
Abstract
The human genome project led to the advancement of genetic technologies and genomic medicine for a variety of human diseases, including monogenic autoimmune and autoinflammatory diseases. As a result, the genome of an individual can now be rapidly sequenced at a low cost, and this technology is beginning to change the practice of rheumatology. In this Perspective, we describe how new sequencing technologies combined with careful clinical phenotyping have led to the discovery of rare rheumatic diseases and their corresponding disease-causing mutations. Additionally, we explore ways in which single-gene mutations, including somatic mutations, are creating opportunities to develop personalized medicines. To illustrate this idea, we focus on diseases affecting the TREX1–cGAS–STING pathway, which is associated with monogenic autoinflammatory diseases and vasculopathies. For many of the affected patients and families, there is an urgent, unmet need for the development of personalized therapies. New innovations related to small molecular inhibitors and gene therapies have the potential to benefit these families, and might help drive further innovations that could prove useful for patients with more common forms of autoimmunity and autoinflammation.
Similar content being viewed by others
Introduction
Genome sequencing is providing increasing insight into a variety of diseases, including autoimmune and autoinflammatory diseases, and is becoming increasingly accessible in the clinic. The human genome project lasted more than a decade and cost ~5 billion US dollars (adjusted for inflation)1, and advancements in this area mean that the genome of an individual can now be rapidly sequenced for under $1,000 US dollars2. Careful clinical phenotyping, combined with sequencing technologies, has enabled the discovery of numerous rare autoinflammatory and autoimmune diseases, together with their corresponding disease-causing mutations, which provides useful insights into these and other rheumatic diseases and opportunities for the development of personalized therapies.
Rheumatic diseases are typically diagnosed using a combination of intuition, laboratory results and judgement. For example, a typical interaction might involve an internationally renowned rheumatologist asking residents and students how to diagnose a patient with systemic lupus erythematosus (SLE). The trainees might respond with a list of diagnostic criteria for SLE3,4, to which the attending physician would answer by explaining that SLE is a clinical diagnosis that should be determined on the basis of a positive anti-nuclear antibody test and expert opinion, rather than by using a list of criteria. Indeed, although laboratory results are important pieces of a diagnostic puzzle, the diagnosis of rheumatic disease is highly dependent on the clinical history and recognition of the clinical features of disease. Nevertheless, all rheumatologists encounter patients with clinical histories and disease phenotypes that are so unique and extraordinary as to defy categorization. In these cases, the integration of genomic medicine into rheumatology has enormous potential to improve diagnosis and treatment.
The study of ultra-rare autoimmune and autoinflammatory diseases might seem like a niche area of medicine, but rare rheumatic syndromes have become increasingly relevant, in part because these monogenic diseases can provide insight into potential aetiologies and mechanisms of more common rheumatic diseases. Unlike infectious diseases, where pathogens are readily identified, defined and targeted, the precise triggers of most rheumatic diseases are still unknown. In this Perspective article, we highlight the ways in which monogenic autoimmune and autoinflammatory diseases are shaping how we think about the aetiology, diagnosis and treatment of rheumatic disease. In particular, we focus on diseases that affect the three prime repair exonuclease 1 (TREX1)–cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway, including various monogenic autoinflammatory diseases and inherited vasculopathies.
Advent of affordable genome sequencing
Historically, DNA was sequenced using a method called classical chain termination sequencing (also known as Sanger sequencing). For this method, a fluorescent dideoxynucleotide (a chain-terminating fluorescently tagged base) is irreversibly attached to the end of a DNA strand during the extension step of a PCR. The fluorescently labelled DNA strands are then separated by size using capillary electrophoresis. Random incorporation of the terminator dideoxynucleotides produces DNA strands of varying lengths. As each chain-terminating base is labelled with a different fluorophore, the DNA sequence can be resolved by measuring the fluorescent signal for each corresponding length. This method of sequencing is very costly and yields relatively short but accurate DNA reads.
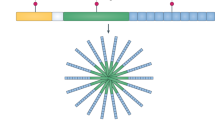
The development of new technology has made genome and exome sequencing much more accessible and affordable for clinical use. Indeed, next-generation sequencing utilizes a very different, error-prone, but low-cost approach5. In one type of next-generation sequencing, individual DNA fragments are randomly attached in clusters to the surface of a flow cell (a specialized channel for adsorbing fragments of DNA). Then, only one base is added sequentially to each cluster. Any of the four bases could be added at each step, but only one base is attached at the 3′ hydroxyl of each cluster. After addition of a fluorescent nucleotide, the flow cell is imaged, and the colour determines which of the four bases was added to each cluster of DNA molecules (Fig. 1). Another cost-lowering approach has been to perform next-generation sequencing of only the exome (the protein-coding portion of the genome) instead of the entire genome. Exome sequencing vastly reduces the number of bases to be sequenced as most of the human genome consists of non-coding DNA.

