
Abstract
Chronic inflammation is accompanied by recurring cycles of tissue destruction and repair and is associated with an increased risk of cancer1,2,3. However, how such cycles affect the clonal composition of tissues, particularly in terms of cancer development, remains unknown. Here we show that in patients with ulcerative colitis, the inflamed intestine undergoes widespread remodelling by pervasive clones, many of which are positively selected by acquiring mutations that commonly involve the NFKBIZ, TRAF3IP2, ZC3H12A, PIGR and HNRNPF genes and are implicated in the downregulation of IL-17 and other pro-inflammatory signals. Mutational profiles vary substantially between colitis-associated cancer and non-dysplastic tissues in ulcerative colitis, which indicates that there are distinct mechanisms of positive selection in both tissues. In particular, mutations in NFKBIZ are highly prevalent in the epithelium of patients with ulcerative colitis but rarely found in both sporadic and colitis-associated cancer, indicating that NFKBIZ-mutant cells are selected against during colorectal carcinogenesis. In further support of this negative selection, we found that tumour formation was significantly attenuated in Nfkbiz-mutant mice and cell competition was compromised by disruption of NFKBIZ in human colorectal cancer cells. Our results highlight common and discrete mechanisms of clonal selection in inflammatory tissues, which reveal unexpected cancer vulnerabilities that could potentially be exploited for therapeutics in colorectal cancer.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
All of the WES and RNA-seq data have been deposited in the European Genome-phenome Archive under accession numbers EGAS00001003801 and EGAS00001003802, respectively. Data for Figs. 1–4 and Extended Data Figs. 3–10 are available as Source Data. All other data are available from the corresponding author on reasonable request.
Change history
20 December 2019
This article was amended to correct Extended Data Fig. 9, which was originally incorrect.
References
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Hunter, P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 13, 968–970 (2012).
Nathan, C. & Ding, A. Nonresolving inflammation. Cell 140, 871–882 (2010).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Martincorena, I. et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Yokoyama, A. et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019).
Burisch, J., Jess, T., Martinato, M. & Lakatos, P. L. The burden of inflammatory bowel disease in Europe. J. Crohns Colitis 7, 322–337 (2013).
Ungaro, R., Mehandru, S., Allen, P. B., Peyrin-Biroulet, L. & Colombel, J. F. Ulcerative colitis. Lancet 389, 1756–1770 (2017).
Eaden, J. A., Abrams, K. R. & Mayberry, J. F. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48, 526–535 (2001).
Kang, H. & Shibata, D. Direct measurements of human colon crypt stem cell niche genetic fidelity: the role of chance in non-Darwinian mutation selection. Front. Oncol. 3, 264 (2013).
Choi, C. R., Bakir, I. A., Hart, A. L. & Graham, T. A. Clonal evolution of colorectal cancer in IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 218–229 (2017).
Eto, A., Muta, T., Yamazaki, S. & Takeshige, K. Essential roles for NF-κB and a Toll/IL-1 receptor domain-specific signal(s) in the induction of IκB-ζ. Biochem. Biophys. Res. Commun. 301, 495–501 (2003).
Yamamoto, M. et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature 430, 218–222 (2004).
Bruno, M. E. C., Frantz, A. L., Rogier, E. W., Johansen, F. E. & Kaetzel, C. S. Regulation of the polymeric immunoglobulin receptor by the classical and alternative NF-κB pathways in intestinal epithelial cells. Mucosal Immunol. 4, 468–478 (2011).
Cao, A. T., Yao, S., Gong, B., Elson, C. O. & Cong, Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J. Immunol. 189, 4666–4673 (2012).
Bruno, M. E. et al. Correlation of biomarker expression in colonic mucosa with disease phenotype in Crohn’s disease and ulcerative colitis. Dig. Dis. Sci. 60, 2976–2984 (2015).
Nakatsuka, Y. et al. Pulmonary Regnase-1 orchestrates the interplay of epithelium and adaptive immune systems to protect against pneumonia. Mucosal Immunol. 11, 1203–1218 (2018).
Mino, T. et al. Regnase-1 and Roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 161, 1058–1073 (2015).
Iwasaki, H. et al. The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat. Immunol. 12, 1167–1175 (2011).
Reznik, B., Clement, S. L. & Lykke-Andersen, J. hnRNP F complexes with tristetraprolin and stimulates ARE-mRNA decay. PLoS One 9, e100992 (2014).
