
Abstract
The current outbreak of coronavirus disease-2019 (COVID-19) poses unprecedented challenges to global health1. The new coronavirus responsible for this outbreak—severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—shares high sequence identity to SARS-CoV and a bat coronavirus, RaTG132. Although bats may be the reservoir host for a variety of coronaviruses3,4, it remains unknown whether SARS-CoV-2 has additional host species. Here we show that a coronavirus, which we name pangolin-CoV, isolated from a Malayan pangolin has 100%, 98.6%, 97.8% and 90.7% amino acid identity with SARS-CoV-2 in the E, M, N and S proteins, respectively. In particular, the receptor-binding domain of the S protein of pangolin-CoV is almost identical to that of SARS-CoV-2, with one difference in a noncritical amino acid. Our comparative genomic analysis suggests that SARS-CoV-2 may have originated in the recombination of a virus similar to pangolin-CoV with one similar to RaTG13. Pangolin-CoV was detected in 17 out of the 25 Malayan pangolins that we analysed. Infected pangolins showed clinical signs and histological changes, and circulating antibodies against pangolin-CoV reacted with the S protein of SARS-CoV-2. The isolation of a coronavirus from pangolins that is closely related to SARS-CoV-2 suggests that these animals have the potential to act as an intermediate host of SARS-CoV-2. This newly identified coronavirus from pangolins—the most-trafficked mammal in the illegal wildlife trade—could represent a future threat to public health if wildlife trade is not effectively controlled.
Similar content being viewed by others


Main
As coronaviruses are common in mammals and birds5, we used the whole-genome sequence of SARS-CoV-2 (strain WHCV; GenBank accession number MN908947) in a Blast search of SARS-related coronavirus sequences in available mammalian and avian viromic, metagenomic and transcriptomic data. We identified 34 closely related contigs in a set of viral metagenomes from pangolins (Extended Data Table 1), and therefore focused our subsequent search on SARS-related coronaviruses in pangolins.
We obtained the lung tissues from 4 Chinese pangolins (Manis pentadactyla) and 25 Malayan pangolins (Manis javanica) from a wildlife rescue centre during March–August 2019, and analysed them for SARS-related coronaviruses using reverse-transcription polymerase chain reaction (RT–PCR) with primers that target a conservative region of betacoronaviruses. RNA from 17 of the 25 Malayan pangolins generated the expected PCR product, whereas RNA from the Chinese pangolins did not amplify. The virus-positive Malayan pangolins were all from the first transport. These pangolins were brought into the rescue centre at the end of March, and gradually showed signs of respiratory disease, including shortness of breath, emaciation, lack of appetite, inactivity and crying. Furthermore, 14 of the 17 pangolins that tested positive for viral RNA died within one and half months of testing. Plasma samples of four PCR-positive and four PCR-negative Malayan pangolins were used in the detection of IgG and IgM antibodies against SARS-CoV-2 using a double-antigen sandwich enzyme-linked immunosorbent assay (ELISA). One of the PCR-positive sample reacted strongly, showing an optical density at 450 nm (OD450) value of 2.17 (cut-off value = 0.11) (Extended Data Table 2). The plasma remained positive at the dilution of 1:80, which suggests that the pangolin was naturally infected with a virus similar to SARS-CoV-2. The other three PCR-positive pangolins had no detectable antibodies against SARS-CoV-2. It is possible that these pangolins died during the acute stage of disease, before the appearance of antibodies. Histological examinations of tissues from four betacoronavirus-positive Malayan pangolins revealed diffuse alveolar damage of varying severity in the lung, compared with lung tissue from a betacoronavirus-negative Malayan pangolin. In one case, alveoli were filled with desquamated epithelial cells and some macrophages with haemosiderin pigments, with considerably reduced alveolar space, leading to the consolidation of the lung. In other cases, similar changes were more focal (Fig. 1, Extended Data Fig. 1). The severe case also had exudate with red blood cells and necrotic cell debris in bronchioles and bronchi. Focal mononuclear-cell infiltration was seen in the bronchioles and bronchi in two of the cases, and haemorrhage was seen in the bronchioles and small bronchi in one case (Extended Data Figs. 1–3). Hyaline membrane and syncytia were not detected in the alveoli of the four cases we examined.

