
Abstract
The ongoing outbreak of viral pneumonia in China and across the world is associated with a new coronavirus, SARS-CoV-21. This outbreak has been tentatively associated with a seafood market in Wuhan, China, where the sale of wild animals may be the source of zoonotic infection2. Although bats are probable reservoir hosts for SARS-CoV-2, the identity of any intermediate host that may have facilitated transfer to humans is unknown. Here we report the identification of SARS-CoV-2-related coronaviruses in Malayan pangolins (Manis javanica) seized in anti-smuggling operations in southern China. Metagenomic sequencing identified pangolin-associated coronaviruses that belong to two sub-lineages of SARS-CoV-2-related coronaviruses, including one that exhibits strong similarity in the receptor-binding domain to SARS-CoV-2. The discovery of multiple lineages of pangolin coronavirus and their similarity to SARS-CoV-2 suggests that pangolins should be considered as possible hosts in the emergence of new coronaviruses and should be removed from wet markets to prevent zoonotic transmission.
Similar content being viewed by others
Main
An outbreak of serious pneumonia disease was reported in Wuhan, China, on 30 December 2019. The causative agent was soon identified as a novel coronavirus1, which was later named SARS-CoV-2. Case numbers grew rapidly from 27 in December 2019 to 3,090,445 globally as of 30 April 20203, leading to the declaration of a public health emergency, and later a pandemic, by the WHO (World Health Organization). Many of the early cases were linked to the Huanan seafood market in Wuhan city, Hubei province, from where the probable zoonotic source is speculated to originate2. Currently, only environmental samples taken from the market have been reported to be positive for SARS-CoV-2 by the Chinese Center for Disease Control and Prevention4. However, as similar wet markets were implicated in the SARS outbreak of 2002–20035, it seems likely that wild animals were also involved in the emergence of SARS-CoV-2. Indeed, a number of mammalian species were available for purchase in the Huanan seafood market before the outbreak4. Unfortunately, because the market was cleared soon after the outbreak began, determining the source virus in the animal population from the market is challenging. Although a coronavirus that is closely related to SARS-CoV-2, which was sampled from a Rhinolophus affinis bat in Yunnan in 2013, has now been identified6, similar viruses have not yet been detected in other wildlife species. Here we identified SARS-CoV-2-related viruses in pangolins smuggled into southern China.
We investigated the virome composition of pangolins (mammalian order Pholidota). These animals are of growing importance and interest because they are one of the most illegally trafficked mammal species: they are used as a food source and their scales are used in traditional Chinese medicine. A number of pangolin species are now regarded as critically endangered on the International Union for Conservation of Nature Red List of Threatened Species. We received frozen tissue samples (lungs, intestine and blood) collected from 18 Malayan pangolins (Manis javanica) during August 2017–January 2018. These pangolins were obtained during anti-smuggling operations performed by Guangxi Customs officers. Notably, high-throughput sequencing of the RNA of these samples revealed the presence of coronaviruses in 6 out of 43 samples (2 lung samples, 2 intestinal samples, 1 lung–intestine mixed sample and 1 blood sample from 5 individual pangolins; Extended Data Table 1). With the sequence read data, and by filling gaps with amplicon sequencing, we were able to obtain six complete or near complete genome sequences—denoted GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L—that fall into the SARS-CoV-2 lineage (within the genus Betacoronavirus of the Coronaviridae) in a phylogenetic analysis (Fig. 1b). The genome sequence of the virus isolate (GX/P2V) has a very high similarity (99.83–99.92%) to the five sequences that were obtained through the metagenomic sequencing of the raw samples, and all samples have similar genomic organizations to SARS-CoV-2, with eleven predicted open-reading frames (ORFs) (Fig. 1a and Extended Data Table 2; two ORFs overlap). We were also able to successfully isolate the virus using the Vero E6 cell line (Extended Data Fig. 1). On the basis of these genome sequences, we designed primers for quantitative PCR (qPCR) detection to confirm that the raw samples were positive for coronavirus. We conducted further qPCR testing on another batch of archived pangolin samples collected between May and July 2018. Among the 19 samples (9 intestine tissues, 10 lung tissues) tested from 12 animals, 3 lung tissue samples from 3 individual pangolins were positive for coronavirus.