Ramirez-Carrozzi, V. et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 12, 1159–1166 (2011).
Robles, A. I. et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology 150, 931–943 (2016).
Fujita, M. et al. Genomic landscape of colitis-associated cancer indicates the impact of chronic inflammation and its stratification by mutations in the Wnt signaling. Oncotarget 9, 969–981 (2018).
Din, S. et al. Mutational analysis identifies therapeutic biomarkers in inflammatory bowel disease-associated colorectal cancers. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-17-3713 (2018).
Baker, A. M. et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 68, 985–995 (2019).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Riddell, R. H. et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum. Pathol. 14, 931–968 (1983).
Yoshida, K. et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011).
Shiraishi, Y. et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 41, e89 (2013).
Okuma, A. et al. Enhanced apoptosis by disruption of the STAT3-IκB-ζ signaling pathway in epithelial cells induces Sjögren’s syndrome-like autoimmune disease. Immunity 38, 450–460 (2013).
el Marjou, F. et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004).
Miyoshi, H. & Stappenbeck, T. S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–2482 (2013).
Katakura, K. et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 115, 695–702 (2005).
Chen, K. et al. IL-17 Receptor signaling in the lung epithelium is required for mucosal chemokine gradients and pulmonary host defense against K. pneumoniae. Cell Host Microbe 20, 596–605 (2016).
Johansen, C. et al. IκBζ is a key driver in the development of psoriasis. Proc. Natl Acad. Sci. USA 112, E5825–E5833 (2015).
Müller, A. et al. IκBζ is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc. Natl Acad. Sci. USA 115, 10088–10093 (2018).
Hildebrand, D. G. et al. IκBζ is a transcriptional key regulator of CCL2/MCP-1. J. Immunol. 190, 4812–4820 (2013).
Goto, N. et al. Distinct roles of Hes1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 77, 3442–3454 (2017).
Blokzijl, F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016).
Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED); the Project for Development of Innovative Research on Cancer Therapeutics (JP19cm0106501 to S.Ogawa) and the Core Research for Evolutional Science and Technology (CREST) (JP19gm1110011 to S. Ogawa); the Ministry of Education, Culture, Sports, Science and Technology of Japan; the High Performance Computing Infrastructure System Research Project (hp160219, hp170227, hp180198 and hp190158 to S. Ogawa and S.M.) (this research used computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project); the Japan Society for the Promotion of Science (JSPS); Scientific Research on Innovative Areas (JP15H05909 to S. Ogawa and S.M.; JP15H05912 to S.M.; JP26115009 to M.S.) and KAKENHI (JP19H05656 to S. Ogawa; JP15H05707 to S.M.; JP17H04157 to H.S.; JP18H05278 to O.T.); the Ministry of Health, Labour and Welfare of Japan; Health and Labour Sciences Research Grants for research on intractable diseases (Investigation and Research for Intractable Inflammatory Bowel Disease) (H.N.); the Takeda Science Foundation (S. Ogawa, H.M. and T.Y.); the Uehara Memorial Foundation (H.S.); and the Foundation for Promotion of Cancer Research (H.S.). S. Ogawa is a recipient of the JSPS Core-to-Core Program A: Advanced Research Networks. We thank M. Nakazawa, M. Nakamura, T. Shirahari, A. Takatsu and A. Ryu for technical assistance; iLAC for sequencing support; the Centre for Anatomical, Pathological and Forensic Medical Research at Kyoto University Graduate School of Medicine for preparing microscope slides; and all staff at the Endoscopy Unit, Kyoto University Hospital for endoscopic sampling.
Author information
Authors and Affiliations
Contributions
N.K., K.Y., H. Marusawa, T.C., H.S. and S. Ogawa designed the study. M.U., K. Kawada, S.N., A. Yokoyama, S.Y., M.M., T. Horimatsu, S. Okamoto, H.O., Y. Sakai, H.I. and H.N. provided specimens. T.K., Y.T., T. Sakurai, H.H. and S.H. performed histological analysis. N.K., Y.I., Y.F. and S.K.K. performed sample preparation. M.S., E.S. and T. Sato performed sequencing. N.K., Y.O., Y. Shiozawa, T.Y. and Y.N. performed mutation calling, validated the results and analysed CNAs, mutational signatures and clonal dynamics. N.K., Y. Shiraishi, K.C., H.T. and S.M. performed bioinformatics analysis. N.K., K.A., T. Hirano, Y.K., Y.W., K. Kataoka, M.M.N., A. Yoda, H. Miyoshi, M.M.T., J.O., R.I., Y.U. and O.T. performed in vitro functional analysis. N.K., N.G., A.K., K. Kataoka and T.M. performed in vivo functional analysis. N.K., A. Yokoyama, Y.N., H. Makishima and S.Ogawa prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Judy Cho, Ramnik Xavier and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Study design.