a–d, Histological changes in the lung tissues are compared between a virus-negative Malayan pangolin (a) and three Malayan pangolins naturally infected with pangolin-CoV (b–d) (original magnification × 1,000). Proliferation and desquamation of alveolar epithelial cells and haemosiderin pigments are seen in tissues from all three infected pangolins and severe capillary congestion is seen in one of them (c). e, Viral particles are seen in double-membrane vesicles in the transmission electron microscopy image taken from Vero E6 cell culture inoculated with supernatant of homogenized lung tissue from one pangolin, with morphology indicative of coronavirus (inserts at the top right corner of e). Scale bar, 200 nm.
To isolate the virus, supernatant from homogenized lung tissue from one dead Malayan pangolin was inoculated into Vero E6 cells. Obvious cytopathogenic effects were observed in cells after a 72-h incubation. Viral particles were detected by transmission electron microscopy: most of these particles were inside double-membrane vesicles, with a few outside of them. They showed typical coronavirus morphology (Fig. 1e). RT–PCR targeting the spike (S) and RdRp genes produced the expected PCR products: these PCR products had approximately 84.5% and 92.2% nucleotide sequence identity, respectively, to the partial S and RdRp genes of SARS-CoV-2.
Illumina RNA sequencing was used to identify viruses in the lung from 12 pangolins (including four that were reported previously15). Mapping sequence data to the reference SARS-CoV-2 WHCV genome identified coronavirus sequence reads in nine samples (Extended Data Table 3). For one sample, higher genome coverage was obtained by remapping the total reads to the reference genome (Extended Data Fig. 4). We obtained the completed coronavirus genome (29,825 bp)—which we designated pangolin-CoV—using the assembled contigs, short sequence reads and targeted PCR analysis. The full S gene was sequenced in six PCR-positive samples, which revealed the presence of only four nucleotide differences in the sequence alignment among these samples (Extended Data Fig. 5); this indicates that only one type of coronavirus was present in the batch of study samples. The predicted S, E, M and N genes of pangolin-CoV are 3,798, 228, 669 and 1,260 bp, respectively, in length and the proteins they encode share 90.7%, 100%, 98.6% and 97.8% amino acid identity to the equivalent proteins of SARS-CoV-2 (Table 1).
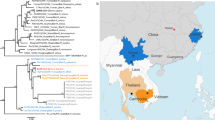
In a Simplot analysis of whole-genome sequences, we found that pangolin-CoV was highly similar to SARS-CoV-2 and RaTG13, with sequence identity between 80 and 98% (except for the S gene) (Fig. 2). Further comparative analysis of the S gene sequences suggests that there were recombination events among some of the SARS-related coronaviruses that we analysed. In the region of nucleotides 1–914, pangolin-CoV is more similar to the bat SARS-related coronaviruses ZXC21 and ZC45, whereas in the remaining part of the gene pangolin-CoV is more similar to SARS-CoV-2 and RaTG13 (Fig. 2). In particular, the receptor-binding domain (RBD) of the S protein of pangolin-CoV has only one amino acid difference with SARS-CoV-2. Overall, these data indicate that SARS-CoV-2 might have originated from the recombination of a virus similar to pangolin-CoV and a virus similar to RaTG13 (Fig. 2). To further support this conclusion, we assessed the evolutionary relationships among betacoronaviruses in the full genome, the RdRp and S genes, and in different regions of the S gene (Fig. 2c, Extended Data Fig. 6). The topologies mostly showed the clustering of pangolin-CoV with SARS-CoV-2 and RaTG13; SARS-CoV-2 and RaTG13 form a subclade within this cluster (Fig. 2c). However, pangolin-CoV and SARS-CoV-2 grouped together in the phylogenetic analysis of the RBD. Conflicts in cluster formation among phylogenetic analyses of different regions of the genome serve as a strong indication of genetic recombination, as has previously been seen for SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV)6,7.

