
Abstract
β-arrestins are critical regulator and transducer proteins for G-protein-coupled receptors (GPCRs). β-arrestin is widely believed to be activated by forming a stable and stoichiometric GPCR–β-arrestin scaffold complex, which requires and is driven by the phosphorylated tail of the GPCR. Here we demonstrate a distinct and additional mechanism of β-arrestin activation that does not require stable GPCR–β-arrestin scaffolding or the GPCR tail. Instead, it occurs through transient engagement of the GPCR core, which destabilizes a conserved inter-domain charge network in β-arrestin. This promotes capture of β-arrestin at the plasma membrane and its accumulation in clathrin-coated endocytic structures (CCSs) after dissociation from the GPCR, requiring a series of interactions with membrane phosphoinositides and CCS-lattice proteins. β-arrestin clustering in CCSs in the absence of the upstream activating GPCR is associated with a β-arrestin-dependent component of the cellular ERK (extracellular signal-regulated kinase) response. These results delineate a discrete mechanism of cellular β-arrestin function that is activated catalytically by GPCRs.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


References
Rosenbaum, D. M., Rasmussen, S. G. F. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G. & Lefkowitz, R. J. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550 (1990).
Kang, D. S., Tian, X. & Benovic, J. L. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 (2014).
Pierce, K. L., Premont, R. T. & Lefkowitz, R. J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 (2002).
Gurevich, E. V. & Gurevich, V. V. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 7, 236 (2006).
Lohse, M. J. & Hoffmann, C. Arrestins–Pharmacology and Therapeutic Potential (Springer, Berlin, Heidelberg, 2014).
Shukla, A. K., Xiao, K. & Lefkowitz, R. J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 (2011).
Gurevich, V. V. & Gurevich, E. V. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 110, 465–502 (2006).
Nuber, S. et al. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664 (2016).
Cahill, T. J. III et al. Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl Acad. Sci. USA 114, 2562–2567 (2017).
Lee, M.-H. et al. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668 (2016).
Kumari, P. et al. Functional competence of a partially engaged GPCR–β-arrestin complex. Nat. Commun. 7, 13416 (2016).
Kumari, P. et al. Core engagement with β-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010 (2017).
Branco, A. F. et al. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc. Toxicol. 11, 191–203 (2011).
Puthenveedu, M. A. & von Zastrow, M. Cargo regulates clathrin-coated pit dynamics. Cell 127, 113–124 (2006).
Santini, F., Gaidarov, I. & Keen, J. H. G protein-coupled receptor/arrestin3 modulation of the endocytic machinery. J. Cell Biol. 156, 665–676 (2002).
Barak, L. S., Ferguson, S. S., Zhang, J. & Caron, M. G. A β-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272, 27497–27500 (1997).
Eichel, K., Jullié, D. & von Zastrow, M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 18, 303–310 (2016).
Mondin, M. et al. Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J. Neurosci. 31, 13500–13515 (2011).
Shukla, A. K. et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141 (2013).
Shukla, A. K. et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 (2014).
Xiao, K., Shenoy, S. K., Nobles, K. & Lefkowitz, R. J. Activation-dependent conformational changes in β-arrestin 2. J. Biol. Chem. 279, 55744–55753 (2004).
Nobles, K. N., Guan, Z., Xiao, K., Oas, T. G. & Lefkowitz, R. J. The active conformation of β-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of β-arrestins1 and -2. J. Biol. Chem. 282, 21370–21381 (2007).
McCorvy, J. D. et al. Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 14, 126–134 (2018).
Gimenez, L. E., Babilon, S., Wanka, L., Beck-Sickinger, A. G. & Gurevich, V. V. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell. Signal. 26, 1523–1531 (2014).
Violin, J. D., Ren, X.-R. & Lefkowitz, R. J. G-protein-coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer. J. Biol. Chem. 281, 20577–20588 (2006).
Peterson, S. M. et al. Elucidation of G-protein and β-arrestin functional selectivity at the dopamine D2 receptor. Proc. Natl Acad. Sci. USA 112, 7097–7102 (2015).
Gurevich, V. V. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J. Biol. Chem. 273, 15501–15506 (1998).
Scheerer, P. & Sommer, M. E. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 45, 160–169 (2017).
Chen, Q. et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 8, 1427 (2017).
Gaidarov, I., Krupnick, J. G., Falck, J. R., Benovic, J. L. & Keen, J. H. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 18, 871–881 (1999).
Lally, C. C. M., Bauer, B., Selent, J. & Sommer, M. E. C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 8, 14258 (2017).
Goodman, O. B. Jr et al. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383, 447–450 (1996).
