
Abstract
Genetic robustness, or the ability of an organism to maintain fitness in the presence of harmful mutations, can be achieved via protein feedback loops. Previous work has suggested that organisms may also respond to mutations by transcriptional adaptation, a process by which related gene(s) are upregulated independently of protein feedback loops. However, the prevalence of transcriptional adaptation and its underlying molecular mechanisms are unknown. Here, by analysing several models of transcriptional adaptation in zebrafish and mouse, we uncover a requirement for mutant mRNA degradation. Alleles that fail to transcribe the mutated gene do not exhibit transcriptional adaptation, and these alleles give rise to more severe phenotypes than alleles displaying mutant mRNA decay. Transcriptome analysis in alleles displaying mutant mRNA decay reveals the upregulation of a substantial proportion of the genes that exhibit sequence similarity with the mutated gene's mRNA, suggesting a sequence-dependent mechanism. These findings have implications for our understanding of disease-causing mutations, and will help in the design of mutant alleles with minimal transcriptional adaptation-derived compensation.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Change history
17 April 2019
In this Article, the Supplementary Tables and Source Data files were originally uploaded in the wrong order.
References
Tautz, D. Redundancies, development and the flow of information. BioEssays 14, 263–266 (1992).
Barabási, A. L. & Oltvai, Z. N. Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113 (2004).
Teng, X. et al. Genome-wide consequences of deleting any single gene. Mol. Cell 52, 485–494 (2013).
Rossi, A. et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233 (2015).
El-Brolosy, M. A. & Stainier, D. Y. R. Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13, e1006780 (2017).
Isken, O. & Maquat, L. E. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 21, 1833–1856 (2007).
Mukherjee, C. et al. Identification of cytoplasmic capping targets reveals a role for cap homeostasis in translation and mRNA stability. Cell Reports 2, 674–684 (2012).
Boehm, V., Gerbracht, J. V., Marx, M. C. & Gehring, N. H. Interrogating the degradation pathways of unstable mRNAs with XRN1-resistant sequences. Nat. Commun. 7, 13691 (2016).
Doi, T. S. et al. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl Acad. Sci. USA 96, 2994–2999 (1999).
Hao, S. & Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288 (2009).
Elkon, R., Zlotorynski, E., Zeller, K. I. & Agami, R. Major role for mRNA stability in shaping the kinetics of gene induction. BMC Genomics 11, 259 (2010).
Rabani, M. et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436–442 (2011).
Sun, M. et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 22, 1350–1359 (2012).
Collins, S. R. et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446, 806–810 (2007).
Berretta, J., Pinskaya, M. & Morillon, A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 22, 615–626 (2008).
Pinskaya, M., Gourvennec, S. & Morillon, A. H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J. 28, 1697–1707 (2009).
Haimovich, G. et al. Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell 153, 1000–1011 (2013).
Ghanbarian, H. et al. Small RNA-directed epigenetic programming of embryonic stem cell cardiac differentiation. Sci. Rep. 7, 41799 (2017).
Modarresi, F. et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30, 453–459 (2012).
Eisensmith, R. C. & Woo, S. L. Molecular basis of phenylketonuria and related hyperphenylalaninemias: mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum. Mutat. 1, 13–23 (1992).
Myerowitz, R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum. Mutat. 9, 195–208 (1997).
Genschel, J. & Schmidt, H. H. Mutations in the LMNA gene encoding lamin A/C. Hum. Mutat. 16, 451–459 (2000).
Chuzhanova, N. A., Anassis, E. J., Ball, E. V., Krawczak, M. & Cooper, D. N. Meta-analysis of indels causing human genetic disease: mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum. Mutat. 21, 28–44 (2003).
Ferec, C. & Cutting, G. R. Assessing the disease-liability of mutations in CFTR. Cold Spring Harb. Perspect. Med. 2, a009480 (2012).
Zhou, Q. et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N. Engl. J. Med. 370, 911–920 (2014).
Dietz, H. C. et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics 17, 468–475 (1993).