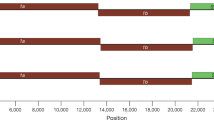
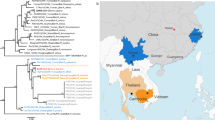
a, Genome organization of coronaviruses including the pangolin coronaviruses obtained in this study, with the predicted ORFs shown in different colours (ORF1a is omitted for clarity). The pangolin coronavirus strain GX/P2V is shown with its sequence length. For comparison, the human sequences NC_045512.2 and NC_004718.3, and bat sequences MG772933.1, GQ153541.1 and KC881006.1 are included (see Extended Data Table 6 for sources). b, Phylogeny of the subgenus Sarbecovirus (genus Betacoronavirus; n = 53) estimated from the concatenated ORF1ab, S, E, M and N genes. Red circles indicate the pangolin coronavirus sequences generated in this study (Extended Data Table 1). GD/P1L is the consensus sequence re-assembled from previously published raw data7. Phylogenies were estimated using a maximum likelihood approach that used the GTRGAMMA nucleotide substitution model and 1,000 bootstrap replicates. Scientific names of the bat hosts are indicated at the end of the sequence names, and abbreviated as follows: C. plicata, Chaerephon plicata; R. affinis, Rhinolophus affinis; R. blasii, Rhinolophus blasii; R. ferrumequinum, Rhinolophus ferrumequinum; R. monoceros, Rhinolophus monoceros; R. macrotis, Rhinolophus macrotis; R. pearsoni, Rhinolophus pearsoni; R. pusillus, Rhinolophus pusillus; R. sinicus, Rhinolophus sinicus. Palm civet (P. larvata, Paguma larvata; species unspecified for Civet007 and PC4-13 sequences) and human (H. sapiens, Homo sapiens) sequences are also shown.
In addition to the animals from Guangxi, after the start of the SARS-CoV-2 outbreak researchers of the Guangzhou Customs Technology Center re-examined five archived pangolin samples (two skin swabs, two unknown tissue samples and one scale) obtained in anti-smuggling operations performed in March 2019. Following high-throughput sequencing, the scale sample was found to contain coronavirus reads, and from these data we assembled a partial genome sequence of 21,505 bp (denoted as GD/P2S), representing approximately 72% of the SARS-CoV-2 genome. Notably, this virus sequence, obtained from a pangolin scale sample, may in fact be derived from contaminants of other infected tissues. Another study of diseased pangolins in Guangdong performed in 2019 also identified viral contigs from lung samples that were similarly related to SARS-CoV-27. Different assembly methods and manual curation were performed to generate a partial genome sequence that comprised 86.3% of the full-length virus genome (denoted as GD/P1L in the phylogeny shown in Fig. 1b).
These pangolin coronavirus genomes have 85.5% to 92.4% sequence similarity to SARS-CoV-2, and represent two sub-lineages of SARS-CoV-2-related viruses in the phylogenetic tree, one of which (comprising GD/P1L and GD/P2S) is very closely related to SARS-CoV-2 (Fig. 1b). It has previously been noted that members of the subgenus Sarbecovirus have experienced widespread recombination8. In support of this, a recombination analysis (Fig. 2) revealed that bat coronaviruses ZC45 and ZXC21 are probably recombinants, containing genome fragments derived from multiple SARS-CoV-related lineages (genome regions 2, 5 and 7) as well as SARS-CoV-2-related lineages, including segments from pangolin coronaviruses (regions 1, 3, 4, 6 and 8).