Laporte, S. A., Oakley, R. H., Holt, J. A., Barak, L. S. & Caron, M. G. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β 2-adrenergic receptor into clathrin-coated pits. J. Biol. Chem. 275, 23120–23126 (2000).
Oakley, R. H., Laporte, S. A., Holt, J. A., Barak, L. S. & Caron, M. G. Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274, 32248–32257 (1999).
Santos, M. de, S., Naal, R. M. Z. G., Baird, B. & Holowka, D. Inhibitors of PI(4,5)P2 synthesis reveal dynamic regulation of IgE receptor signaling by phosphoinositides in RBL mast cells. Mol. Pharmacol. 83, 793–804 (2013).
Manley, S. et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 5, 155–157 (2008).
Hammond, G. R. V., Sim, Y., Lagnado, L. & Irvine, R. F. Reversible binding and rapid diffusion of proteins in complex with inositol lipids serves to coordinate free movement with spatial information. J. Cell Biol. 184, 297–308 (2009).
Liu, A. P., Loerke, D., Schmid, S. L. & Danuser, G. Global and local regulation of clathrin-coated pit dynamics detected on patterned substrates. Biophys. J. 97, 1038–1047 (2009).
Latorraca, N. R. et al. Molecular mechanism of GPCR-mediated arrestin activation. Nature https://doi.org/10.1038/s41586-018-0077-3 (2018).
O’Hayre, M. et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal. 10, eaal3395 (2017).
Cao, T. T., Deacon, H. W., Reczek, D., Bretscher, A. & von Zastrow, M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature 401, 286–290 (1999).
Temkin, P. et al. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 13, 715–721 (2011).
Yudowski, G. A., Puthenveedu, M. A. & von Zastrow, M. Distinct modes of regulated receptor insertion to the somatodendritic plasma membrane. Nat. Neurosci. 9, 622–627 (2006).
Oakley, R. H., Laporte, S. A., Holt, J. A., Caron, M. G. & Barak, L. S. Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 (2000).
Bouvier, M. et al. Removal of phosphorylation sites from the β2-adrenergic receptor delays onset of agonist-promoted desensitization. Nature 333, 370–373 (1988).
Krasel, C., Bünemann, M., Lorenz, K. & Lohse, M. J. β-Arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 280, 9528–9535 (2005).
Merrifield, C. J., Feldman, M. E., Wan, L. & Almers, W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4, 691–698 (2002).
Stauffer, T. P., Ahn, S. & Meyer, T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8, 343–346 (1998).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Nair, D. et al. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 33, 13204–13224 (2013).
Rossier, O. et al. Integrins β1 and β3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat. Cell Biol. 14, 1057–1067 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 27–38 (1996).
Acknowledgements
We thank J. Benovic, V. Gurevich, A. Inoue and S. Gutkind for sharing reagents and discussions; T. Balla, J. Fraser, B. Kobilka, R. Lefkowitz, M. Lohse, A. Manglik, B. Shoichet, M. Sommer, R. Sunahara and B. Lobingier, K. Varandas and other von Zastrow laboratory members for discussions; and W. Huynh and R. Vale for contributions to protein purification. All live-cell imaging experiments were performed in the Nikon Imaging Center at UCSF. These studies were supported by grants from the U.S. National Institutes of Health (to R.O.D. and M.v.Z.). J.-B.S. received support from Aquitaine grant Dynascreen, LabEx BRAIN and the IdEx Bordeaux. K.E. and N.R.L. are recipients of National Science Foundation Graduate Research Fellowships. M.M. is a recipient of an American Heart Association Postdoctoral Fellowship.
Reviewer information
Nature thanks D. Calebiro, C. Hoffmann and B. Roth for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
K.E., D.J., B.B.-R., N.R.L., R.O.D. and M.v.Z designed the research. D.J. performed and analysed single-molecule imaging experiments. B.B.-R. performed and analysed FRAP imaging experiments. K.E. performed and analysed all other imaging and biochemical experiments. N.R.L. performed and analysed molecular dynamics simulations and provided a structural interpretation of experimental data, with guidance from R.O.D. M.M. provided important advice and assistance with in vitro studies. J.-B.S provided software and advice for single molecule analysis. K.E. and M.v.Z. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Verification of GPCR-specificity of the discrete β-arrestin trafficking mechanism, demonstration that this mechanism produces super-stoichiometric β-arrestin accumulation in CCSs and that its activation does not require the GPCR tail.
