
Abstract
Organic chemistry has largely been conducted in an ad hoc manner by academic laboratories that are funded by grants directed towards the investigation of specific goals or hypotheses. Although modern synthetic methods can provide access to molecules of considerable complexity, predicting the outcome of a single chemical reaction remains a major challenge. Improvements in the prediction of ‘above-the-arrow’ reaction conditions are needed to enable intelligent decision making to select an optimal synthetic sequence that is guided by metrics including efficiency, quality and yield. Methods for the communication and the sharing of data will need to evolve from traditional tools to machine-readable formats and open collaborative frameworks. This will accelerate innovation and require the creation of a chemistry commons with standardized data handling, curation and metrics.
Similar content being viewed by others
Main
The preparation of oxalic acid and urea by Wöhler almost 200 years ago established the field that we call organic synthesis1. Human insight from reactivity explored in the interim can now lead to beautifully organized campaigns of complex natural products and bioactive molecules, which represent the pinnacle of synthetic design2. The idea of a synthesis machine that can build any molecule dates from the 1960s. However, although the first computer programs to design organic syntheses emerged around this time3,4, they failed to capture the imagination of chemists. Synthesis laboratories have remained sceptical of the ability of computer programs to learn the ‘art’ of organic chemistry, and have continued their tried and true approaches in their laboratories.

Now, the scepticism of synthetic chemists seems to be on the verge of changing. Using computer-aided synthesis planning (CASP), it is now possible to take the molecular structure of a desired product and output a detailed list of reaction schemes that connect the target molecule to known and often purchasable starting materials through a sequence of intermediates that are likely to be unknown5,6 (Box 1). For example, the decision-tree-like search engine Chematica—which has a user-friendly graphical user interface and has been coded with human-curated rules over the past decade—has received laboratory validation of the predicted synthesis of medicinally relevant targets7. Approaches towards such programs usually reflect the priorities and prejudices of the programmers, and others have used different approaches—for example, using machine-learning algorithms or Monte Carlo Tree Search (as in AlphaGo8) to guide the search, and a filter network to pre-select the most promising retrosynthetic steps that is trained on essentially all reactions ever published in organic chemistry9,10,11. In the future, it will be substantially faster for such programs to learn automatically from the primary data rather than rely on extracted rules and hand-designed heuristics, in analogy to the differences in strategy between Stockfish and AlphaZero in learning chess12.
The digitization of multistep organic synthesis is fast approaching, and the automation of the synthesis planning is just the first component that must be considered before automated reaction prediction can become a reality. The selection of reaction conditions is a key element of automated reaction prediction and is potentially a far more challenging task13 (Fig. 1). This Perspective surveys the current prospects for the prediction of above-the-arrow conditions and addresses the challenges that are involved in integrating them into optimal methods of synthesis. For one, it has been stated that “syntheses are reported in prose”14. Not only are the reactions conditions often poorly communicated, but details are also omitted when explaining exactly how operations were carried out, meaning that many assumptions are made about the skills of the researcher repeating the synthesis. The prediction problem must then consider an even broader range of variables in order to master or fully execute a synthesis or optimization, depending on the context of academic research and medicinal or process chemistry.

To perform an organic chemical reaction in a laboratory, the conditions listed above the arrow are required to run the synthesis and isolate the desired product.

Challenges in culture and data reporting
Proposing specific reactions to a given target on the basis of the literature and canonical rules may seem to be a mysterious and daunting task to most, but it is considered a routine activity for practitioners of organic synthesis who begin to grasp the principles as chemistry undergraduates15. Throughout a career these skills are improved, and the well-trained chemist often uses rules and patterns of chemical reactivity that they have developed by immersion in the field. With a new synthetic problem at hand, the chemist tries to compare it to a known one before making sense of it—a similar concept to that used by deep-learning algorithms. Historically, having spent a day reading the literature or conducting database searches, the chemist absorbs the precedents and sets off for the laboratory. Within a modern chemistry setting, predicting the starting point for experimentation—especially for complex molecular environments—is now challenging for even the best-educated of chemists. The yield and the selectivity (chemo-, regio-, diastereo- and enantioselectivity) of any transformation in the field of catalysis can be controlled by millions of permutations—including temperature, solvent, ligand and ancillary reagents—even before other metrics of quality are applied. Simply using a large number of experiments in (electronic) notebooks to select the above-the-arrow conditions has been unsuccessful so far, as the data are fractured and collected without diversity of starting materials—often because organizations have experience with molecules that were influenced by a target area in biology. Another obstacle related to human nature is that when reactions fail, the experimentalist is often not concerned with complete documentation and moves onto another task. In the area of medicinal chemistry, in which enormous numbers of experiments are performed, it has been stated that there are only two yields that matter: enough and not enough16. Overall, the current approaches used to record experiments fail to capture the ‘messiness’ of organic synthesis, as well as the continuous nature of the solutions in the real world.