a, Sliding window analysis of changing patterns of sequence similarity between human SARS-CoV-2, pangolin and bat coronaviruses. The potential recombination breakpoints are shown in pink dash lines, and regions separated by the breakpoints are alternatively shaded in yellow. These potential breakpoints subdivide the genomes into eight regions (regions with fewer than 200 bp were omitted), indicated by the red bars at the bottom of the analysis boxes. The names of the query sequences are shown vertically to the right of the analysis boxes. The similarities to different reference sequences are indicated by different colours. Guangdong pangolin-CoV GD/P1L and pangolin-CoV GD/P2S were merged for this analysis. The blue arrows at the top indicate the position of the ORFs in the alignment. b, Phylogenetic trees of different genomic regions. SARS-CoV- and SARS-CoV-2-related lineages are shown in blue and red tree branches, respectively. Branch supports obtained from 1,000 bootstrap replicates are shown. Branch scale bars are shown as 0.1 substitutions per site.
More notable, however, was the observation of putative recombination signals between the pangolin coronaviruses, bat coronavirus RaTG13 and human SARS-CoV-2 (Fig. 2). In particular, SARS-CoV-2 exhibits very high sequence similarity to the Guangdong pangolin coronaviruses in the receptor-binding domain (RBD) (97.4% amino acid similarity, indicated by red arrow in Fig. 2a; the alignment is shown in Fig. 3a), even though it is most closely related to bat coronavirus RaTG13 in the remainder of the viral genome. Indeed, the Guangdong pangolin coronaviruses and SARS-CoV-2 possess identical amino acids at the five critical residues of the RBD, whereas RaTG13 only shares one amino acid with SARS-CoV-2 (residue 442, according to numbering of the human SARS-CoV9) and these latter two viruses have only 89.2% amino acid similarity in the RBD. Notably, a phylogenetic analysis of synonymous sites only from the RBD revealed that the topological position of the Guangdong pangolin is consistent with that of the remainder of the viral genome, rather than being the closest relative of SARS-CoV-2 (Fig. 3b). Therefore, it is possible that the amino acid similarity between the RBD of the Guangdong pangolin coronaviruses and SARS-CoV-2 is due to selectively mediated convergent evolution rather than recombination, although it is difficult to differentiate between these scenarios on the basis of the current data. This observation is consistent with the fact that the sequence similarity of ACE2 is higher between humans and pangolins (84.8%) than between humans and bats (80.8–81.4% for Rhinolophus sp.) (Extended Data Table 3). The occurrence of recombination and/or convergent evolution further highlights the role that intermediate animal hosts have in the emergence of viruses that can infect humans. However, all of the pangolin coronaviruses identified to date lack the insertion of a polybasic (furin-like) S1/S2 cleavage site in the spike protein that distinguishes human SARS-CoV-2 from related betacoronaviruses (including RaTG13)10 and that may have helped to facilitate the emergence and rapid spread of SARS-CoV-2 through human populations.

a, Sequence alignment showing the RBD in human, pangolin and bat coronaviruses. The five critical residues for binding between SARS-CoV RBD and human ACE2 protein are indicated in red boxes, and ACE2-contacting residues are indicated by yellow boxes as previously described9. In the Guangdong pangolin-CoV sequence, the codon positions encoding the amino acids Pro337, Asn420, Pro499 and Asn519 have ambiguous nucleotide compositions, resulting in possible alternative amino acids at these sites (threonine, glycine, threonine and lysine, respectively). Sequence gaps are indicated with dashes. The short black lines at the top indicate the positions of every 10 residues. GD, Guangdong; GX, Guangxi. b, Phylogenetic trees of the SARS-CoV-2-related lineage estimated from the entire RBD region (top) and synonymous sites only (bottom). Branch supports obtained from 1,000 bootstrap replicates are shown. Branch scale bars are shown as 0.1 substitutions per site.