a, Mean β-arrestin-2–GFP enrichment at CCSs in cells expressing Flag–β1AR after the following treatments: 10 µM isoproterenol (green, n = 14 cells), 15-min pretreatment with 10 µM CGP 20712 A and treatment with 10 µM isoproterenol (red, n = 12 cells), or 10 µM CGP 20712 A alone (grey, n = 12 cells). Data shown for the 10 µM isoproterenol condition are replotted from Fig. 1b. b, Maximum β-arrestin-2–GFP enrichment at CCSs in HEK 293 cells transfected with the indicated receptor or empty vector and treated with 10 µM isoproterenol. c, β-arrestin-2–GFP enrichment at CCSs in H9c2 cells without GPCR overexpression and treated with 10 µM isoproterenol or 10 µM dobutamine (n = 5 or 4 cells, respectively, from two independent experiments). d, β-arrestin-2–GFP enrichment at CCSs in H9c2 cells without GPCR overexpression and treated as indicated (n = 12 cells). e, Live-cell TIRF microscopy images (representative of n = 3 independent experiments) showing Flag–β1AR (blue), β-arrestin-1–mVenus (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. f, Enrichment into CCSs (n = 7 cells from three independent experiments). g, Maximum β-arrestin-1–mVenus enrichment at CCSs in HEK 293 cells transfected with Flag–β1AR or empty vector and treated with 10 µM isoproterenol (n = 7 and 11 cells, respectively, from three independent experiments; P = 0.0023 using an unpaired t-test with Welch’s correction). h, Mean β-arrestin-2–GFP enrichment at CCSs in cells expressing Flag–β2AR after the following treatments: 10 µM isoproterenol (green, n = 15 cells), 15-min pretreatment with 10 µM ICI 118,551 and then treatment with 10 µM isoproterenol (red, n = 14 cells) or 10 µM ICI 118,551 (grey, n = 12). Data shown for the 10 µM isoproterenol condition are replotted from Fig. 1d. i, Fluorescence intensity profiles from lines shown in Fig. 1e. j, Time-dependent correlation coefficient of line scans across cells derived from immobilization experiments shown in Fig. 1e, f (n = 3). k, Live-cell TIRF microscopy images (representative of n = 3 independent experiments) showing Flag–β2AR–GFP and clathrin-light-chain–DsRed (red) before and after treatment with 10 µM isoproterenol. Fluorescence from the Alexa Fluor 647-conjugated Flag antibody shown in blue and GFP fluorescence shown in green. l, Difference in GFP and Alexa Fluor 647 fluorescence enrichment at CCSs in cells co-expressing Flag–β1ARs (red), Flag–β2ARs (blue) and β-arrestin-2–GFP or Flag–β2AR-GFP (black). Cells were labelled with Alexa Fluor 647-conjugated Flag antibody for 10 min before live-cell imaging. Data were derived from the experiments shown in Fig. 1a, b (blue line, n = 14 cells from three independent experiments), Fig. 1c, d (red line, n = 15 cells from three independent experiments) and Extended Data Fig. 1k (black line n = 12 cells from three independent experiments). m, Plot of β-arrestin–GPCR stoichiometry calculated from the data displayed in k, calibrated according to the double-labelled Flag–β2AR–GFP reference construct, which defines 1:1 stoichiometry (For β1AR and β2AR, n = 14 and 15 cells, respectively, from three independent experiments). A correction index was calculated by dividing GFP fluorescence by Alexa Fluor 647 (Flag) fluorescence in CCSs. This correction index was then applied to receptor and β-arrestin-2 enrichment in CCSs to determine β-arrestin-2–GPCR stoichiometry throughout the time course. Images were captured continuously at 0.5 Hz and stoichiometry values over the time course were calculated using a rolling average with 50-frame window size. Scale bar, 5 µm. Scatter plots show overlay of mean and s.e.m. a, d, h, l and l show data as mean ± s.e.m. **P < 0.01.
Extended Data Fig. 2 Additional demonstration that multiple GPCRs can activate the discrete β-arrestin trafficking mechanism.