a, Similarity plot of the full-length genomes and S gene sequences of pangolin-CoV against sequences of SARS-CoV-2 strain WIV02, as well as RaTG13, ZC45 and ZXC21. Although pangolin-CoV has a high sequence identity to SARS-CoV-2 and RaTG13 in most regions of the S gene, it is more similar to ZXC21 and ZC45 at the 5′ end. SARS-rCoV, SARS-related coronavirus. Parameters for the similarity plots are: window, 500 bp; step, 50 bp; gap strip, on; Kimura (2 parameter); T/t 2.0. b, Because of the presence of genetic recombination, there is discrepancy in cluster formation among the outcomes of phylogenetic analyses of different regions of the S gene. c, Phylogeny of coronaviruses closely related to SARS-CoV-2, based on full genome sequences. The phylogenetic tree was constructed using RAxML with the substitution model GTRGAMMAI and 1,000 bootstrap replicates. Numbers (>70) above or below branches are percentage bootstrap values for the associated nodes. The scale bar represents the number of substitutions per site. Red circles indicate the pangolin coronavirus sequences generated in this study, and blue triangles indicate SARS-CoV-2 sequences from humans.
As the S proteins of both SARS-CoV and SARS-CoV-2 have previously been shown to specifically recognize angiotensin-converting enzyme 2 (ACE2) during the entry of host cells2,8, we conducted molecular binding simulations of the interaction of the S proteins of the four closely related SARS-related coronaviruses with ACE2 proteins from humans, civets and pangolins. As expected, the RBD of SARS-CoV binds efficiently to ACE2 from humans and civets in the molecular binding simulation. In addition, this RBD appears to be capable of binding ACE2 of pangolins. By contrast, the S proteins of SARS-CoV-2 and pangolin-CoV can potentially recognize only the ACE2 of humans and pangolins (Extended Data Fig. 7).
SARS-CoV-2 is one of three known zoonotic coronaviruses (the others are SARS-CoV and MERS-CoV) that infect the lower respiratory tract and cause severe respiratory syndromes in humans7,9. Thus far, SARS-CoV-2 has been more contagious, but less deadly, than SARS-CoV10: the total number of human infections by SARS-CoV-2 far exceeds those of SARS-CoV11. Epidemiological investigations of the SARS-CoV-2 outbreak have shown that some of the initial patients were associated with the Huanan seafood market, where live wildlife was also sold10. No animals thus far have been implicated as carriers of the virus. SARS-CoV-2 forms a cluster with SARS-CoV and bat SARS-related coronaviruses (Fig. 2c). In addition, a bat coronavirus (RaTG13) has about 96% sequence identity to SARS-CoV-2 at the whole-genome level2. Therefore, it is reasonable to assume that bats are the native host of SARS-CoV-2, as has previously been suggested for SARS-CoV and MERS-CoV12,13. The SARS-related coronavirus identified in the present study and the metagenomic assemblies of viral sequences from Malayan pangolins14 is genetically related to SARS-CoV-2, but is unlikely to be directly linked to the current outbreak because of its substantial sequence differences from SARS-CoV-2. However, a virus related to pangolin-CoV appears to have donated the RBD to SARS-CoV-2. SARS-related coronavirus sequences have previously been detected in dead Malayan pangolins15. These sequences appear to be from the same virus (pangolin-CoV) that we identified in the present study, as judged from their sequence similarity. Here we provide evidence for the potential for pangolins to act as the zoonotic reservoir of SARS-CoV-2-like coronaviruses. However, the pangolins we studied here showed clinical signs of disease. In general, a natural reservoir host does not show severe disease, whereas an intermediate host may have clinical signs of infection16. Although a SARS-CoV-2-like coronavirus was detected in the lungs of these pangolins, a direct association between the clinical signs or pathology and active virus replication is not available as we lack evidence from immunohistochemistry or in situ hybridization experiments. The experimental infection of healthy pangolins with pangolin-CoV would provide more definitive answers; however, as pangolins are protected it is difficult to carry out such experiments. Further studies are needed to confirm the role of pangolins in the transmission of SARS-related coronaviruses.