To advance the field of machine learning in organic synthesis, enormous improvements will be required to enable the prediction of the discrete and continuous variables in the reaction conditions that appear above the arrow (Fig. 1). This will be possible only if it is accompanied by advances in the reporting of cases in which syntheses are captured in the form of digital code that can be published, versioned and transferred flexibly between platforms to enhance reproducibility. Despite the abundant incentives for academic and industrial scientists to share synthetic data via publication, the data published in most journal articles represents only a fraction of the raw data collected in a given research project. As a community we rely on outdated means that are mere facsimiles rather than machine-readable formats. A stumbling block is not only how to uniformly collect, clean and label data that are of use for training inside an organization or laboratory, but also how to align incentives to make data broadly available via new data intermediaries.
Further challenges for machine learning concern the identification and scoring of the criteria for the efficiency of the overall synthetic sequence, as there are currently no clear criteria on which this can be judged. It is already impossible for a human to assess all available options from either the recalling of synthetic methods or searching online. The formulation of such rules has primarily occurred in an academic setting around the definition of an ideal synthesis17,18,19. The ‘fit-for-purpose’ rule of academia or medicinal chemistry will certainly be unacceptable in the fine- and commodity-chemical sectors of the industry, in which efficiency, quality and safety are all a necessity. The reaction steps, time to a workable answer, speed and throughput, availability of diverse raw materials, process economics, sustainability and energy consumption all need to be included in assessing digitization of the multistep synthesis to define an answer that is beyond the output of a detailed list of potential reaction schemes.
Complexity in the execution of synthesis
The total synthesis of maoecrystal V (Fig. 2) is a good illustration of the level of above-the-arrow complexity in contemporary natural-product synthesis. In the preparation of this compound, which was completed by the Baran laboratory20, what is essentially an aldol reaction—taught in first-year organic chemistry classes—proved to be the most challenging step.

The natural product is prepared in a longest linear sequence of 11 steps. Step 7 is a reaction of enone 1 and formaldehyde to provide hydroxymethylketone 2. In order to perform this reaction in a laboratory, at least 16 conditions—including workup procedures—are listed above the arrow.
The enolate-based installation of the hydroxymethyl group overcame the challenges of chemo- and regioselectivity. Over 1,000 experiments were carried out in order to optimize the reaction conditions, changing every conceivable variable possible; as a result, the optimized reaction has at least 16 conditions listed above the arrow. Conditions such as solvent and temperature changes and those used in workups are rarely considered in this context, but are essential for the successful repetition of the experiment. The desired product 2 was obtained with complete chemoselectivity, although the diastereoselectivity (2:1) and the yield (84%) remained intransigent to further improvement. Although far from optimal, the intermediate hydroxymethylketone 2 was processed onto maoecrystal V to provide sufficient material to answer the key biological questions presented by this molecule.
Different challenges prevail in the field of medicinal chemistry, in which molecules are designed to engage with increasingly more complex biological targets. Hundreds or thousands of molecules are required to advance from a hit compound to a drug candidate, and the synthetic route provides a platform from which to optimize for molecular function and explore biology. A consideration for any reaction used in medicinal chemistry is its level of tolerance to the polar functional groups and nitrogen heteroatoms that are typically found in biologically active molecules. As artificial intelligence and big data are increasingly used in medicinal chemistry for compound prediction and prioritization, it will become even more important to make the right compound the first time21. It is clear that even for well-precedented reactions and obvious retrosynthetic disconnections (that is, breaking a molecule up into simpler starting materials), there are fundamental practical limitations when considering the conditions needed to make sufficient material for biological testing22. Even within the context of the late-stage functionalization of a drug-like molecule, the individual conditions in that single step can still profoundly affect selectivity23.