a, For first generation DNA sequencing (classical chain termination sequencing), chain-terminating fluorescently tagged bases are randomly attached to the end of a DNA strand during a polymerase chain reaction (PCR), resulting in DNA strands of varying sizes. The DNA strands are separated by size using capillary electrophoresis and the signal for each corresponding length is used to determine the overall sequence of the DNA. Compared with next-generation sequencing technology, this approach has a higher cost and is less efficient. b, Next-generation sequencing technology has a low cost because numerous DNA strands can be sequenced in parallel on the surface of a flow cell. The most commonly use next-generation approach, reversible terminator sequencing (as utilized by the Illumina platform), is shown. For this approach, a DNA library is first prepared, involving attachment of adaptors to each end of fragmented DNA. The DNA is hybridized to a flow cell via these adaptors, and amplified in a process known as bridge amplification to generate clusters. To sequence these clusters, fluorescently-tagged reversible terminators (bases that have a blocking group at the 3’OH to prevent further sequence extension) are added, the first base is incorporated and any excess bases are washed away, before the fluorescent signal is measured. The blocking group is then removed so this process can be repeated for another round. Although next-generation sequencing has a higher error rate than chain termination sequencing, repeated sequencing of the same DNA can be performed at a low cost, allowing reliable reporting of a consensus sequence. The original version of this figure was created with BioRender.com. dNTP, deoxyribose nucleotide; ddNTP, dideoxyribonucleotide.
What makes next-generation sequencing remarkable is that once a fluorescent base has been imaged, that fluorophore can be removed, leaving the base attached. The 3′ hydroxyl is subsequently regenerated, and another base can be added to each cluster, followed by repeat imaging. This process is repeated over and over, resulting in numerous sequences of individual DNA molecules. This type of sequencing is more error-prone than terminator sequencing by capillary electrophoresis, which might seem counterintuitive; however, the cost savings and efficiency of next-generation sequencing provides a huge advantage. Next-generation sequencing is inexpensive because many DNA strands can be sequenced in parallel on the surface of the flow cell, causing the price of genome sequencing to plummet. To compensate for next-generation sequencing-associated sequencing errors or artefacts, each sequence must be covered repeatedly to generate a reliable result, which is why next-generation sequencing results include an indicator of the depth or fold-coverage of sequencing. Various kinds of errors and biases can occur during exome sequencing, including PCR bias and binding kinetics bias. If a base is only sequenced once, then there is no way of knowing whether a resulting mutation is a PCR error, a somatic mutation or a variant. However, with a 50-fold average coverage, the likelihood that every base will have been covered at least 30 times is extremely high. Higher coverage increases the reliability and interpretability of the results.
Interpreting the results of genetic testing can be challenging without specialized expertise. All patients have unique mutations and variants, as well as common polymorphisms. The American College of Medical Genetics and Genomics recommends the use of standard terminology (pathogenic, likely pathogenic, uncertain significance, likely benign and benign)6 to describe variants on the basis of various evidence, including population and functional data. However, in many cases, additional experiments in the research laboratory, including cell culture and animal model studies, are necessary to demonstrate the functional or pathogenic effects of mutations. As interpreting genetic information can be difficult, genetic counselling and medical genetics expertise can help to prepare patients and families before undergoing genetic testing. Consulting with experts in genetic diagnosis can also help to prevent families and practitioners from over-interpreting the results of genetic testing. For families known to be affected by rare, disease-causing mutations, genetic counselling prior to testing is often critically important. Patients might have concerns related to life insurance as well as reproductive decisions, and genetic counsellors have an important role in preparing patients for the decision to undergo genetic testing.