a, Live-cell TIRF microscopy images showing Flag–MOR (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with the opioid peptide DAMGO (10 µM). b, Mean Flag–MOR and β-arrestin-2–GFP enrichment at CCSs after treatment with 10 µM DAMGO (n = 12 cells). c, Maximum β-arrestin-2–GFP enrichment at CCSs for HEK 293 cells expressing Flag–MOR or empty vector and treated with 10 µM DAMGO (n = 12 cells per condition from three independent experiments; P < 0.0001 using a two-tailed unpaired t-test with Welch’s correction). d, Live-cell TIRF microscopy images showing Flag–KOR (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM dynorphin. e, Enrichment into CCSs after bath application of 10 µM dynorphin (n = 18 cells). f, Maximum β-arrestin-2–GFP enrichment at CCSs in HEK 293 cells expressing Flag–KOR or empty vector and treated with 10 µM dynorphin (n = 18 and 13 cells, respectively, from three independent experiments; P = 0.0028 using a two-tailed unpaired t-test with Welch’s correction). g, Live-cell TIRF microscopy images showing Flag–DRD2 (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red), before and after treatment with 10 µM quinpirole. h, Enrichment into CCSs after bath application of 10 µM quinpirole (n = 12 cells). i, Maximum β-arrestin-2–GFP enrichment at CCSs in cells expressing Flag–DRD2 or untransfected and treated with 10 µM quinpirole (n = 11 and 12 cells from three independent experiments; P = 0.0095 using a two-tailed unpaired t-test with Welch’s correction). a, d and g show representative images from three independent experiments. b, e and h show mean ± s.e.m. Scatter plots show overlay of mean and s.e.m. Scale bars, 5 µm. *P < 0.05; **P < 0.01; ***P < 0.001.
Extended Data Fig. 3 Direct interaction with the GPCR, but not the GPCR cytoplasmic tail, is required for activation of β-arrestin trafficking.
a, Live-cell TIRF microscopy images showing Flag–β1AR(415T) (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. b, Maximum β-arrestin-2–GFP enrichment at CCSs after treatment with 10 µM isoproterenol for cells co-expressing the indicated Flag–β1AR receptor (n = 10 and 12 cells, respectively, from three independent experiments; P = 0.5825 calculated using a two-tailed unpaired t-test). c, Live-cell TIRF microscopy images showing Flag–β2AR(365T) (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. d, Maximum β-arrestin-2–GFP enrichment at CCSs in HEK 293 cells treated with 10 µM isoproterenol and transfected with either Flag–β2AR or empty vector (n = 11 and 13 cells, respectively, from three independent experiments; P = 0.0269 calculated using a two-tailed unpaired t-test with Welch’s correction). e, Maximum β-arrestin-2–GFP enrichment at CCSs for cells co-expressing the indicated Flag–β2AR receptor and treated with 10 µM isoproterenol (n = 12 cells from three independent experiments; P = 0.0606 calculated using a two-tailed unpaired t-test). f, Live-cell TIRF microscopy images showing Flag–DRD2 (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM quinpirole. g, h, Initial enrichment in CCSs before treatment with 10 µM quinpirole (g) and maximum enrichment after treatment with 10 µM quinpirole (h) (n = 12 cells from three independent experiments; P = 0.19 and 0.4873, respectively, using a two-tailed unpaired t-test). i, Live-cell TIRF microscopy images showing Flag–β1AR (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 5 µM forskolin (fsk). j, k, Initial enrichment in CCSs before treatment with 5 µM forskolin (j) and maximum enrichment after treatment with 5 µM forskolin (k) (n = 12 cells from three independent experiments; P = 0.6325 and 0.0971, respectively, using a two-tailed unpaired t-test). l, Live-cell TIRF microscopy images showing Flag–β2AR (blue), β-arrestin-2–GFP KNC mutant (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. m, n, Initial enrichment in CCSs before treatment with 10 µM isoproterenol (m) and maximum enrichment after treatment with 10 µM isoproterenol (n) (n = 9 (wild-type) or 8 (KNC mutant) cells from three independent experiments; P = 0.6681(m) and P = 0.001 (n) using a two-tailed unpaired t-test with Welch’s correction). a, c, f, i and l show representative images from three independent experiments. Scatter plots show overlay of mean and s.e.m. Scale bars, 5 µm. *P < 0.05, **P < 0.01.
Extended Data Fig. 4 Additional verification that charge mutations in the finger-loop-proximal region of β-arrestin produce a constitutive activation phenotype.