As with natural-product synthesis, process chemistry has often been described as an ‘art’24. Well-trained organic chemists read literature and generate the reaction sequence that in their best estimate meets their goals; however, these estimates are often biased by cultural- and company-based specific information on route selection, approaches to reject impurities, and the preparation of salts to improve the crystallinity, solubility and stability of intermediates or the active pharmaceutical compound. Process chemists have developed an intuition as to how well a reaction is likely to scale to obtain a high yield, high concentration, and low catalyst loading with good impurity rejection, and this informs the choice of a synthesis. This informal knowledge, acquired over many years by real-world reinforcement, is rarely captured in any form besides institutional knowledge.
Additionally, only a few well-conceived ideas can currently be pursued by process chemists in the laboratory. Commercial and regulatory pressures ensure that, among the range of potential routes identified early on, a single approach will be taken forward for validation and commercialization. These decisions are made largely with contradictory—or, at best, missing—data concerning the future potential efficiency of the route. This critical selection process is performed in the absence of quantitative efficiency data, and is often influenced by judgements on risk mitigation to product filing, or by broad assumptions around supply chain and tax and treasury. Although a considerable financial impact can be achieved by minimizing the costs of reagents and solvents and by optimizing the conditions for small improvements in yield or product quality, this impact cannot overcome the selection of a suboptimal route. It is highly desirable to understand all of the viable options before beginning full-scale development25.
Further predictions of conditions that are not historically included above the arrow must be used to narrow the range of options for further exploration. In process chemistry, the crystallinity and solubility, physical attributes of crystallization kinetics, particle-size reduction, flow ability, and solid-state stability are all key to understanding chemical intermediates and pharmaceutical properties. Thus, machine-learning algorithms should ideally be tailored to different criteria than those in other areas of synthesis. We will need to advance our ability to predict the organic-solvent solubility, the crystal phase and the morphology of compounds if we are to develop viable options without a priori knowledge.
Emerging examples of innovation using enhanced data
The goal of building a synthesis machine that can provide high-quality reagents for biology—beyond peptides and oligonucleotides—has been championed as a way of freeing up chemists for creative thinking by removing the bottleneck of synthesis26. However, a general commoditization of synthetic medicinal chemistry is not likely to emerge until we have made these orders-of-magnitude improvements in above-the-arrow prediction. Ultimately, machine learning will enable the field to predict individual conditions by moving along the spectrum of individual chemistry experiments, run one at a time, through large data assimilation and then back to individual conditions. A chemist can then, with a high degree of confidence, guarantee that sufficient product will be obtained in a single experiment to test the function of a molecule.
Scientists at Merck recognized this problem and systematically built tools, using high-throughput experimentation and analysis, to address the gaps in data27. Using the ubiquitous palladium-catalysed Suzuki–Miyaura cross-coupling reaction as a test case, they developed automation-friendly reactions that could operate at room temperature by using robotics employed in biotechnology coupled with emerging high-throughput analysis techniques. More than 1,500 chemistry experiments can be carried out in a day with this setup, using as little as 0.02 mg of starting material per reaction. This has since been expanded to allow for the in situ analysis of structure–activity relationships (nanoSAR)28. The authors note that, in the future, machine learning may aid the navigation of both reaction conditions and biological activity. Complementary approaches, such as inverse molecular design using machine learning, may also generate models for the rational design of prospective drugs29,30.
In order to reduce analysis time, ultra-high-throughput chemistry can be coupled to an advanced mass spectrometry method (such as matrix-assisted laser desorption ionization–time-of-flight spectrometry; MALDI–TOF) to enable the classification of thousands of experiments in minutes31. This classification approach may at first be slightly uncomfortable for synthetic chemists who hold stock in obtaining a hard yield, but it will surely become commonplace as more statistical methods and predictive models are deployed.
Machine learning has recently been used to predict the performance of a reaction on a given substrate in the widely used Buchwald–Hartwig C–N coupling reaction32. The Doyle laboratory used a robot-enabled simultaneous evaluation method with three 1,536-well plates that consisted of a full matrix of aryl halides, Buchwald ligands, bases and additives, giving a total of 4,608 reactions. The yields of these reactions were used as the model output and provided a clean, structured dataset containing substantially more reaction dimensions than have previously been examined with machine learning. Approximately 30% of the reactions failed to deliver any product, with the remainder spread relatively evenly over the range of non-zero yields. Using concepts popularized by the Sigman group33, scripts were built to compute and extract atomic, molecular and vibrational descriptors for the components of the cross-coupling. Using these descriptors as inputs and reaction yield as the output, a random forest algorithm was found to afford high predictive performance. This model was also successfully applied to sparse training sets and out-of-sample reaction outcome prediction, suggesting that a systematic reaction-profiling capability and machine learning will have general value for the survey and navigation of reaction space for other reaction types.