As the RBD of pangolin-CoV is nearly identical to that of SARS-CoV-2, the virus in pangolins presents a potential future threat to public health. Pangolins and bats are both nocturnal animals, eat insects and share overlapping ecological niches17,18, which make pangolins an ideal intermediate host for some SARS-related coronaviruses. Therefore, more systematic and long-term monitoring of SARS-related coronaviruses in pangolins and related animals should be implemented to identify the potential animal source of SARS-CoV-2 in the current outbreak.
Our findings support the call for stronger enforcement of regulations against the illegal trade in pangolins. Owing to the demand for their meat as a delicacy and their scales for use in traditional medicine in China, the illegal smuggling of pangolins from Southeast Asia to China is widespread18. International co-operation in the implementation of stricter regulations against illegal wildlife trade and consumption of game meat should be encouraged, as this will increase the protection of endangered animals and help to prevent future outbreaks of diseases caused by SARS-related coronaviruses.
Methods
No statistical methods were used to predetermine sample size.
Metagenomic analysis and viral genome assembly
We collected viromic, metagenomic and transcriptomic data of different mammals and birds in public databases—including NCBI Sequence Read Archive (SRA) and European Nucleotide Archive (ENA)—for searching potential coronavirus sequences. The raw reads from the public databases and some in-house metagenomic datasets were trimmed using fastp (v.0.19.7)19 to remove adaptor and low-quality sequences. The clean reads were mapped to the SARS-CoV-2 reference sequence (MN908947) using BWA-MEM (v.0.7.17)20 with >30% matches. The mapped reads were collected for downstream analyses. Contigs were de novo-assembled using Megahit (v.1.0.3)21 and identified as related to SARS-CoV-2 using BLASTn with E–values < 1 × 10−5 and sequence identity >90%.
Samples
Pangolins used in the study were confiscated by Customs and Department of Forestry of Guangdong Province in March and August 2019. They included four Chinese pangolins (M. pentadactyla) and 25 Malayan pangolins (M. javanica). The first transport confiscated contained 21 Malayan pangolins, and the second transport contained 4 Malayan pangolins and 4 Chinese pangolins. These pangolins were sent to the wildlife rescue centre, and were mostly inactive and crying, and eventually died in custody despite exhaustive rescue efforts. Tissue samples were taken from the lung of pangolins that had just died for histological and virological examinations.
Pathological examinations
Histological examinations were performed on lung tissues from five Malayan pangolins. In brief, the tissues collected were cut into small pieces and fixed in 10% buffered formalin for 24 h. They were washed free of formalin, dehydrated in ascending grades of ethanol and cleared with chloroform, and then embedded with molten paraffin wax in a template. The tissue blocks were sectioned with a microtome. The sections were transferred onto grease-free glass slides, deparaffinized and rehydrated through descending grades of ethanol and distilled water. They were stained with a haematoxylin and eosin staining kit (Baso Diagnostics, Wuhan Servicebio Technology). Finally, the stained slides were mounted with coverslips and examined under an Olympus BX53 equipped with an Olympus PM-C 35 camera.
Virus isolation and RT–PCR analysis
Lung tissue extract from pangolins was inoculated into Vero E6 cells for virus isolation. The cell line was tested free of mycoplasma contamination using LookOut Mycoplasma PCR Detection Kit (SIGMA), and was authenticated by microscopic morphologic evaluation. Cultured cell monolayers were maintained in Dulbecco’s Modified Eagle Medium (DMEM) and Ham’s F-12. The inoculum was prepared by grinding the lung tissue in liquid nitrogen, diluting it 1:2 with DMEM, filtering it through a 0.45-μm filter (Merck Millipore), and treating it with 16 μg/ml trypsin solution. After incubation at 37 °C for 1 h, the inoculum was removed from the culture and replaced with fresh culture medium. The cells were incubated at 37 °C and observed daily for cytopathic effects.