a, Live-cell TIRF microscopy images showing Flag–β2AR, clathrin light chain–DsRed (red) and the polar core mutant of β-arrestin-2–GFP (green) in the absence of agonist treatment. b, Clustering index of β-arrestin-2–GFP for the indicated construct in the absence of agonist treatment. Statistical significance was calculated using a two-tailed unpaired t-test with Welch’s correction (polar core mutant: n = 12 cells from three independent experiments, P < 0.0001; finger-loop-proximal mutant: n = 16 cells from three independent experiments, P < 0.0001; R77A: n = 12 cells from three independent experiments, P = 0.0403; K78A: n = 12 cells from three independent experiments, P = 0.0016). Wild-type and finger-loop-proximal mutant data is replotted from Fig. 3b. c, Association of β-arrestin-2–GFP constructs with the β-adaptin subunit of AP-2 in the absence of agonist treatment. Molecular mass markers (in kDa) are shown on the right side of blots. For gel source data, see Supplementary Fig. 1. The western blots in c are representative of three independent experiments, quantified in d, and shown as AP-2/GFP intensity in the immunoprecipitation conditions (n = 3 independent experiments, P = 0.0218 using a two-tailed unpaired t-test). e, β-arrestin-2–GFP expression in cell lysates from c. f–i, Live-cell TIRF microscopy images showing Flag–β2AR, clathrin light chain–DsRed (red) and β-arrestin-2–GFP with the indicated point mutations (green) in the absence of agonist treatment. Detailed descriptions of β-arrestin mutations are provided in Extended Data Table 1. a and f–i show representative images from three independent experiments. Scatter plots show overlay of mean and s.e.m. Scale bars, 5 µm. *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Fig. 5 Molecular dynamics simulations suggest that finger-loop-proximal charged residues stabilize β-arrestin in an inactive state.
a, Crystal structures of β-arrestin-1 (left) in an inactive (middle) and active (right) conformation reveal an extensive network of polar residues proximal to the finger loop involving residues R76, K77 and D78. b, Histogram of distances between K77 and E313 in simulations of inactive β-arrestin-1 (blue) and active β-arrestin-1 (pink), showing frequency of K77–E313 salt-bridge formation. The K77–E313 distance corresponds to the minimum distance between polar heavy atoms on the two residues’ side chains. A separation distance of less than 3.0 Å corresponds to formation of the salt bridge. For the six simulations started from the inactive state, the salt bridge formed 1.1%, 5.7%, 6.3%, 17.6%, 1.0% and 2.0% of the time, respectively (simulation lengths were 4.7 μs, 3.1 μs, 2.9 μs, 5.1 μs, 5.2 μs and 5.7 μs, respectively). For the six simulations started from the active state, the salt bridge formed 0.02%, 0.04%, 0.0%, 1.1%, 0.0% and 0.0% of the time, respectively (simulation lengths were 5.0 μs, 5.0 μs, 4.7 μs, 4.8 μs, 4.8 μs and 5.0 μs, respectively). c, Inactive-state crystal structure of β-arrestin-1 in which E313 interacts with R188 on a different β-arrestin-1 molecule in the crystal lattice. d, Sequence alignment of arrestins showing conservation of residues R76, K77, D78 and E313. Detailed description of β-arrestin mutations are provided in Extended Data Table 1.
Extended Data Fig. 6 Verification that the conserved phosphoinositide binding determinant in the β-arrestin C domain is specifically required for the catalytic trafficking mechanism and operates upstream of clathrin- and AP-2-binding interactions.
a, b, Graphical representation of β-arrestin interaction domains without (a) and with (b) βAR activation by isoproterenol. c, Live-cell TIRF microscopy images showing Flag–β2AR (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. d, Live-cell TIRF microscopy images showing Flag–β2AR (blue), β-arrestin-2–GFP lipid mutant (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. e, Representative western blot (from four independent experiments) of purified wild-type and lipid mutant versions of β-arrestin-1(1–393) immunoprecipitation with PtdIns(4,5)P2-coated agarose beads and quantified in f as per cent of input protein (n = 4 independent experiments, P = 0.0142 using a two-tailed unpaired t-test). For gel source data, see Supplementary Fig. 1. g, Live-cell TIRF microscopy images showing Flag–β2AR (blue), β-arrestin-2(F191G/L192G)–GFP lipid anchor mutant (green), and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. h, Maximum β-arrestin-2–GFP enrichment at CCSs in cells expressing the indicated