Hall, G. W. & Thein, S. Nonsense codon mutations in the terminal exon of the β-globin gene are not associated with a reduction in β-mRNA accumulation: a mechanism for the phenotype of dominant β-thalassemia. Blood 83, 2031–2037 (1994).
Sulem, P. et al. Identification of a large set of rare complete human knockouts. Nat. Genet. 47, 448–452 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Gerri, C. et al. Hif-1α regulates macrophage-endothelial interactions during blood vessel development in zebrafish. Nat. Commun. 8, 15492 (2017).
Rossi, A. et al. Regulation of Vegf signaling by natural and synthetic ligands. Blood 128, 2359–2366 (2016).
Lawson, N. D. & Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307–318 (2002).
Proulx, K., Lu, A. & Sumanas, S. Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Dev. Biol. 348, 34–46 (2010).
Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. & Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272–W276 (2016).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82 (2011).
Doyle, E. L. et al. TAL effector specificity for base 0 of the DNA target is altered in a complex, effector- and assay-dependent manner by substitutions for the tryptophan in cryptic repeat −1. PLoS One 8, e82120 (2013).
Gagnon, J. A. et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 9, e98186 (2014).
Vejnar, C. E., Moreno-Mateos, M. A., Cifuentes, D., Bazzini, A. A. & Giraldez, A. J. Optimized CRISPR–Cas9 system for genome editing in zebrafish. Cold Spring Harb. Protoc. https://doi.org/10.1101/pdb.prot086850 (2016).
Theodosiou, M. et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. eLife 5, e10130 (2016).
Gapuzan, M. E., Yufit, P. V. & Gilmore, T. D. Immortalized embryonic mouse fibroblasts lacking the RelA subunit of transcription factor NF-κB have a malignantly transformed phenotype. Oncogene 21, 2484–2492 (2002).
Ran, F. A. et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Thakore, P. I. et al. Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015).
Martin, L. et al. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 74, 3104–3113 (2014).
Rädle, B. et al. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J. Vis. Exp. 78, e50195 (2013).
Sun, W. & Chen, W. Metabolic labeling of newly synthesized RNA with 4sU to in parallel assess RNA transcription and decay. Methods Mol. Biol. 1720, 25–34 (2018).
Vanhollebeke, B. et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. eLife 4, e06489 (2015).
Kwon, H. B. et al. In vivo modulation of endothelial polarization by Apelin receptor signalling. Nat. Commun. 7, 11805 (2016).
Blecher-Gonen, R. et al. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 8, 539–554 (2013).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Davis, M. P., van Dongen, S., Abreu-Goodger, C., Bartonicek, N. & Enright, A. J. Kraken: a set of tools for quality control and analysis of high-throughput sequence data. Methods 63, 41–49 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wintersinger, J. A. & Wasmuth, J. D. Kablammo: an interactive, web-based BLAST results visualizer. Bioinformatics 31, 1305–1306 (2015).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 (2011).
Acknowledgements
We thank V. Serobyan, F. Mueller, Z. Jiang, A. Beisaw and F. Gunawan for discussion and comments on the manuscript; J. Pestel for the alcama mutant; A. Atzberger for support with cell sorting; and N. Gehring and V. Böhm for providing the XRN1-resistant sequence plasmid. M.A.E.-B. was supported by a Boehringer Ingelheim Fonds PhD fellowship. Research in the D.Y.R.S. laboratory is supported by the Max Planck Society, the EU, the DFG and the Leducq Foundation.
Reviewer information
Nature thanks Miles Wilkinson and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
M.A.E.-B. designed and performed most of the experiments, analysed the data and wrote the manuscript; Z.K. and A.R. designed and performed mESC experiments and some imaging and edited the manuscript; C.K. performed bioinformatics analyses; S.G. performed ATAC-seq and RNA-seq; N.F. generated some zebrafish mutants and performed some qPCR experiments; K.K. performed some qPCR experiments; G.L.M.B. performed some imaging; C.M.T. generated the upf1 mutant, under the supervision of A.J.G.; S.-L.L., R.F. and C.G. provided unpublished mutants; and D.Y.R.S. helped to design the experiments and analyse data, supervised the work and wrote the manuscript. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Schematic illustration of the mutant alleles generated for this study.