It has been suggested by Chuang and Keiser that this experimental design failed classical controls in machine learning, as it cannot distinguish chemically trained models from those trained on random features34. As they noted, flexible and powerful machine-learning models have become widespread, and their use can become problematic without some understanding of the underlying theoretical frameworks behind the models. The ability to distinguish peculiarities of the layout of an experiment from those that extract meaningful and actionable patterns also need to developed. Regardless, it is clear that the approach taken by Doyle—publishing a complete dataset and aligned code on GitHub—enables a clear demonstration of the scientific method of testing and generating hypotheses in independent laboratories.
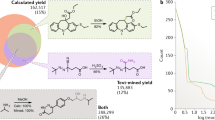
The application of machine learning to the prediction of reactions has also been demonstrated for the conversion of alcohols to fluorides, the products of which are high-value targets in medicinal chemistry35 (Fig. 2). In order to train a model for this reaction, descriptors for the substrates and reagents used in 640 screening reactions were tabulated. These included computed atomic and molecular properties as well as binary categorical identifiers (such as primary, secondary, cyclic). A random forest algorithm was used and was trained on 70% of the screening entries. The model was evaluated using a test set comprising the remaining 192 reactions and was validated on five structurally different substrates from outside the training set. The yields of these reactions were predicted with reasonable accuracy, which is more than sufficient to enable synthetic chemists to evaluate the feasibility of a reaction and to select initial reaction conditions. In comparison to previous studies, this training set was 80% smaller, encompassed much broader substrate diversity and incorporated multiple mechanisms. The expansion of the training set for this deoxyfluorination reaction to include additional variables (that is, stoichiometry, concentration, solvent and temperature) could lead to more accurate and comprehensive coverage of the complex reaction space.
Flow chemistry presents another opportunity for accelerated reaction development36. A recent publication by a Pfizer team37 demonstrated high-throughput reaction screening of the Suzuki–Miyaura coupling with multiple discrete (catalyst, ligand and base) and continuous (temperature, residence time and pressure) variables (5,760 reactions in total), overcoming a common problem in which limited amounts of material do not allow for the application of flow reaction screening in medicinal chemistry (Fig. 3a, b). Quinolines (3a–g) and indazole acids (4a–d) were used to validate the platform. In an important demonstration of the capability of the platform for the preparation of useful quantities of material, the team programmed the injection of 100 consecutive segments based on optimal conditions from screening, enabling the preparation of approximately 100 mg of a target molecule per hour.

Six hundred and forty screening reactions were performed to train a machine-learning model (yields presented as a heat map). This was used for the successful prediction of the yield and conditions for structurally different substrates that do not appear in the training set. This figure was adapted with permission from ref. 35, copyright 2018 American Chemical Society.
The Jamison and Jensen groups have described an automated flow-based platform38 to optimize above-the-arrow conditions to improve the yield, selectivity and reaction scope of a diverse range of reactions; this is typically a tedious and labour-intensive task in the laboratory. By using feedback from online analytics, the system converges on optimal conditions that can then be repeated or transferred with high fidelity as needed. These automated systems in academic laboratories may also play a part in the rapid collection of large, standardized datasets39.
Chemical synthesis may no longer be solely a human activity. In a recent study, the Cronin laboratory demonstrated that a robotic reaction-handling system controlled by a machine-learning algorithm might be able to explore organic reactions an order of magnitude faster than a manual process40. The robotic approach enabled the capture of information on failed or non-reactive experiments in a structured fashion, making it useful for reaction mapping. The powerful machine-learning algorithm was able to predict the reactivity of 1,000 reaction combinations from the above Pfizer dataset (Fig. 4a), with greater than 80% accuracy, after considering the outcomes of around 10% of the dataset.
In this machine-learning analysis of the Pfizer work, one-hot encoding of the reaction conditions—in which the variables were assigned binary representations—and the clean standardized yield data were used to explore the prediction of yields by a neural network (catalyst loading and temperature were not included). In this approach, a random selection of 10% (n = 576) of the Suzuki–Miyaura reactions is used to train the neural net, and the remaining reactions are then scored by the model (Fig. 4b). The candidates with the highest predicted yield are then added to the performed reactions, and the performance of the neural network is evaluated by calculating the mean of the true yield and the standard deviation of the yield. The neural network is then retrained, and the whole cycle is repeated until the entire space is explored in panels of 100 to demonstrate the alignment with the high-throughput experimentation as well as to evaluate the performance of the neural net. Such rapid evaluation is markedly enabled by the publication of reliable clean data.