To our knowledge, pangolins are the only mammals in addition to bats that have been documented to be infected by a SARS-CoV-2-related coronavirus. It is notable that two related lineages of coronaviruses are found in pangolins that were independently sampled in different Chinese provinces and that both are also related to SARS-CoV-2. This suggests that these animals may be important hosts for these viruses, which is surprising as pangolins are solitary animals that have relatively small population sizes, reflecting their endangered status11. Indeed, on the basis of the current data it cannot be excluded that pangolins acquired their SARS-CoV-2-related viruses independently from bats or another animal host. Therefore, their role in the emergence of human SARS-CoV-2 remains to be confirmed. In this context, it is noteworthy that both lineages of pangolin coronaviruses were obtained from trafficked Malayan pangolins, which originated from Southeast Asia, and that there is a marked lack of knowledge of the viral diversity maintained by this species in regions in which it is indigenous. Furthermore, the extent of virus transmission in pangolin populations should be investigated further. However, the repeated occurrence of infections with SARS-CoV-2-related coronaviruses in Guangxi and Guangdong provinces suggests that this animal may have an important role in the community ecology of coronaviruses.
Coronaviruses, including those related to SARS-CoV-2, are present in many wild mammals in Asia5,6,7,12. Although the epidemiology, pathogenicity, interspecies infectivity and transmissibility of coronaviruses in pangolins remains to be studied, the data presented here strongly suggests that handling these animals requires considerable caution and their sale in wet markets should be strictly prohibited. Further surveillance of pangolins in their natural environment in China and Southeast Asia are necessary to understand their role in the emergence of coronaviruses and the risk of future zoonotic transmissions.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Ethics statement
The animals studied here were rescued and treated by the Guangxi Zhuang Autonomous Region Terrestrial Wildlife Medical-aid and Monitoring Epidemic Diseases Research Center under the ethics approval (wild animal treatment regulation No. [2011] 85). The samples were collected following the procedure guideline (Pangolins Rescue Procedure, November, 2016).
Sample collection, viral detection and sequencing of pangolins in Guangxi
We received frozen tissue samples of 18 pangolins (M. javanica) from Guangxi Medical University, China, that were collected between August 2017 – January 2018. These pangolins were seized by the Guangxi Customs during their routine anti-smuggling operations. All animal individuals comprised samples from multiple organs including lungs, intestine and blood, with the exception of six individuals for which only lung tissues were available, five with mixed intestine and lung tissues only, one with intestine tissues only, and one comprising two blood samples. Using the intestine–lung mixed sample we were able to isolate a novel Betacoronavirus using the Vero-E6 cell line (from ATCC; Extended Data Fig. 1). The cell line was subjected to species identification and authentication by microscopic morphologic evaluation and growth curve analysis, and was tested free of mycoplasma contamination. The cell line was not on the list of common misidentified cell lines by ICLAC. A High Pure Viral RNA Kit (Roche) was used for RNA extraction on all 43 samples. For RNA sequencing (GX/P2V and GX/P3B), a sequencing library was constructed using an Ion Total RNA-Seq Kit v2 (Thermo Fisher Scientific), and the library was subsequently sequenced using an Ion Torrent S5 sequencer (Thermo Fisher Scientific). For other samples, reverse transcription was performed using an SuperScript III First-Strand Synthesis System for RT–PCR (Thermo Fisher Scientific). DNA libraries were constructed using the NEBNext Ultra II DNA Library Prep Kit and sequenced on a MiSeq sequencer. The NGS (next-generation sequencing) QC Toolkit V2.3.3 was used to remove low-quality and short reads. Both BLASTn and BLASTx were used to search against a local virus database, using the data available at NCBI/GenBank. Genome sequences were assembled using the CLC Genomic Workbench v.9.0. To fill gaps in high throughput sequencing and obtain the whole viral genome sequence, amplicon primers based on the bat SARS-like coronavirus ZC45 (GenBank accession number MG772933) sequence and the coronavirus contigs obtained in the initial sequencing were designed for further amplicon-based sequencing.