β-arrestin-2–GFP construct and treated with 10 µM isoproterenol (n = 12 cells from three independent experiments; P = 0.9227 calculated using a two-tailed unpaired t-test). i, Live-cell TIRF microscopy images showing Flag–β2AR (blue), β-arrestin-2–GFP CCS mutant (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. j, Representative images of HEK 293 cells co-expressing Flag–β2AR (blue), β-arrestin-2–GFP lipid and CCS mutant (green) and clathrin light chain–DsRed (red) before and after treament with 10 µM isoproterenol. Representative β-arrestin images false coloured to indicate fluorescence intensity, maximum fluorescence enrichment at CCSs and normalized average plasma membrane (PM) β-arrestin-2–GFP fluorescence (mean ± s.e.m.), respectively, from cells co-expressing Flag–β1ARs (n = 12 cells per condition) without isoproterenol treatment (k–m), and the following β-arrestin-2–GFP constructs with treatment with 10 µM isoproterenol: wild-type (n–p), lipid mutant (q–s), CCS mutant (t–v) and CCS and lipid mutant (w–y). Wild-type β-arrestin-2–GFP maximum enrichment at CCSs shown in r, u and x is replotted from o. z–ac, Live-cell TIRF microscopy images showing cells before and after treatment with 10 µM isoproterenol and co-expressing Flag–β1AR (blue), clathrin light chain–DsRed (red) and the following GFP-labelled versions of β-arrestin-2 (green): wild-type (z), lipid mutant (aa), CCS mutant (ab), and CCS and lipid mutant (ac). ad, Live-cell TIRF microscopy images showing Flag–β2AR and the indicated β-arrestin-2–GFP construct in the absence of agonist treatment. ae, Clustering index of β-arrestin-2–GFP for the indicated construct in the absence of agonist treatment. Detailed description of β-arrestin mutations are provided in Extended Data Table 1. c, d, g, i–k, n, q, t, w and z–ad show representative images from three independent experiments. In r, u and x, n = 12 cells from three independent experiments; unpaired t-test with Welch’s correction, P = 0.0007, 0.0018 and 0.0012, respectively. In ae, n = 12 (wild-type) and 16 (finger-loop-proximal mutant) from three independent experiments; unpaired t-test with Welch’s correction, P < 0.0001; n = 12 (wild-type) and 15 (finger loop proximal & lipid mutant) from three independent experiments; P = 0.5464. Wild-type and finger-loop-proximal mutant data replotted from Fig. 3b. Scatter plots show overlay of mean and s.e.m. Scale bars, 5 μm. **P < 0.01, ***P < 0.001.
Extended Data Fig. 7 Phosphoinositide binding is essential for catalytic activation of β-arrestin trafficking but is dispensable for trafficking mediated by the scaffold mechanism.
a, Live-cell microscopy images of HEK 293 cells co-expressing Flag–β2AR–V2R C tail (blue), β-arrestin-2–GFP CCS mutant (green) and clathrin light chain–DsRed (red) before and after treatment with 10 µM isoproterenol. b, Normalized plasma membrane fluorescence of β-arrestin-2–GFP lipid mutant in cells co-expressing Flag–β2AR–V2R (n = 12 cells from three independent experiments) when treated with 10 µM isoproterenol. c, Maximum β-arrestin-2–GFP enrichment at CCSs in cells expressing indicated β-arrestin-2–GFP before and after activation of Flag–β2AR–V2R chimaera with 10 µM isoproterenol (n = 10 and 12 cells, respectively, from three independent experiments; P = 0.6433, two-tailed unpaired t-test). d, Live-cell microscopy images of COS-1 cells co-expressing Flag–β2AR (blue), β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) that have been pre-treated for 1 h with 1 µM PAO or vehicle (DMSO) before treatment with 10 µM isoproterenol. e, Normalized mean fold increase over initial β-arrestin-2–GFP fluorescence in cells co-expressing Flag–β2AR when pre-treated for 1 h with 1 µM PAO following treatment with 10 µM isoproterenol (n = 12 cells from three independent experiments). f, Live-cell microscopy images of COS-1 cells co-expressing Flag–β2AR–V2R C tail (blue), β-arrestin-2–GFP (green), and clathrin light chain (CLC)–dsRed (red) that have been pre-treated for 1 h with 1 μM PAO before before treatment with 10 µM isoproterenol. g, Normalized average fold increase over initial β-arrestin-2–GFP fluorescence in cells co-expressing Flag–β2AR or Flag–β2AR–V2R when pre-treated for 1 h with 1 µM PAO following treatment with 10 µM isoproterenol (n = 12 cells from three independent experiments). a, d and f show representative images from three independent experiments. b, e and g show mean ± s.e.m. Scatter plots show overlay of mean and s.e.m.
Extended Data Fig. 8 sptPALM controls, MSD plots and cellular model.