A common theme in these three machine-learning examples is that predictions can be made with relatively small datasets: in some cases, with only 10% of the total number of reactions it is possible to predict the outcomes of the remaining 90%, without the need to physically conduct the experiments (Fig. 4). The high-fidelity data can originate from ultra-high-throughput screening, from flow chemistry or from an individual scientist, but the most important feature is the contextualized, internally consistent source that provides effective, secure and accurate data. This is important because it is currently not known how large these datasets need to be in order to predict across the molecules that represent drug-like space. Naturally, some reactivity trends may be reflective of how the individual experiments are conducted and not truly informative of a particular catalyst or ligand. A diagnostic approach using small libraries of curated drug-like molecules—known as ‘informer libraries’—has been presented as a way to better capture reaction scope and evolve synthetic models, but this should be viewed as an intermediary step as the field moves forward22.

a, A Suzuki–Miyaura reaction optimized in flow. A heat map of yields of the 5,760 reactions run is shown (3a–d with 4a–c and the reaction of 3e–g with 4d), evaluated across a matrix of 11 ligands (plus one blank) × 7 bases (plus one blank) × 4 solvents (ref. 37). b, These data were used for one-hot encoding of reactants 3, reactants 4, ligands, bases and solvents as a test set for prediction of yield from the test set (30% of the reactions). Predictions for the full dataset are also shown. Panel a is adapted from ref. 37, reprinted with permission from AAAS; panel b is adapted from ref. 40.
There have also been important advances in predictive catalysis41,42. This is an exciting, emerging field that uses parameterization and analysis of catalysts to enable the forecast of an attainable improvement—for example, the enantioselectivity of a transformation or improved turnover in a biocatalytic reaction43,44,45—to provide confidence for route selection. For example, in the synthesis of letermovir46, a series of new catalysts was identified that provided the desired product in improved enantioselectivity and facilitated faster route optimization. The models are currently limited in scope, requiring a focused solvent screen on the best-performing catalysts, and process optimization had already taken place for the desired starting material. However, these models will greatly improve with the availability of enhanced datasets, which encompass a full range of activity from diverse sources47.
Extending these early successes to the prediction of the impurity profile of a reaction becomes especially difficult for catalysis, because many on-cycle and off-cycle events can markedly alter the optimum yield and because impurities do not always track with conversion. The current machine-learning systems do not yet take the mechanism of byproduct formation into account. However, process chemists will need information in order to predict and understand both the fate of impurities formed during each step in the process and where impurities are removed in the overall sequence; this is necessary not only to improve performance but also, and often more importantly, to meet regulatory requirements. Almost all of this information currently resides with corporations and is elusive internally and hidden externally. The messiness of data in our broad field of organic synthesis remains a challenge, and we should seek more engagement and demand more focused attention than we have in the past 50 years48.
Accelerating future innovation
There is a recent trend for organic chemists to publish ever larger numbers of examples in methodology papers. However, these reports remain focused on the knowledge and the dataset published in a journal article, which represents only a small portion of the raw data collected. These data have not yet been collected in a standardized manner, and highly complex substrates are often not included. In more general terms, in the 200-year history of organic synthesis, we have not yet developed methods to collect, clean and label data in a way that makes it useful for training in the context of new reaction optimization, especially in the areas of catalysis design and development. Existing datasets in the public or private domain have simply not been built with this in mind.
We have seen that large datasets or even ultra-high-throughput experimentation are not a prerequisite to machine learning. Biopharma deals with hundreds of millions of documents—including laboratory data and clinical trial reports, publications and patent filings, as well as billions of database records. Companies and not-for-profit alliances are working to provide solutions to data management. Despite the quantity of data, chemical structure information is essentially captured as an image in a book—it is essentially unusable, whereas above-the-arrow and other data are currently considered out of scope and the vast amounts of historic data in paper and electronic notebooks remains orphaned. Consequently, to avoid repeating current synthetic methods in the field we need to embrace modern approaches and pay attention to future needs. This will avoid simply restating the master data problem. Metrics for similarity calculations49 use the fingerprints of molecules to compare how similar they are to each other and will ensure that we avoid bias introduced by human-curated examples for machine learning. We need our data to emerge beyond the positive results and the publication- or career-driven biases. A published data point should be one click away from raw experimental data, all the way from the weighing of materials to analytical data, enabled by the Internet of Things50.