Partial DNA sequences of the different mutant alleles generated for this study, and images of gels providing evidence for deletions in RNA-less alleles. Red indicates mutation (asterisks, deletion; upper-case letters, substitution; lower-case letters, insertion); green indicates stop codon in alleles with a PTC; arrows indicate genotyping primers.
Extended Data Fig. 2 Transcriptional adaptation is independent of the loss of protein function.
a, qPCR analysis of epas1a, epas1b, vegfab, emilin3a and alcamb mRNA expression levels in wild-type and hif1ab, vegfaa, egfl7 and alcama mutant embryos injected (inj.) with eGFP mRNA (control; ctrl) or wild-type hif1ab, vegfaa, egfl7 or alcama mRNA. b, qPCR analysis of vclb, epas1a, epas1b and emilin3a mRNA expression levels in vcla, hif1ab and egfl7 wild-type, heterozygous and mutant zebrafish. c, qPCR analysis of hbegfa, hif1ab, vegfaa and alcama mRNA expression levels in hbegfa, hif1ab, vegfaa and alcama wild-type and heterozygous zebrafish, using primers specific for the wild-type allele. d, qPCR analysis of Fermt1 and Rel mRNA expression levels in wild-type and Fermt2 and Rela knockout cells transfected with empty vectors (control) or plasmids encoding wild-type FERMT2 or RELA. e, Western blot analysis of FERMT2 and ACTB levels in Fermt2 knockout cells transfected with empty vectors (control) or plasmids encoding wild-type FERMT2. f, Western blot analysis of RELA and ACTB levels in Rela knockout cells transfected with empty vectors (control) or plasmids encoding wild-type RELA. g, qPCR analysis of Actg1 mRNA expression levels in wild-type and heterozygous Actb mESCs. n = 3 biologically independent samples. Wild-type or control expression levels were set at 1 for each assay. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values (a–d, g). The experiments in e, f were performed only once. For the source data for western blots, see Supplementary Fig. 1.
Extended Data Fig. 3 Transcriptional adaptation involves enhanced transcription and is independent of the DNA lesion itself.
a, qPCR analysis of hbegfb and emilin3a mRNA and pre-mRNA expression levels in hbegfa and egfl7 wild-type and mutant zebrafish. b, qPCR analysis of Fermt1 and Rel mRNA and pre-mRNA expression levels in Fermt2 and Rela wild-type and knockout cells. c, IGV tracks of the Fermt1 locus showing ATAC-seq signals in wild-type and Fermt2 knockout cells. d, qPCR analysis of hbegfa, hbegfb, egfl7 and emilin3a mRNA expression levels in hbegfa and egfl7 wild-type and Δ3 mutant zebrafish. e, qPCR analysis of vegfaa, vegfab, egfl7 and emilin3a mRNA expression levels in vegfaa and egfl7 wild-type and 5′UTR mutant zebrafish. f, qPCR analysis of vcla and vclb mRNA expression levels in vcla wild-type and last exon (exon 22) mutant zebrafish. n = 3 biologically independent samples. Wild-type expression levels were set at 1 for each assay. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
Extended Data Fig. 4 Reduction in mutant transcript levels is caused by mRNA decay.