a, Representative image of a clathrin mask (green) generated from a CLC–GFP image (red). b–e, Representative diffusion maps overlaid with the clathrin mask for HEK 293 cells treated with 10 µM isoproterenol and expressing PAmCherry–β1AR (b), PAmCherry–β2AR (c), β-arrestin-2–PAmCherry and Flag–β1AR (d), and β-arrestin-2–PAmCherry and Flag–β2AR (e). f, Distribution of diffusion coefficients (D) of false-positive detections from HEK 293 cells expressing Flag–β2AR and imaged under standard sptPALM acquisition conditions to determine contribution of false-positive detections in the experimental setup and analysis. g, Distribution of diffusion coefficients of PAmCherry–β2AR, PAmCherry–PLCδ1-PH and β-arrestin-2–PAmCherry in live cells imaged at 37 °C after treatment with 10 µM isoproterenol (n = 13, 21 and 8 cells, respectively). Black lines show diffusion coefficient profiles that have not been corrected for false-positive detections, showing limited contribution to the profiles. β-arrestin-2–PAmCherry and PAmCherry–PLCδ1-PH were co-expressed individually with Flag–β2AR. h, Average MSD plots derived from sptPALM analysis of PAmCherry–β1AR and PAmCherry–β2AR trajectories in HEK 293 cells treated with 10 µM isoproterenol (n = 8 and 13 cells, respectively). i, Distribution of diffusion coefficients of wild-type and CCS mutant β-arrestin-2–PAmCherry when co-expressed with Flag–β1AR in live HEK 293 cells imaged at 37 °C after treatment with 10 µM isoproterenol (n = 13 and 17 cells, respectively). Black lines show diffusion coefficient profiles that have not been corrected for false-positive detections, showing limited contribution to the profiles. j, Average MSD plots derived from sptPALM analysis of wild-type and CCS mutant β-arrestin-2–PAmCherry trajectories in cells co-expressing Flag–β1AR and treated with 10 µM isoproterenol (n = 13 and 17 cells, respectively). k, Distribution of diffusion coefficients of wild-type and CCS mutant β-arrestin-2–PAmCherry when co-expressed with Flag–β2AR in live cells imaged at 37 °C after treatment with 10 µM isoproterenol (n = 21 and 10 cells, respectively). Black lines show diffusion coefficient profiles that have not been corrected for false-positive detections, showing limited contribution to the profiles. β-arrestin-2–PAmCherry diffusion coefficient profiles when activated by the β2AR are replotted from g. l, Average MSD plots derived from sptPALM analysis of wild-type and CCS mutant β-arrestin-2–PAmCherry trajectories in HEK 293 cells co-expressing Flag–β2AR and treated with 10 µM isoproterenol (n = 21 and 10 cells, respectively). m, n, Immobile (m) and mobile(n) β-arrestin-2–PAmCherry trajectory detections overlaid with a clathrin marker (red) in live cells co-expressing Flag–β1AR after treatment with 10 µM isoproterenol. o, p, Immobile (o) and mobile (p) β-arrestin-2–PAmCherry trajectory detections overlaid with a clathrin marker (red) in live cells co-expressing Flag–β2AR after treatment with 10 µM isoproterenol. Trajectory detections are false coloured based on the density of detections at each pixel. Error bars represent s.e.m; in some cases, error bars are smaller than the height of the symbol and are therefore not shown. Scale bars, 500 nm for sptPALM images. q, Proposed cellular pathway for catalytic activation of β-arrestin. r, Representative microscopy images of COS-1 cells co-expressing Flag–β2AR, β-arrestin-2–GFP (green) and clathrin light chain–DsRed (red) that were treated with 10 µM isoproterenol for 3 min. Then, β-arrestin-2–GFP was photobleached in the indicated yellow region (inset; insets are also shown in Fig. 5h). a–e, m–p and r show representative examples from at least three independent experiments. f–l show data as mean ± s.e.m.; in some cases, error bars are smaller than the height of the symbol and are, therefore, not shown. Scale bars; 500 nm for sptPALM images, 5 µm for larger FRAP images, 0.5 µm for inset FRAP images.
Extended Data Fig. 9 Differences in the bioenergetics of catalytic versus scaffold mechanisms of regulated β-arrestin trafficking and β-arrestin-dependent activation of ERK1/2 promoted by catalytic activation.