Viral RNA was extracted from the lung tissue using the QIAamp Viral RNA Mini kit (Qiagen) following the manufacturer-recommended procedures, and examined for coronavirus by RT–PCR using a pair of primers (F: 5′-TGGCWTATAGGTTYAATGGYATTGGAG-3′, R: 5′-CCGTCGATTGTGTGWATTTGSACAT-3′) designed to amplify the S gene of betacoronavirus.
Transmission electron microscopy
Cell cultures that showed cytopathic effects were examined for the viral particles using transmission electron microscopy. Cells were collected from the culture by centrifugation at 1,000g for 10 min, and fixed initially with 2.5% glutaraldehyde solution at 4 °C for 4 h, and again with 1% osmium tetroxide. They were dehydrated with graded ethanol and embedded with PON812 resin. Sections (80 nm in thickness) were cut from the resin block and stained with uranyl acetate and lead citrate sequentially. The negative stained grids and ultrathin sections were observed under a HT7800 transmission electron microscope (Hitachi).
Serological test
Plasma samples from eight Malayan pangolins were tested for anti-SARS-CoV-2 antibodies using a double-antigen ELISA kit for the detection of antibodies against SARS-CoV-2 by Hotgen, following manufacturer-recommended procedures. The assay was designed for the detection of both IgG and IgM antibodies against SARS-CoV-2 in humans and animals, and marketed as supplementary diagnostic tool for COVID-19. It uses the capture of antibodies against SARS-CoV-2 by the S1 antigen precoated on ELISA plates, and the detection of the antibodies through the use of horseradish peroxidase-conjugated RBD. Both the S1 antigen and RBD fragment were expressed in eukaryotic cells. Data generated by the test developer have shown a 95% detection rate in the analysis of sera from over 200 patients with COVID-19s. The assay has an inter-test variation of ≤15%, and no cross-reactivities with sera or plasma from patients positive for SARS-CoV, common and avian influenza viruses, mycoplasma and chlamydia. Fifty microlitres of plasma was analysed in duplicate, together with two negative controls and one positive control. The reaction was read on a Synergy HTX Multi-Mode Microplate Reader (BioTek) at 450/630 nm, with optical density (OD) values being calculated. The cut-off OD value for positivity was 0.105 + mean OD from the negative controls, and the cut-off value for OD for the positive control was set at ≥ 0.5. Positive samples were tested again with serial-diluted plasma.
Metagenomic sequencing
The lung tissue was homogenized by vortex with silica beads in 1 ml of phosphate-buffered saline. The homogenate was centrifuged at 10,000g for 5 min, with the supernatant being filtered through a 0.45-μm filter (Merck Millipore) to remove large particles. The filtrate or virus culture supernatant was used in RNA extraction with the QIAamp Viral RNA Mini kit. cDNA was synthesized from the extracted RNA using PrimeScriptScript II reverse transcriptase (Takara) and random primers, and amplified using Klenow Fragment (New England Biolabs). Sequencing libraries were prepared with NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs), and sequenced paired-end (150-bp) on an Illumina NovaSeq 6000. Specific PCR assays were used to fill genome sequence gaps, using primers designed based on sequences flanking the gap.
Phylogenetic analysis
Multiple sequence alignments of all sequence data were constructed using MAFFT v.7.22122. The phylogenetic relationship of the viral sequences was assessed using RAxML v.8.0.1423. The best-fit evolutionary model for the sequences in each dataset was identified using ModelTest24. Potential recombination events and the location of possible breakpoints in betacoronavirus genomes were detected using Simplot (version 3.5.1)25 and RDP 4.9926.
Molecular simulation of interactions between RBD and ACE2
The interaction between the RBD of the S protein of SARS-related coronavirus and the ACE2 of humans, civets, and pangolins was examined using molecular dynamic simulation. The crystal structure of SARS-CoV RBD domain binding to human ACE2 protein complex was downloaded from Protein Data Bank (PDB code 2AJF27). The structures of the complexes formed by ACE2 of civets or pangolins and the RBD of SARS-CoV-2, RaTG13 and pangolin-CoV were made using the MODELLER program28, and superimposed with the template (PDB code 2AJF). The sequence identity of SARS-CoV RBD (PDB code 6ACD) to the RBD of SARS-CoV-2, RaTG13 and pangolin-CoV was 76.5%, 76.8% and 74.2%, respectively, and the sequence identity of the human ACE2 protein to that of pangolins and civets was 85.4% and 86.9%, respectively.