A total of six samples (including the virus isolate) contained reads that matched members of the genus Betacoronavirus (Extended Data Table 1). We obtained near complete viral genomes from these samples (98%, compared to SARS-CoV-2), which were designated GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L. Their average sequencing coverage ranged from approximately 8.4X to 8,478X (Extended Data Fig. 2a-f). On the basis of these genome sequences, we designed primers for qPCR to confirm the positivity of the original tissue samples (Extended Data Table 4). This revealed an original lung tissue sample that was also qPCR positive, in addition to the six original samples with coronavirus reads. We further tested an additional 19 samples (nine intestine tissues and ten lung tissues), from 12 smuggled pangolins sampled between May–July 2018 by the group from Guangxi Medical University. The genome sequences of GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L have been submitted to GISAID database and assigned accession numbers EPI_ISL_410538 - EPI_ISL_410543.
Sample collection, viral detection and sequencing of pangolins in Guangdong
After the start of the SARS-CoV-2 outbreak, the Guangzhou Customs Technology Center re-examined their five archived pangolin samples (two skin swabs, two unknown tissue and one scale) obtained in anti-smuggling operations undertaken in March 2019. RNA was extracted from all five samples (Qiagen), and was subjected to high-throughput RNA sequencing on the Illumina HiSeq platform by Vision medicals. The scale sample was found to contain coronavirus reads using a BLAST-based approach. These reads were quality assessed, cleaned and assembled into contigs by both de novo (MEGAHIT v1.1.313) and using reference (BWA v0.7.1314) assembly methods, using BetaCoV/Wuhan/WIV04/2019 as a reference. The contigs were combined, and approximately 72% of the coronavirus genome (21,505 bp) was obtained. This sequence has about 6.6× sequencing coverage (Extended Data Fig. 2g) and denoted pangolin-CoV GD/P2S. This sequence has been deposited on GISAID with accession number EPI_ISL_410544.
A recently published meta-transcriptomic study of pangolins7 deposited 21 RNA-seq raw files on the SRA database (https://www.ncbi.nlm.nih.gov/sra). We screened these raw read files using BLAST methods and found that five (SRR10168374, SRR10168376, SRR10168377, SRR10168378 and SRR10168392) contained reads that mapped to SARS-CoV-2. These reads were subjected to quality assessment, cleaning and then de novo assembly using MEGAHIT13 and reference assembly using BWA14. These reads were then merged and curated in a pileup alignment file to obtain the consensus sequences. This combined consensus sequence is 25,753 bp in length (about 86.3% of BetaCoV/Wuhan/WIV04/2019; about 6.9× coverage) and denoted pangolin-CoV GD/P1L (available in the Supplementary Information Dataset). Notably, it has 66.8% overlap and a sequence identity of 99.79% with the GD/P2S sequence. As the genetic distance between these viruses is very low, for the recombination analysis we merged the GD/P1L and GD/P2S sequences into a single consensus sequence to minimize gap regions within any sequences.
The viral genome organizations of the Guangxi and Guangdong pangolin coronaviruses were similar to SARS-CoV-2. They possessed nine non-overlapping open reading frames (ORFs) plus two overlapping ORFs, and shared the same gene order of ORF1ab replicase, envelope glycoprotein spike (S), envelope (E), membrane (M), nucleocapsid (N), plus other predicted ORFs. A detailed comparison of the ORF length and similarity with SARS-CoV-2 and bat coronavirus RaTG13 is provided in Extended Data Table 2.
