Vaccines Assay Development Team for their assistance during this study. Staffing services were supported in part by an NYU CTSA grant (UL1 TR001445) from the National Center for Advancing Translational Sciences, National Institutes of Health. BioNTech is the sponsor of the study. Pfizer was responsible for the design, data collection, data analysis, data interpretation and writing of the report. The corresponding authors had full access to all of the data in the study and had final responsibility for the decision to submit the data for publication. All study data were available to all authors.
Author information
Authors and Affiliations
Contributions
K.U.J., P.R.D., W.C.G., N.K., S.L., A.G., R.B., O.T. and U.Ş. were involved in the design of the overall study and strategy. K.N., M.J.M., E.E.W., R.F. and A.R.F. provided feedback on the study design. W.K., D.C., K.A.S., K.R.T., C.F.-G. and P.-Y.S. performed the immunological analyses. M.J.M., K.N., E.E.W., R.F., A.R.F., K.E.L. and V.R. collected data as study investigators. P.L. and K.K. developed the statistical design and oversaw the data analysis. J.A., K.U.J., P.R.D. and W.C.G. drafted the initial version of the manuscript. All authors reviewed and edited the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
N.K., J.A., A.G., S.L., R.B., K.A.S., P.L., K.K., W.K., D.C., K.R.T., P.R.D., W.C.G. and K.U.J. are employees of Pfizer and may hold stock options. U.Ş. and Ö.T. are stock owners, management board members and employees at BioNTech and are inventors on patents and patent applications related to RNA technology. M.J.M., K.E.L., K.N., E.E.W., A.R.F., R.F. and V.R. received compensation from Pfizer for their role as study investigators. C.F.-G. and P.-Y.S. received compensation from Pfizer to perform the neutralization assay.
Additional information
Peer review information Nature thanks Barbra Richardson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Post vaccination changes in lymphocyte count over time.
The following time points are shown: dose 1/day 1–3, around 1 day after dose 1; dose 1/day 6–8, around 7 days after dose 1; pre-dose 2, before dose 2; dose 2/day 6–8, around 7 days after dose 2. Symbols denote group means; circle, placebo; plus, 10 μg; cross, 30 μg; triangle, 100 μg. The box-and-whisker plots show the median (centre), first and third quartiles (lower and upper edges), and minimum and maximum values (lower and upper whiskers).
Supplementary information
Supplementary Information
Redacted clinical trial protocol.
Rights and permissions
About this article

Cite this article
Mulligan, M.J., Lyke, K.E., Kitchin, N. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020). https://doi.org/10.1038/s41586-020-2639-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2639-4
This article is cited by
-
Computational design and engineering of self-assembling multivalent microproteins with therapeutic potential against SARS-CoV-2
Journal of Nanobiotechnology (2024)
-
Enhancing Immunological Memory: Unveiling Booster Doses to Bolster Vaccine Efficacy Against Evolving SARS-CoV-2 Mutant Variants
Current Microbiology (2024)
-
Overview of diagnostic tools and nano-based therapy of SARS-CoV-2 infection
Chemical Papers (2024)
-
The use of RNA-based treatments in the field of cancer immunotherapy
Molecular Cancer (2023)
-
Effect of anxiety and depression on self-reported adverse reactions to COVID-19 vaccine: a cross-sectional study in Shanghai, China
BMC Public Health (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