The molecular dynamic simulations of RBD–ACE2 complexes were carried out using the AMBER 18 suite29 and ff14SB force field30. After two-stage minimization, NVT and NPT-MD, a 30-ns production molecular dynamics simulation was applied, with the time step being set to 2 fs and coordinate trajectories being saved every 3 ps. The MM-GBSA31 approach was used to calculate the binding free energy of each ACE2 protein to the RBD of the S protein, using the python script MMPBSA.py32 in the build-in procedure of AMBER 18 suite. The last 300 frames of all simulations were extracted to calculate the binding free energy that excludes the contributions of disulfide bond.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequence reads generated in this study are available in the NCBI SRA database under the BioProject accession PRJNA607174. The complete genome sequence of pangolin-CoV has been deposited in GISAID with the accession number EPI_ISL_410721.
Change history
11 November 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-03838-z
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Cui, J., Li, F. & Shi, Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Banerjee, A., Kulcsar, K., Misra, V., Frieman, M. & Mossman, K. Bats and coronaviruses. Viruses 11, 41 (2019).
Masters, P. S. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292 (2006).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Sabir, J. S. M. et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351, 81–84 (2016).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Song, Z. et al. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 11, E59 (2019).
Li, Q. et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020).
WHO. Coronavirus Disease 2019 (COVID-19) Situation Report – 79, https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200408-sitrep-79-covid-19.pdf?sfvrsn=4796b143_4 (WHO, 2020).
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005).
Lam, T. T.-Y. et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature https://doi.org/10.1038/s41586-020-2169-0 (2020).
Liu, P., Chen, W. & Chen, J. P. Viral metagenomics revealed sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 11, 979 (2019).
Wu, D. et al. Civets are equally susceptible to experimental infection by two different severe acute respiratory syndrome coronavirus isolates. J. Virol. 79, 2620–2625 (2005).
Nowak, R. M. Walker’s Bats of the World (Johns Hopkins Univ. Press, 1994).
Shepherd, C. R. Overview of the pangolin trade in southeast Asia. In Proc. Workshop on Trade and Conservation of Pangolins Native to South and Southeast Asia: 30 June-2 July 2008, Singapore Zoo (eds Pantel, S. & Chin, S. Y.) 6–12 (TRAFFIC Southeast Asia, 2009).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Posada, D. & Crandall, K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Lole, K. S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003 (2015).
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Webb, B. & Sali, A. Comparative protein structure modeling Using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.32 (2014).
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S. & Walker, R. C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Miller, B. R. III et al. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 31822056 and 31820103014), National Key R & D Program of China (2017YFD0500404), Fund for the Key Program and Creative Research Group of the Department of Education of Guangdong province (2019KZDXM004 and 2019KCXTD001), Guangdong Science and Technology Innovation Leading Talent Program (2019TX05N098), the Major Program of Guangdong Basic and Applied Research, the 111 Project (D20008), Chinese Academy of Engineering (2020-KYGG-04-01), Department of Science and Technology of Guangdong Province (2020B1111320002) and Department of Agriculture of Guangdong province.
Author information
Authors and Affiliations
Contributions
Y.S., L.X. and W.C. conceived the study; J.-J.Z., F.-H.H., Y.-J.W., S.-M.P., M.H., W.-J.X., Q.-H.C. and W.C. collected samples; J.Z., N.Z., X.Z., N.L., Y.G., X.L., X.S., Zhipeng Zhang, F.S. and W.H. performed virus isolation and sequencing; K.X., Y.F., Y.L., Ziding Zhang and Y.S. contributed to the analysis; Y.S. and L.X. wrote the manuscript; Y.F. and R.-A.C. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Andrew Ward and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Pathological changes in the lungs of pangolins.
a–d, Lungs of three Malayan pangolins naturally infected with pangolin-CoV (b–d, original amplification × 100) in comparison with the lung from a virus-negative Malayan pangolin (a). Different degrees of consolidation are seen in the lung tissues from three infected pangolins (b–d). Exudate is seen in the bronchi of one infected pangolin (b). Severe congestion is seen in the lung of one pangolin (d).