Before we do that, we need to provide a framework in which to enable the collection and publication of new data as it is generated, much like the Bermuda Accord51. This established that all the DNA sequence information from large-scale human genomic projects should be freely available and in the public domain. With increasing exploration of new research areas that cross disciplines—for example, chemical biology or proteomics—it is becoming common for very different traditions towards data sharing to coexist in the same laboratory52. In the field of organic synthesis the intensity of the work, the amount of capital allocation required and the degree of specialization in data rather than ‘art’ will lead to the creation of a new kind of chemist—one whose principal objective is the generation of high-quality datasets. These datasets will go on to be the foundation for a new partnership of hypothesis-driven and hypothesis-free discovery based on big data in chemistry53. This distinction exists today in biology as the number of data-generating projects advances, in which medical breakthroughs such as CRISPR often emerge from unpredictable origins. The field has adapted, and enables academic data-generating researchers to continue obtaining grant funding for their work as well as advancing their careers through publication and peer recognition. Governmental agencies and large independent global charities can clearly influence the funding of the new data-generation projects, science policy, intellectual property and regulation.
Synthetic chemistry has emerged relatively unscathed from the narrative of poor reproducibility in science and has not yet faced a crisis of confidence54. There have been important calls regarding reproducibility55 and the discussion will remain contemporary as it essential that the quality, reproducibility and traceability of the raw data and models. As in several of the machine-learning examples discussed above32,40, the availability of reliable data and the code enables others to verify and retest alternative hypotheses. This helps to demonstrate the effect of data that is findable, accessible, interoperable and reusuable (FAIR)56. For example, the Cronin group was able to rapidly model the data for the Suzuki–Miyaura reaction presented from the Pfizer flow platform40. As we report more complete datasets that include reactions that fail to give products in expected yield or quality, we need to be cautious. A failure may not represent a true reflection of the reactivity profile of a current method, and we need to ensure that it does not limit exploration or utilization of a newly developed reaction. In the future, machine learning will therefore need to become a partner in order to elucidate reaction concepts, elusive high-value transformations and problems of which chemists are not currently aware (unknown unknowns), as well as to rapidly identify unanticipated observations or spare events.
It is exciting to consider the potential societal impact of innovations similar to AlphaGo Zero in the chemical space. Commercial software packages are emerging and, although it is clear that these approaches will advance in sophistication, it is not necessary for the end user to understand the underlying complexity as long as the answers satisfy their needs. Unlike in closed systems such as chess or Go, there are no clearly defined rules for winning, and explainable artificial intelligence will be an ongoing issue57,58. It remains to be seen whether the machines can become experts or merely expert tools.
Future advances in the digitization of chemistry will not come at an equal pace, and some areas of organic synthesis will be affected much sooner than others. Computing power is no longer a limitation, and there are much more sophisticated algorithms that can handle fuzzy datasets developing in fields that have more direct monetization. Although the technology is not yet reliable enough, it is clear that the field of synthesis and optimization in applications such as medicinal and process chemistry will become a more evidence-led practice. Some organic chemists will ignore the signals of this transformation, some will improve and make incremental progress, and some will be the innovators, embracing these tools to augment their scientific intuition and creativity.
References
Wöhler, F. Ueber künstliche bildung des harnstoffs. Ann. Phys. 88, 253–256 (1828).
Whitesides, G. M. Complex organic synthesis: structure, properties, and/or function? Isr. J. Chem. 58, 142 (2018).
Corey, E. J. & Wipke, W. T. Computer-assisted design of complex organic syntheses. Science 166, 178–192 (1969).
Corey, E. J., Wipke, W. T., Cramer, R. D. III & Howe, W. J. Computer-assisted synthetic analysis. Facile man–machine communication of chemical structure by interactive computer graphics J. Am. Chem. Soc. 94, 421–430 (1972).
Szymkuć, S. et al. Computer-assisted synthetic planning: the end of the beginning. Angew. Chem. Int. Ed. 55, 5904–5937 (2016).
Coley, C. W., Green, W. H. & Jensen, K. F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 51, 1281–1289 (2018).