a–c, Summary of sampling types, histology of samples, subjects and sequencing platforms for single crypts (a), bulk crypts (b) and colorectal cancers (c). COAD, TCGA Colon Adenocarcinoma data collection; READ, TCGA Rectum Adenocarcinoma data collection. d, Summary of independent clones as determined by PyClone analysis of bulk-crypt samples from patients with UC.
Extended Data Fig. 2 Sampling methods.
a, For preparation of epithelial samples, after treating intestinal mucosa with 20 mM EDTA in PBS at 4 °C for 20 min, the epithelium was dissociated from the lamina propria. Then, a single crypt or epithelium (for bulk crypts) was collected. b, For geographical tracking of clones in a cluster of single crypts in colorectal mucosa from control individuals, acrylic adhesive was put on the mucosal surface, followed by a treatment with 20 mM EDTA in PBS. After peeling off the epithelium upside down, single crypts that were attached to the adhesive were collected one by one under a stereomicroscope and their positions recorded (top). To isolate crypts from patients with UC, after colorectal mucosa was treated with 20 mM EDTA in PBS, a gentle pressure was applied to the whole mucosal tissue from both sides to push up crypts, and each single crypt was isolated in a manner similar to that described above. c, For single-crypt sequencing, genomic DNA was extracted from single crypts by direct cell lysis and heat denaturation and split into two aliquots, each of which was subjected to WGA using Φ29 DNA polymerase. One of the two WGA products was subjected to WES, and the other was stocked for validation.
Extended Data Fig. 3 Mutations detected in single crypts.
a, b, Representative histograms of the VAFs of somatic mutations detected by WES in single crypts from control individuals (10 out of 43; a) and patients with UC (10 out of 58; b). c, Frequency of driver mutations among single crypts from control individuals (n = 43) and patients with UC (n = 58). Genes with a significant difference in mutation frequency between non-UC and UC crypts are indicated by asterisks (two-sided Fisher’s exact test with Benjamini–Hochberg adjustment; q < 0.1). d, The landscape of driver mutations for single crypts (n = 58) from non-dysplastic UC samples. The number of mutations in each single crypt is shown above, and the disease duration and age of subjects are shown below.
Extended Data Fig. 4 Phylogenetic trees in the colorectal epithelium.
a–d, Phylogenetic structure of single crypts clustered in an area of 0.25–4 mm2 of epithelia from two control individuals (an 84-year-old man (CR40; a) and an 88-year-old woman (CR43; b)) and two patients with UC (a 56-year-old man (HCM28; c) and a 42-year-old woman (UC3; d)). Phylogenetic trees of clones (right or bottom) are projected onto the positions of crypts, as indicated in the images (left or top). The colours of the branches in the trees correspond to those of the circles for the positions of crypts. The estimated time of UC onset is indicated on the basis of the mutation rate in non-UC crypts (see Fig. 1a). The estimated age of the most-recent common ancestral crypt is also indicated for patients with UC (c, d). Branches that contain driver mutations are indicated. e, Histograms of the number of mutations (or height) of branch points in phylogenetic trees are plotted for control individuals (n = 3, top) and patients with UC (n = 3, bottom).
Extended Data Fig. 5 Mutations detected in colorectal epithelium from control individuals and patients with UC.
a, MCF of mutations that were detected with WES in isolated bulk-crypt samples from non-UC (n = 136) and UC (n = 133) colorectal epithelia. Samples belonging to each subject are separated by blank columns. MCFs of driver mutations are indicated by coloured triangles, and disease state by colour, as shown at the top. The colon segment that was sampled, mutation status of driver genes, number of mutations, duration of UC and age of subjects are shown at the bottom. b, c, The average number of mutations (b) and average value of maximum MCFs (c) detected by WES of bulk-crypt samples within each individual is plotted against the duration of UC. Control individuals are plotted at zero on the x axis. Regression lines with (b) or without (c) assuming zero intercept are shown with R2 values. The calculated P value in c represents the significance of the correlation between the duration of UC and the average value of maximum MCFs (F test). d, e, The average number of mutations (d) and average value of maximum MCFs (e) of bulk-crypt samples from caecum to transverse colon (right side) and descending colon to rectum (left side) within the same individual is plotted for patients with UC (n = 22). Two-sided paired t-test.