Sequence, phylogenetic and recombination analyses
The human SARS-CoV-2 and bat RaTG13 coronavirus genome sequences were downloaded from Virological.org (http://virological.org) and the GISAID (https://www.gisaid.org) databases in January 2020, with the data kindly shared by the submitters (Extended Data Table 5). Other coronaviruses (subgenus Sarbecovirus) were downloaded from GenBank (Extended Data Table 6) and compared to those obtained here. We constructed a multiple sequence alignment of their complete genomes and individual genes using MAFFT v.7.27315. Maximum likelihood phylogenies were estimated using RAxML v.8.2.1216 from 100 inferences, using the GTRGAMMA model of nucleotide substitution with 1,000 bootstrap replicates. To investigate potential recombination events, we used SimPlot v.3.5.117 to conduct a window sliding analysis to determine the changing patterns of sequence similarity and phylogenetic clustering between the query and the reference sequences. A full plot for the recombination analysis is provided in Extended Data Fig. 3. We also examined phylogenetic clusters performed directly from the multiple sequence alignment. Maximum likelihood trees were estimated from each window extraction (that is, genome regions 1 to 8) using RAxML as described above.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Data that support the findings of this study have been deposited in the GISAID database (https://www.gisaid.org) with accession numbers EPI_ISL_410538–EPI_ISL_410544 and the SRA database under BioProject accession number PRJNA606875. The data are also available as Supplementary Information.
References
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
World Health Organization. Coronavirus disease 2019 (COVID-19) Situation Report – 101 https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200430-sitrep-101-covid-19.pdf v.101 (30 April 2020).
Cohen, J. Mining coronavirus genomes for clues to the outbreak’s origins. Science https://www.sciencemag.org/news/2020/01/mining-coronavirus-genomes-clues-outbreak-s-origins (31 January 2020).
Wang, M. et al. SARS-CoV infection in a restaurant from palm civet. Emerg. Infect. Dis. 11, 1860–1865 (2005).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Liu, P., Chen, W. & Chen, J. P. Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 12, 11 (2019).
Hon, C. C. et al. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826 (2008).
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127-20 (2020).
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 176, 104742 (2020).
Heinrich, S. et al. The Global Trafficking of Pangolins: A Comprehensive Summary of Seizures and Trafficking Routes from 2010–2015 (TRAFFIC, 2017).
Wang, W. et al. Discovery of a highly divergent coronavirus in the Asian house shrew from China illuminates the origin of the alphacoronaviruses. J. Virol. 91, e00764-17 (2017).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M. & Katoh, K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10 (2019).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Lole, K. S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999).
He, R. et al. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 316, 476–483 (2004).
Snijder, E. J. et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331, 991–1004 (2003).
Marra, M. A. et al. The genome sequence of the SARS-associated coronavirus. Science 300, 1399–1404 (2003).
Song, H. D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005).
Han, Y. et al. Identification of diverse bat alphacoronaviruses and betacoronaviruses in China provides new insights into the evolution and origin of coronavirus-related diseases. Front. Microbiol. 10, 1900 (2019).
Tao, Y. & Tong, S. Complete genome sequence of a severe acute respiratory syndrome-related coronavirus from Kenyan bats. Microbiol. Resour. Announc. 8, e00548-19 (2019).
Hu, D. et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 7, 1–10 (2018).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Wu, Z. et al. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 213, 579–583 (2016).
Wu, Z. et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 10, 609–620 (2016).
Yang, L. et al. Novel SARS-like betacoronaviruses in bats, China, 2011. Emerg. Infect. Dis. 19, 989–991 (2013).
Xu, L. et al. Detection and characterization of diverse alpha- and betacoronaviruses from bats in China. Virol. Sin. 31, 69–77 (2016).
He, B. et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J. Virol. 88, 7070–7082 (2014).
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005).
Drexler, J. F. et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 84, 11336–11349 (2010).
Lau, S. K. et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84, 2808–2819 (2010).
Yuan, J. et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 91, 1058–1062 (2010).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Tang, X. C. et al. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 80, 7481–7490 (2006).
Yeh, S. H. et al. Characterization of severe acute respiratory syndrome coronavirus genomes in Taiwan: molecular epidemiology and genome evolution. Proc. Natl Acad. Sci. USA 101, 2542–2547 (2004).