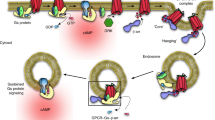
a, Schematic depicting the proposed co-existence of catalytic and scaffolding mechanisms of β-arrestin trafficking tuned according to tail binding affinity, emphasizing the difference in tail versus core interactions (shaded boxes). The tail interaction, which requires GPCR phosphorylation (Rp), drives scaffold formation by stabilizing GPCR–β-arrestin complex formation. The core interaction mediates catalysis by providing a kinetically favourable path for β-arrestin to remain at the plasma membrane irrespective of whether it is associated with a GPCR. The core interaction requires phosphoinositide binding to the β-arrestin C domain, which explains why the phosphoinositide requirement is specific to the catalytic mechanism and can be overcome by formation of a sufficiently stable scaffold complex requiring the phosphorylated GPCR tail. Primary energy inputs maintaining each proposed trafficking cycle are indicated by red arrows. The present results identify a specific requirement of the catalytic mechanism for phosphoinositide binding to the C domain but they do not exclude binding in the scaffold complex (which we think is likely). We also cannot presently rule out the possible existence of additional interaction(s) in the catalytic mechanism, such as phosphoinositide binding to the β-arrestin N domain, which has the potential to displace the β-arrestin C terminus22. b, Representative TIRF microscopy images of live cells (from three independent experiments) before and after treatment of cells expressing chimaeric Flag-tagged β1AR-V2Rs with 10 µM isoproterenol. Profiles of Flag-β2AR and β-arrestin-2–GFP average enrichment into CCSs in COS-1 cells expressing either an empty vector construct (c) or GRK2 (d) and treated with 10 µM isoproterenol (n = 15 or 12 cells, respectively, from three independent experiments). e, Difference in enrichment between β-arrestin-2–GFP and β2AR in cells shown in c and d, showing the effect of GRK2 overexpression. f, Representative western blot showing phosphorylated ERK1/2 and total ERK1/2 signal in extracts prepared from parental or β-arrestin-CRISPR-knockout HEK 293 cells expressing Flag–β1AR and exposed to 10 μM isoproterenol for the indicated time period. g, Quantification of ERK1/2 activation from the western blots in f (n = 5 independent experiments, P = 0.004 using a one-way ANOVA). h, Representative western blot showing phosphorylated ERK1/2 and total ERK1/2 signal in extracts prepared from parental or β-arrestin-CRISPR-knockout HEK 293 cells expressing Flag–β2AR and exposed to 10 μM isoproterenol for the indicated time period. i, Quantification of ERK1/2 activation from the western blots in h (n = 5 independent experiments). f and h show representative western blots from five independent experiments. Data are mean ± s.e.m. For gel source data, see Supplementary Fig. 1. Error bars represent s.e.m. **P < 0.01.
Supplementary information
Supplementary Figure 1
This file contains the uncropped western blots with molecular weight markers and indication of the cropped region.
Video 1: sptPALM video of PA-mCherry-β1AR
HEK 293 cells were co-transfected with PA-mCherry-β1AR and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of PA-mCherry-β1AR. This is a representative video from 2 independent experiments.
Video 2: Representative sptPALM video of PA-mCherry-β2AR
HEK 293 cells were co-transfected with FLAG–β1AR, PA-mCherry-β2AR, and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of PA-mCherry-β2AR. This is a representative video from at least 3 independent experiments.
Video 3: Representative sptPALM video of β-arrestin-2-PA-mCherry after activation of β1AR
HEK 293 cells were co-transfected with FLAG-β1AR, β-arrestin-2-PA-mCherry, and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of β-arrestin-2-PA-mCherry. This is a representative video from at least 3 independent experiments.
Video 4: Representative sptPALM video of β-arrestin-2-PA-mCherry after activation of β2AR
HEK 293 cells were co-transfected with FLAG-β2AR, β-arrestin-2-PA-mCherry, and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of β-arrestin-2-PA-mCherry. This is a representative video from at least 3 independent experiments.
Video 5: Representative sptPALM video of β-arrestin-2-PA-mCherry (CCS mutant) after activation of β1AR
HEK 293 cells were co-transfected with FLAG-β1AR, β-arrestin-2-PA-mCherry (CCS mutant), and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of β-arrestin-2-PA-mCherry (CCS mutant). This is a representative video from at least 3 independent experiments.
Video 6: Representative sptPALM video of β-arrestin-2-PA-mCherry (CCS mutant) after activation of β2AR
HEK 293 cells were co-transfected with FLAG-β2AR, β-arrestin-2-PA-mCherry (CCS mutant), and CLC-GFP and treated with 10 μM isoproterenol. Shown are single-molecule images of β-arrestin-2-PA-mCherry (CCS mutant). This is a representative video from at least 3 independent experiments.
Rights and permissions
About this article

Cite this article
Eichel, K., Jullié, D., Barsi-Rhyne, B. et al. Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386 (2018). https://doi.org/10.1038/s41586-018-0079-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0079-1
This article is cited by
-
Molecular mechanism of biased signaling at the kappa opioid receptor
Nature Communications (2023)
-
Biophysics in tumor growth and progression: from single mechano-sensitive molecules to mechanomedicine
Oncogene (2023)
-
Distinct activation mechanisms of β-arrestin-1 revealed by 19F NMR spectroscopy
Nature Communications (2023)
-
Location bias contributes to functionally selective responses of biased CXCR3 agonists
Nature Communications (2022)
-
Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying β-arrestin transducer bias
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