a, qPCR analysis of hbegfa, egfl7 and alcama mRNA and pre-mRNA expression levels in hbegfa, egfl7 and alcama wild-type and mutant zebrafish. b, qPCR analysis of Fermt2 and Rela mRNA and pre-mRNA expression levels in Fermt2 and Rela wild-type and knockout cells. c, qPCR analysis of 4sU-labelled Fermt2, Rela and Actg1 mRNA and pre-mRNA expression levels in Fermt2, Rela and Actg1 wild-type and knockout cells. d, Fitted exponential decay curves of Fermt2 mRNA expression levels in wild-type and Fermt2 knockout cells. t1/2, half-life. e, Fitted exponential decay curves of Rela mRNA expression levels in wild-type and Rela knockout cells. f, Fitted exponential decay curves of Actg1 mRNA expression levels in wild-type and Actg1 knockout cells. n = 3 (a, b, d–f) or n = 2 (c) biologically independent samples. Wild-type expression levels were set at 1 for each assay (a–c). Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
Extended Data Fig. 5 RNA decay induces transcriptional adaptation.
a, qPCR analysis of hbegfa, vegfaa and vcla mRNA expression levels in upf1;hbegfa, upf1;vegfaa and upf1;vcla double mutant zebrafish. b, qPCR analysis of Rela mRNA expression levels after siRNA-mediated knockdown of the indicated proteins in Rela knockout cells. c, qPCR analysis of Actb mRNA expression levels after siRNA-mediated knockdown of the indicated proteins in Actb knockout cells. d, qPCR analysis of hbegfa mRNA expression levels in 6 dpf hbegfa mutants treated with NMD inhibitor (NMDi). e, qPCR analysis of hbegfb mRNA expression levels in 6 dpf hbegfa mutants treated with NMDi. f, qPCR analysis of Rela mRNA expression levels in Rela knockout cells treated with cycloheximide (CHX). g, qPCR analysis of Rel mRNA expression levels in Rela knockout cells treated with CHX. H, qPCR analysis of endogenous hif1ab and vegfaa mRNA expression levels in 6 hpf wild-type embryos injected with uncapped hif1ab or vegfaa RNA. I, qPCR analysis of Actg1 mRNA expression levels in mESCs transfected with uncapped Actb RNA at different times after transfection. j, qPCR analysis of injected hif1ab, epas1a, injected vegfaa and vegfab RNA expression levels in 6 hpf wild-type embryos injected with uncapped hif1ab or vegfaa transcripts with or without a 5′ XRN1-resistant (xr) sequence. k, qPCR analysis of epas1a and vegfab mRNA expression levels in 6 hpf wild-type zebrafish embryos injected with uncapped sense or antisense (rev) hif1ab or vegfaa RNA; the same eGFP uncap. control samples were used for the epas1a experiments. Wild-type or control expression levels were set at 1 for each assay (a–d, f, h–k). n = 3 biologically independent samples. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
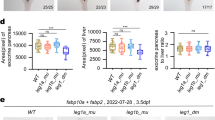
Extended Data Fig. 6 Mutant mRNA decay helps confer genetic robustness.
a, qPCR analysis of Fermt2 and Fermt1 mRNA expression levels following CRISPR interference-mediated knockdown of Fermt2 transcription in Fermt2 knockout cells. b, qPCR analysis of emilin3a, emilin3b and emilin2a mRNA expression levels in 20 hpf wild types, egfl7Δ4 mutants and egfl7full locus del. mutants. c, Number of central arteries (CtAs) connecting to the basilar artery (BA) in 58 hpf vegfaaΔ10 and vegfaapromoter-less mutants. d, Blood-flow velocity in 78 hpf wild types, hbegfaΔ7 mutants and hbegfafull locus del. mutants. e, Quantification of the cardiac ventricle length in 100 hpf wild types, alcama Δ8 mutants and alcamapromoter-less mutants. Wild-type or control expression levels were set at 1 for each assay (a, b). n = 3 (a, b); n = 13 (vegfaaΔ10−/−) and 19 (vegfaapromoter-less−/−) (c); n = 25 (d); and n = 18 (wild-type siblings of alcamaΔ8−/−), 7 (alcamaΔ8−/−), 22 (wild-type siblings of alcamapromoter-less−/−) and 15 (alcamapromoter-less−/−) (e) animals. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
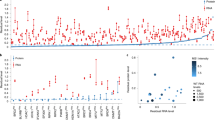
Extended Data Fig. 7 Analysis of sequence similarity parameters in models of transcriptional adaptation.