Extended Data Fig. 6 Functional analysis of driver genes in UC mucosa.
a, Distribution of driver gene mutations in non-dysplastic UC samples (58 isolated single crypts and 399 bulk crypts, top) and CAC samples (n = 99, bottom). Amino acid sequences of different species around hotspot mutations of HNRNPF are shown. Completely conserved amino acids across all species are indicated by asterisks. b, Expression levels of Nfkbiz in colon epithelial organoids (n = 3 biological replicates) treated as indicated for 1 or 24 h. Data are normalized by the levels of PBS (control) and shown as mean ± s.d. Asterisks indicate P < 0.05 between the indicated treatment and treatment with PBS (two-sided Student's t-test). LTα1β2, lymphotoxin α1/β2; ODN, CpG oligodeoxynucleotides; OSM, oncostatin M. c, Immunoblot analysis of HCT116 cells that express empty vector, Myc-tagged wild-type (WT) ZC3H12A or the indicated ZC3H12A mutants after stimulation with IL-1β for the indicated times, using anti-Myc and β-actin antibodies. d, Immunoblot analysis of ZC3H12A, Flag-tagged dominant-negative BTRC(ΔF) and β-actin in HeLa cells that express doxycycline-inducible BTRC(ΔF) after stimulation with IL-17A or IL-1β for the indicated times with or without doxycycline (Dox). Data are representative of two independent experiments. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 7 Clonal expansion in UC rectums.
a, Expansion of clones within the rectum from the remaining four out of the six analysed patients with UC (two are presented in Fig. 2d). All of the representations follow those in Fig. 2d. Driver mutations including those found in the two samples in Fig. 2d are summarized in Supplementary Table 9. b, Area occupied by each clone and total number of mutations estimated by PyClone for each clone detected in the UC rectums. Colours indicate the highest pathological grade within each clone. c–h, Phylogenetic trees of representative clones (6 out of 41) that were found in two or more rectum samples surgically obtained from six patients with UC. Imputed clonal structures are depicted in violin plots at the bottom, in which the estimated cellular prevalence of imputed mutational clusters (x axis) are shown as the distribution of posterior probabilities (width of the violin plots) calculated according to a PyClone model. Colours correspond to those in the associated branches of trees. The estimated time of UC onset is indicated on the basis of the mutation rate in non-UC crypts (Fig. 1a). Branches that contain driver mutations are also indicated.
Extended Data Fig. 8 Effect of Nfkbiz on clonal expansion in a mouse model of chronic DSS-induced colitis.
a, Experimental schedule for induction of colitis in R26LacZ/WTNfkbizWT/WTVil1-creER (control) and R26LacZ/WTNfkbizfl/flVil1-creER (Nfkbiz cKO) mice. Cre-mediated recombination was induced by administration of tamoxifen (TAM), followed by induction of colitis by treatment with DSS. b, e, Images of whole-mount X-gal staining of the large intestine from control (top) and Nfkbiz cKO (bottom) mice after tamoxifen administration without (b) or with (e) subsequent treatment with DSS. c, f, Fraction of LacZ-positive areas in the middle to distal colon before (c) or after (f) induction of colitis with DSS according to genotypes. Two-sided Mann–Whitney U test. d, Density of LacZ-positive areas (number per cm2) having indicated sizes in control and Nfkbiz cKO mice before induction of colitis with DSS. g, Colon length (left) and histological score (right) after chronic DSS-induced colitis for the evaluation of severity of colitis. Two-sided Mann–Whitney U test. h, Relative change in weight of Nfkbiz cKO and control mice during three courses of DSS-induced colitis. Data are shown as mean ± s.d. i, Significant enrichment of NFKBIZ target genes (left) and genes associated with the IL-17 signalling pathway (right) among the downregulated genes in rectum epithelium from Nfkbiz cKO mice compared with control mice four days after the start of treatment with DSS. Data were generated from n = 3 biologically independent mice for both genotypes. Two-sided nominal P values were calculated by GSEA.