Klucznik, T. et al. Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 4, 522–532 (2018).
Silver, D. et al. Mastering the game of Go without human knowledge. Nature 550, 354–359 (2017).
Schwaller, P., Gaudin, T., Lanyi, D., Bekas, C. & Laino, T. “Found in translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 9, 6091–6098 (2018).
Segler, M. H. S. & Waller, M. P. Modelling chemical reasoning to predict and invent reactions. Chem. Eur. J. 23, 6118–6128 (2017).
Segler, M. H. S., Preuss, M. & Waller, M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018).
Kasparov, G. Chess, a Drosophila of reasoning. Science 362, 1087 (2018).
Cernak, T. A machine with chemical intuition. Chem 4, 401–403 (2018).
Steiner, S. et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 363, eaav2211 (2019).
Garg, N. K. Empowering students to innovate: engagement in organic chemistry teaching. Angew. Chem. Int. Ed. 57, 15612–15613 (2018).
Engkvist, O. et al. Computational prediction of chemical reactions: current status and outlook. Drug Discov. Today 23, 1203–1218 (2018).
Gaich, T. & Baran, P. S. Aiming for the ideal synthesis. J. Org. Chem. 75, 4657–4673 (2010).
Trost, B. M. The atom economy—a search for synthetic efficiency. Science 254, 1471–1477 (1991).
Burns, N. Z., Baran, P. S. & Hoffmann, R. W. Redox economy in organic synthesis. Angew. Chem. Int. Ed. 48, 2854–2867 (2009).
Cernijenko, A., Risgaard, R. & Baran, P. S. 11-step total synthesis of (−)-maoecrystal V. J. Am. Chem. Soc. 138, 9425–9428 (2016).
Griffen, E. J., Dossetter, A. G., Leach, A. G. & Montague, S. Can we accelerate medicinal chemistry by augmenting the chemist with Big Data and artificial intelligence? Drug Discov. Today 23, 1373–1384 (2018).
Kutchukian, P. S. et al. Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci. 7, 2604–2613 (2016).
Yao, H. et al. Enabling efficient late-stage functionalization of drug-like molecules with LC-MS and reaction-driven data processing. Eur. J. Org. Chem. 2017, 7122–7126 (2017).
Yasuda, N. (ed.) The Art of Process Chemistry (Wiley-VCH, 2010).
Li, J., Albrecht, J., Borovika, A. & Eastgate, M. D. Evolving green chemistry metrics into predictive tools for decision making and benchmarking analytics. ACS Sustainable Chem. Eng. 6, 1121–1132 (2018).
Trobe, M. & Burke, M. D. The molecular industrial revolution: automated synthesis of small molecules. Angew. Chem. Int. Ed. 57, 4192–4214 (2018).
Buitrago Santanilla, A. et al. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 347, 49–53 (2015).
Gesmundo, N. et al. Nanoscale synthesis and affinity ranking. Nature 557, 228–232 (2018).
Sanchez-Lengeling, B. & Aspuru-Guzik, A. Inverse molecular design using machine learning: generative models for matter engineering. Science 361, 360–365 (2018).
Schneider, G. Automating drug discovery. Nat. Rev. Drug Discov. 17, 97–113 (2018).
Lin, S. et al. Mapping the dark space of chemical reactions with extended nanomole synthesis and MALDI-TOF MS. Science 361, eaar6236 (2018).
Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. & Doyle, A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 360, 186–190 (2018). This article demonstrates machine learning in prediction of the performance of a catalytic reaction using data obtained via high-throughput experimentation.
Zhao, S. et al. Enantiodivergent Pd-catalyzed C–C bond formation enabled through ligand parameterization. Science 362, 670–674 (2018).
Chuang, K. V. & Keiser, M. J. Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”. Science 362, eaat8603 (2018). This article illustrates the need to incorporate random-control procedures when applying machine learning to new scientific domains and the importance of experimental design.
Nielsen, M. K., Ahneman, D. T., Riera, O. & Doyle, A. G. Deoxyfluorination with sulfonyl fluorides: navigating reaction space with machine learning. J. Am. Chem. Soc. 140, 5004–5008 (2018). This paper demonstrates the use of machine learning on a relatively small dataset obtained by traditional laboratory experimentation.
Reizman, B. J. & Jensen, K. F. Feedback in flow for accelerated reaction development. Acc. Chem. Res. 49, 1786–1796 (2016).