Vega, V. B. et al. Mutational dynamics of the SARS coronavirus in cell culture and human populations isolated in 2003. BMC Infect. Dis. 4, 32 (2004).
Acknowledgements
We thank the staff of the Guangxi and Guangdong Custom Bureau for their laborious anti-smuggling operations and all of the scientists who shared their genomic sequences of the coronaviruses used in this study. The computations were performed using research computing facilities offered by Information Technology Services, the University of Hong Kong. This work was supported by research grants from The Natural Science Foundation of China (NSFC; 81621005), the State Key Research Development Program of China (2019YFC1200401), NSFC Excellent Young Scientists Fund (Hong Kong and Macau) (31922087), National Key Plan for Scientific Research and Development of China (2016YFD0500302 and 2017YFE0190800), funding for Guangdong–Hongkong–Macau Joint Laboratory (2019B121205009), Li Ka Shing Foundation, National Institutes of Health (HHSN272201400006C), Guangxi Scientific and Technological Research (2020AB39264) and Guangxi Medical University Training Program for Distinguished Young Scholars, and the Australian Research Council (FL170100022).
Author information
Authors and Affiliations
Contributions
W.-C.C., N.J., J.-C.H., Y.G. and Y.-L.H. designed and supervised the research. J.-F.J., B.-G.J., W.W., T.-T.Y., K.Z., L.-F.L. and X.-M.C. collected samples. Y.-W.Z., Y.-X.S., W.-J.L., W.W., T.-T.Y., J.L. and L.-F.L. prepared materials for sequencing. Y.-W.Z., Y.-X.S., G.-Q.P., X.Q., F.-F.S. and S.Q. performed genome sequencing. Y.-W.Z., M.H.-H.S., X.-B.N. and T.T.-Y.L. performed genome assembly and annotation. Y.-G.T., T.T.-Y.L., M.H.-H.S., Y.-W.Z., X.-B.N., E.C.H., Y.-S.L. and N.J. performed the genome analysis and interpretation. T.T.-Y.L., N.J., E.C.H. and W.-C.C. wrote the paper. All authors took part in data interpretation and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Paul Kellam, W. Ian Lipkin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Microscopy image of the cytopathic effect of the virus in Vero E6 cells.
a, Negative control. Uninfected cells of the Vero E6 cell line. b, Cytopathic effect seen in viral culture (five days after inoculation). The experiment was performed twice independently in two laboratories and produced similar results.
Extended Data Fig. 2 Read coverage depth of each pangolin coronavirus analysed in this study.
a, GX/P1E. b, GX/P2V. c, GX/P3B. d, GX/P4L. e, GX/P5L. f, GX/P5E. g, GD/P1L. h, GD/P2S.
Supplementary information
Supplementary Data
Partial genomic sequence of the Guangdong pangolin coronavirus (GD/P1L) reconstructed from the sequencing data of Liu et al. Viruses 11, (2019). Gaps were placed based on the alignment with BetaCoV/bat/Yunnan/RaTG13/2013.
Rights and permissions
About this article

Cite this article
Lam, T.TY., Jia, N., Zhang, YW. et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285 (2020). https://doi.org/10.1038/s41586-020-2169-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2169-0
This article is cited by
-
An RNA-Seq analysis of coronavirus in the skin of the Pangolin
Scientific Reports (2024)
-
“Long COVID” and Its Impact on The Environment: Emerging Concerns and Perspectives
Environmental Management (2024)
-
Emergence of SARS and COVID-19 and preparedness for the next emerging disease X
Frontiers of Medicine (2024)
-
Isolation and characterization of a pangolin-borne HKU4-related coronavirus that potentially infects human-DPP4-transgenic mice
Nature Communications (2024)
-
Cepharanthine analogs mining and genomes of Stephania accelerate anti-coronavirus drug discovery
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