Extended Data Fig. 2 Pathological changes in the bronchiole of pangolins.
a–d, Three Malayan pangolins positive for pangolin-CoV (b–d, original amplification × 100) in comparison with a virus-negative Malayan pangolin (a). Red blood cells are seen in the bronchioles of two infected pangolins (b, d). Mononuclear cell infiltration is seen in the bronchiole wall of one infected pangolin (c). Severe congestion is seen in the alveolar tissue (in close proximity to the bronchiole) of one pangolin (d). The respiratory epithelium in the bronchioles is intact.
Extended Data Fig. 3 Pathological changes in the bronchus of pangolins.
a–d, Three Malayan pangolins positive for pangolin-CoV (b–d, original amplification × 100) in comparison with a virus-negative Malayan pangolin (a). Exudate with red blood cells is seen in the bronchus of one infected pangolin (b). Macrophages with haemosiderin pigments and mononuclear cell infiltration are seen in the bronchus wall of two infected pangolins (c and d, respectively). The respiratory epithelium in the bronchi is intact.
Extended Data Fig. 4 Mapping raw reads from pangolin lung to pangolin-CoV genome.
Results of the mapping of raw reads from the high-throughput sequencing of the lung tissue of lung08 (M4 in our study) to the assembled pangolin-CoV genome.
Extended Data Fig. 5 Sequence polymorphism among nucleotide sequences of the full S gene among virus-positive lung samples from six pangolins.
Dots denote nucleotide identity to the reference sequence.
Extended Data Fig. 6 Phylogeny of coronaviruses closely related to SARS-CoV-2.
a, Based on nucleotide sequences of the S gene. b, Based on nucleotide sequences of the RdRp gene. The phylogenetic trees were constructed by RAxML with the substitution model GTRGAMMAI and 1,000 bootstrap replicates. Numbers (>70) above or below branches are percentage bootstrap values for the associated nodes. The scale bar represents the number of substitutions per site. Red circles indicate the pangolin coronavirus sequences generated in this study, and blue triangles indicate SARS-CoV-2 sequences from humans.
Extended Data Fig. 7 Molecular binding simulations of the interaction of the S proteins of four closely related SARS-related coronaviruses with the ACE2 proteins of humans, civets and pangolins.
a, Free energy (kcal mol−1) for the binding of the RBD of S proteins of four SARS-related coronaviruses to the ACE2 of potential hosts. b, Alignment of the RBD sequences (key amino acids involved in interactions with ACE2 are boxed) of the S proteins from several genetically related SARS-related coronaviruses. c, Alignment of partial ACE2 amino acid sequences (key amino acids involved in interactions with RBD are marked with arrowheads) from humans, pangolins and civets at their interface with the RBD of S proteins.
Supplementary information
Rights and permissions
About this article

Cite this article
Xiao, K., Zhai, J., Feng, Y. et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289 (2020). https://doi.org/10.1038/s41586-020-2313-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2313-x
This article is cited by
-
Addressing biodiversity conservation, disease surveillance, and public health interventions through One Health approach in Hainan’s tropical rainforest
One Health Advances (2024)
-
An RNA-Seq analysis of coronavirus in the skin of the Pangolin
Scientific Reports (2024)
-
Recombination-aware phylogenetic analysis sheds light on the evolutionary origin of SARS-CoV-2
Scientific Reports (2024)
-
The neurobiology of SARS-CoV-2 infection
Nature Reviews Neuroscience (2024)
-
Effect of polyphenols against complications of COVID-19: current evidence and potential efficacy
Pharmacological Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