Perera, D. et al. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 359, 429–434 (2018). This article illustrates that a flow apparatus can accelerate reaction optimization earlier in the drug-discovery process and also provides reliable data that enables other laboratories to build machine-learning algorithms.
Bedard, A.-C. et al. Reconfigurable system for automated optimization of diverse chemical reactions. Science 361, 1220–1225 (2018).
Caramelli, D. et al. Networking chemical robots for reaction multitasking. Nat. Commun. 9, 3406 (2018).
Granda, J. M., Donina, L., Dragone, V., Long, D.-L. & Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 559, 377–381 (2018). This article predicts the reactivity of about 1,000 reaction combinations with accuracy greater than 80 per cent after considering the outcomes of slightly over 10 per cent of the dataset and, notably, the approach was also used to calculate the reactivity of published datasets.
Harper, K. C. & Sigman, M. S. Predicting and optimizing asymmetric catalyst performance using the principles of experimental design and steric parameters. Proc. Natl Acad. Sci. USA 108, 2179–2183 (2011).
Zahrt, A. F. et al. Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning. Science 363, eaau5631 (2019).
Matsuda, T. (ed.) Future Directions in Biocatalysis 2nd edn (Elsevier, 2017).
Kan, S. B. J., Russell, D., Lewis, R. D., Chen, K. & Arnold, F. H. Directed evolution of cytochrome c for carbon–silicon bond formation: bringing silicon to life. Science 354, 1048–1051 (2016).
Arnold, F. H. Innovation by evolution: bringing new chemistry to life – Nobel lecture. Nobel Media AB 2019 https://www.nobelprize.org/prizes/chemistry/2018/arnold/lecture/ (2019).
Metsänen, T. T. et al. Combining traditional 2D and modern physical organic-derived descriptors to predict enhanced enantioselectivity for the key aza-Michael conjugate addition in the synthesis of Prevymis™ (letermovir). Chem. Sci. 9, 6922–6927 (2018).
Gedeck, P., Skolnik, S. & Rodde, S. Developing collaborative QSAR models without sharing structures. J. Chem. Inf. Model. 57, 1847–1858 (2017).
Donoho, D. 50 years of data science. J. Comput. Graph. Stat. 26, 745–766 (2017).
Bajusz, D., Racz, A. & Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 7, 20 (2015).
Martinot, T. Could Internet-of-Things be the next step in the evolution of chemistry. TetraScience Blog https://blog.tetrascience.com/blog/could-internet-of-things-be-the-next-step-in-the-evolution-of-chemistry/ (2016).
Contreras, J. L. Bermuda’s legacy: policy, patents, and the design of the genome commons. Minn. J. Law Sci. Technol. 12, 61–125 (2011).
Amann, R. I. et al. Toward unrestricted use of public genomic data. Science 363, 350–352 (2019).
Lander, E. S. The heroes of CRISPR. Cell 164, 18–28 (2016).
Baker, M. Is there a reproducibility crisis? Nature 533, 452–454 (2016).
Bergman, R. G. & Danheiser, R. L. Reproducibility in chemical research. Angew. Chem. Int. Ed. 55, 12548–12549 (2016).
Brock, J. “A love letter to your future self”: what scientists need to know about FAIR data. Nature Index https://www.natureindex.com/news-blog/what-scientists-need-to-know-about-fair-data (2019).
Preece, A., Harborne, D., Braines, D., Tomsett, R. & Chakraborty, S. Stakeholders in explainable AI. Preprint at https://arxiv.org/abs/1810.00184 (2018).
Caliskan, A., Bryson, J. J. & Narayanan, A. Semantics derived automatically from language corpora contain human-like biases. Science 356, 183–186 (2017).
Reviewer information
Nature thanks Ian Churcher, Jacob Janey and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Davies, I.W. The digitization of organic synthesis. Nature 570, 175–181 (2019). https://doi.org/10.1038/s41586-019-1288-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1288-y
This article is cited by
-
Prediction of chemical reaction yields with large-scale multi-view pre-training
Journal of Cheminformatics (2024)
-
An integrated self-optimizing programmable chemical synthesis and reaction engine
Nature Communications (2024)
-
An intriguing vision for transatlantic collaborative health data use and artificial intelligence development
npj Digital Medicine (2024)
-
A deep learning framework for accurate reaction prediction and its application on high-throughput experimentation data
Journal of Cheminformatics (2023)
-
Automate and digitize
Nature Synthesis (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