a, Numbers of differentially expressed genes in the different knockout cell line models; P ≤ 0.05; these genes are distributed throughout the genome (data not shown). b, Venn diagram of genes upregulated in the three different cell line models with L2F knockout > wild-type and P ≤ 0.05. c, KEGG pathway enrichment analysis for genes commonly upregulated in Fermt2, Actg1 and Actb knockout compared to wild-type cells. The top ten pathways based on P value are displayed. The dashed line marks a P value of 0.05. Circle sizes provide an estimation of scale; outer grey circles represent the total number of genes in the pathway; and centred coloured circles represent the number of genes in the pathway that are commonly upregulated. d, Impact of various values of three different BLASTn alignment-quality parameters (alignment length, bit score and E value) on the significance of the observed correlation between upregulation and sequence similarity, and therefore the identification or prediction of putative adapting genes. The E value describes the probability of the match resulting from chance (a lower value corresponds to a lower probability), and the bit score evaluates the combination of alignment quality and length (a higher value corresponds to a better alignment).The y axis of each diagram shows the negative log10 of the P value and the x axis shows the respective parameter value. A P value of 0.05 is marked with a black horizontal line. The E value thresholds used in our analyses are highlighted with a circle. Lines ending preliminarily indicate a lack of any remaining alignments after that point. The first row of diagrams explores large variations of thresholds, in an attempt to identify the total range, whereas the second row focuses on the most relevant window for the three genes investigated. The optimal thresholds differ considerably depending on the gene analysed. n = 2 biologically independent samples. P value was computed by bootstrapping random subsamples (see the ‘Sequence similarity and subsampling analyses’ section of the Methods). P values were not corrected for multiple testing.
Extended Data Fig. 8 Expression level of genes exhibiting sequence similarity in the different mouse cell line models.
a–c, RNA-seq analysis of genes exhibiting sequence similarity with Fermt2 (a), Actg1 (b) or Actb (c) in knockout compared to wild-type cells. Bold, significantly upregulated in knockout relative to wild-type cells; red, L2F>0, blue, L2F<0; green, P value or adjusted P value ≤ 0.05; purple; genes exhibiting sequence similarity with the mutated gene's mRNA in their promoter region; yellow, genes exhibiting sequence similarity with the mutated gene's mRNA in their 3′UTR region. Other non-coloured genes exhibit sequence similarity with the mutated gene's mRNA in their exons or introns. Boxed, upregulated in knockout but not RNA-less cells; no Fermt2 RNA-less allele was analysed. d, qPCR analysis of Ubapl, Fmnl2, Cdk12 and Actr1a pre-mRNA expression levels in Actg1 knockout relative to wild-type cells. e, qPCR analysis of actb1 mRNA expression levels in 6 hpf wild-type zebrafish injected with uncapped mouse Actb RNA. f, Schematic representation of regions of sequence similarity between hif1ab mRNA and the epas1a locus. Grey shaded triangles represent the alignments; intensity represents the alignment quality; and width at the base represents the length of the similarity region. g, qPCR analysis of epas1a mRNA expression levels in 6 hpf wild-type zebrafish embryos injected with uncapped RNA composed solely of the hif1ab sequences similar to the epas1a promoter, exons, introns or 3′UTR; the same eGFP uncap. control samples were used for all comparisons. n = 2 (a–c) or n = 3 (d, e, g) biologically independent samples. DESeq2 tests were used to test for significance of coefficients in a negative binomial generalized linear model with the Wald test (a–c). P values were not corrected for multiple testing. Wild-type or control expression levels were set at 1 for each assay (d, e, g). Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
Extended Data Fig. 9 Transcriptional adaptation involves chromatin remodelling that is dependent on the activity of decay factors.