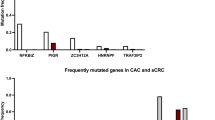
Extended Data Fig. 9 Mutational profiles and CNAs in control individuals and patients with UC.
a, Landscape of driver mutations in CAC samples (n = 99) and dysplastic UC samples (n = 13). Genes with asterisks are drivers in CACs (Methods). CNAs of known oncogenes are depicted only for the 34 CACs that were newly analysed in this study. b, Frequency of driver mutations among clones identified in non-dysplastic samples from patients with UC (n = 183) versus sCRC samples from the Japanese cohort (n = 525). Asterisks indicate q < 0.1 (two-sided Fisher’s exact test with Benjamini–Hochberg adjustment). c, Number (middle) and relative frequency (bottom) of mutations allocated to signatures A and B in samples of single crypts from control individuals (n = 43) and non-dysplastic (n = 241; 58 single crypts and 183 clones), dysplastic (n = 13 clones) and non-hypermutated CAC (n = 92 samples) from patients with UC. Information about disease state, pathology and sample type is indicated at the top. d, Comparison of the number of mutations detected by WES in clones identified in non-dysplastic samples, dysplastic samples and CAC samples without hypermutation from patients with UC. Two-sided Mann–Whitney U test. e, Mutational signatures of colorectal epithelium from control individuals and patients with UC. f, Fraction of signature B mutations in single crypts from control individuals, and clones identified in non-dysplastic (ND), dysplastic (D) and non-hypermutated CAC samples containing ≥ 10 mutations from patients with UC. Two-sided Mann–Whitney U test. g, Box plots of the fractions of genomic regions that contain CNAs in clones identified in non-dysplastic (n = 183), dysplastic (n = 13) and CAC (n = 34) samples from patients with UC. Two-sided Mann–Whitney U test. h, Bottom, colour-gradient maps of CNAs and UPDs as detected by WES data are shown for clones identified in non-dysplastic (n = 183), dysplastic (n = 13) and CAC (n = 34) samples from patients with UC. Top, pathological grade, fractions of genomes that exhibit CNAs and UPDs and TP53 mutation status are also shown. Box plots as in Fig. 4.
Extended Data Fig. 10 Effect of NFKBIZ knockout in CRC cell lines and an AOM–DSS-induced colorectal tumour model.
a, Experimental strategy for enrichment analysis using CRISPR–Cas9 (Methods). MOI, multiplicity of infection. b, Experimental schedule for induction of CAC in Nfkbizfl/flVil1-cre (Nfkbiz cKO) and Nfkbizfl/fl (control) mice using AOM followed by three cycles of DSS. c, Box plots of tumour numbers relative to tumour size (maximum diameter). Two-sided Mann–Whitney U test. d, Colon length (left) and histological score (right) for the evaluation of inflammation in the AOM–DSS-induced tumour model. No significant difference was observed between Nfkbiz cKO and control mice (two-sided Mann–Whitney U test). e, Relative change in weight of mice during three courses of treatment with DSS in the AOM–DSS-induced tumour model. Data are shown as mean ± s.d. f, Histological images of haematoxylin and eosin (H&E) staining (left), and immunohistochemistry for β-catenin (middle) and Ki-67 (right). Tumours in Nfkbiz cKO and control mice were indistinguishable in terms of histology and immunochemistry. Representative images are shown from n = 3 biologically independent samples for both genotypes. Box plots as in Fig. 4.
Supplementary information
Supplementary Figures
Supplementary Figure 1: Uncropped scans with size marker indications.
Supplementary Tables
This file contains Supplementary Tables 1-13.
Source data
Rights and permissions
About this article

Cite this article
Kakiuchi, N., Yoshida, K., Uchino, M. et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 577, 260–265 (2020). https://doi.org/10.1038/s41586-019-1856-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1856-1
This article is cited by
-
Regulation of inflammatory diseases via the control of mRNA decay
Inflammation and Regeneration (2024)
-
Tissue mosaicism following stem cell aging: blood as an exemplar
Nature Aging (2024)
-
Decoding the basis of histological variation in human cancer
Nature Reviews Cancer (2024)
-
NFKBIZ regulates NFκB signaling pathway to mediate tumorigenesis and metastasis of hepatocellular carcinoma by direct interaction with TRIM16
Cellular and Molecular Life Sciences (2024)
-
Colitis-associated carcinogenesis: crosstalk between tumors, immune cells and gut microbiota
Cell & Bioscience (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