a, qPCR analysis of Rel mRNA expression levels after siRNA-mediated knockdown of the indicated proteins in Rela knockout cells. b, ChIP–qPCR analysis of H3K4me3 occupancy at non-promoter regions (as a control) of Fermt1, Rel and Actg2 in Fermt2, Rela and Actg1 knockout cells, respectively, compared to wild-type cells. c, ChIP–qPCR analysis of H3K4me3 occupancy near the Rel TSS and a non-promoter region (as a control) after siRNA-mediated knockdown of the indicated proteins in Rela knockout cells. d, Current expanded model of transcriptional adaptation to mutations. RNA decay fragments may act as intermediates to bring decay factors and chromatin remodellers to adapting gene loci, thereby triggering increased gene expression. Alternatively, RNA decay fragments may function by repressing antisense RNAs at the adapting gene loci, thus allowing for increased sense mRNA expression. It is, however, likely that additional mechanisms are involved in transcriptional adaptation, and possibly in a gene-dependent manner. n = 3 (a) or n = 2 (b, c) biologically independent samples. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
Extended Data Fig. 10 The potential role of antisense transcripts in the transcriptional adaptation response.
a, qPCR analysis of Cdk9 and Sox9 mRNA expression levels in cells transfected with uncapped Cdk9 or Sox9 RNA. b, qPCR analysis of BDNF and BDNF-AS mRNA expression levels in HEK293T cells transfected with uncapped BDNF RNA. c, Integrated genome viewer tracks of vclb and hbegfb loci, showing the location of the annotated antisense transcripts. Two alignments of 105 and 147 bp were observed between vcla mRNA and vclb antisense RNAs, and an alignment of 39 bp was observed between hbegfa mRNA and hbegfb antisense RNA. Antisense transcripts shown were acquired from the datasets in GSE32898. d, qPCR analysis of vclb and hbegfb antisense (AS) RNA expression levels in vcla and hbegfa wild-type and mutant zebrafish at 24 and 72 hpf, respectively. Control expression levels were set at 1 for each assay. n = 3 biologically independent samples. Data are mean ± s.d., and a two-tailed Student’s t-test was used to calculate P values.
Supplementary information
Supplementary Information
This file contains a Supplementary Discussion; Supplementary Data and Supplementary Figures. The merged supplementary information PDF includes: 1. Supplementary Discussion, 2. Supplementary Data: Includes (1) MUSCLE alignment of zebrafish actb1 and mouse Actb coding sequences, (2) MUSCLE alignment of the synthetic hif1ab transcript which consists of sequences similar to epas1a, and the full hif1ab transcript, (3) BLASTn alignment of hif1ab transcript and epas1a and (4) sequence of the synthetic transcript composed of sequences of hif1ab mRNA not exhibiting similarity with epas1a genomic locus, 3. Supplementary Figure 1: uncropped images of western blots.
Supplementary Table
This file contains Supplementary Table 1: Commonly upregulated genes in Fermt2, Actg1 and Actb K.O. cells.
Supplementary Table
This file contains Supplementary Table 2: siRNAs used in the study.
Supplementary Table
This file contains Supplementary Table 3: Genotyping primers.
Supplementary Table
This file contains Supplementary Table 4: qPCR primers.
Supplementary Table
This file contains Supplementary Table 5: gRNAs used in the study.
Source data
Rights and permissions
About this article

Cite this article
El-Brolosy, M.A., Kontarakis, Z., Rossi, A. et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197 (2019). https://doi.org/10.1038/s41586-019-1064-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1064-z
This article is cited by
-
miR-430 regulates zygotic mRNA during zebrafish embryogenesis
Genome Biology (2024)
-
dCas13-mediated translational repression for accurate gene silencing in mammalian cells
Nature Communications (2024)
-
Cell-type-specific mRNA transcription and degradation kinetics in zebrafish embryogenesis from metabolically labeled single-cell RNA-seq
Nature Communications (2024)
-
Inborn errors of immunity: an expanding universe of disease and genetic architecture
Nature Reviews Genetics (2024)
-
A CRISPRi/a screening platform to study cellular nutrient transport in diverse microenvironments
Nature Cell Biology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