Rare diseases
The human stories behind rare rheumatic diseases illustrate how genetic information can benefit patients. In 1988, a team of physicians met a patient with a unique disease of multiple organs, including the kidneys, liver, brain and eyes7. No immunosuppressive therapy was effective, and the patient and several of his family members passed away with severe multi-organ damage. The histological assessment at autopsy revealed vasculopathy involving multiple organs, including the brain and eye, as well as brain lesions that resemble radiation necrosis8,9. A careful family history revealed that about half of the patients’ family members suffered from a similar condition, although the family had previously received other diagnoses, including multiple sclerosis and SLE. Two decades later, an international team of scientists reported that this disease, now known as retinal vasculopathy with cerebral leukoencephalopathy (RVCL; also known as RVCL-S or HERNS), is caused by autosomal-dominant C-terminal frameshift mutations in the gene TREX1 (ref. 10). One hundred percent of patients with RVCL have similar, autosomal-dominant mutations in the carboxy (C)-terminal region of TREX1, and all of these patients develop multi-organ damage beginning around the age of 40 years8. Furthermore, all patients with RVCL die prematurely from the disease, often within 5–10 years of the onset of symptoms8. RVCL is clinically distinct from an autosomal-recessive autoinflammatory disease known as Aicardi–Goutières syndrome (AGS), although both RVCL and AGS are characterized by mutations in TREX1. Whereas AGS can also be caused by mutations in other genes11, RVCL is only caused by mutations in TREX1. Unlike RVCL, which is caused by a single truncation in one TREX1 allele, AGS can result from the complete loss of TREX1 function12,13. TREX1 encodes a DNA exonuclease14 and loss of TREX1 function in AGS leads to accrual of dsDNA in the cytosol and unabated activation of the cGAS-STING pathway, as TREX1 negatively regulates the expression of type I interferon and interferon-stimulated genes12,13,15 (Fig. 2). The amino terminal domain of the TREX1 enzyme contains all of the structural elements for full exonuclease activity, whereas the C-terminal region controls localization of TREX1 at the perinuclear space. The precise immunological and molecular mechanism by which TREX1 frameshift mutations cause RVCL is less well understood, although it might be related to mislocalization of a functional TREX1 enzyme10 or, alternatively, dysregulation of cGAS–STING signalling16.

Different single-gene mutations in the same pathway can cause unique clinical phenotypes, including organ pathology resembling that of common rheumatic diseases. TREX1 is a DNase that degrades cytosolic DNA to prevent activation of the cGAS–STING pathway, which induces production of cytokines, including type I interferon. Mutations in different regions of TREX1 cause entirely distinct clinical phenotypes. Loss of TREX1 function causes disease of the central nervous system, whereas mutations in STING or mutations in COPA that result in STING activation trigger lung disease in patients with SAVI or COPA syndrome. These pathways are increasingly being studied in common rheumatic diseases. The original version of this figure was created with BioRender.com. ANCA, anti-neutrophil cytoplasmic antibody; CNS, central nervous system; ER, endoplasmic reticulum.
What is most concerning for patients with RVCL and their families is the fact that no effective treatment is yet available for this disease. As a consequence, these patients undergo relentless disease progression leading to blindness, chronic renal insufficiency, liver damage, as well as dementia, strokes, osteonecrosis, thyroid disease, gastrointestinal disease, chronic pain, disability and premature death8. Nevertheless, despite our incomplete understanding of the molecular mechanisms that underlie RVCL, the discovery of disease-causing TREX1 mutations has transformed the lives of these patients and their families (Fig. 3). Now, patients with RVCL have the option to undergo genetic testing in early adulthood—long before disease onset. This testing enables patients and their families to prepare and plan for the future. Patients with RVCL can now consider in vitro fertilization combined with genetic testing, which prevents transmission of the mutant TREX1 allele to the next generation. Furthermore, the patients also have the option of participating in longitudinal studies and clinical trials17,18. Most importantly, patients with RVCL and their families now feel more hopeful, as mutations in TREX1 pinpoint this protein as a key therapeutic target. Physicians and scientists are now working to define molecular mechanisms of RVCL pathogenesis, and to develop gene therapies to correct the disease-causing mutation, as well as small molecule drugs that preferentially correct defects elicited by the mutant TREX1 protein.

Retinal vasculopathy with cerebral leukoencephalopathy (RVCL) was discovered more than three decades ago, in 1988. The identification of disease-causing mutations in the gene TREX1 has enabled patients to plan ahead, to participate in research studies, and to choose in vitro fertilization with genetic testing to prevent the next generation from inheriting the dominant disease-causing mutation, which results in premature death in 100% of cases. Now researchers are developing personalized medicines for RVCL, including gene therapies and small molecules that target the mutant TREX1 protein. *J.J.M., unpublished work. SLE, systemic lupus erythematosus.
In 2014, another rare disease known as STING-associated vasculopathy with onset in infancy (SAVI) was discovered and reported by the laboratory of Raphaela Goldbach-Mansky at the National Institutes of Health19. Patients with SAVI develop severe Raynaud syndrome, vasculopathy with autoamputation of digits, skin rash and pulmonary fibrosis, often within the first year of life19. The disease-causing mutation renders STING constitutively active. STING is an important player in the cell-intrinsic innate immune response against viruses and other pathogens20. Introduction of the SAVI mutation into animal models using CRISPR/Cas9 technology has confirmed that these STING gain-of-function mutations are indeed pathogenic21,22,23. The peripheral blood mononuclear cells of patients with SAVI exhibit constitutive upregulation of type I interferon-stimulated genes, which are one of the most prominent pathways activated downstream of STING19. These findings led to the use of JAK inhibitors as a treatment for patients with SAVI, which can reduce signalling downstream of the type I interferon receptor24. Unfortunately, JAK inhibition does not always control the progression of this disease19,24,25,26. Studies in mouse models of SAVI have shown that disease progresses normally even in animals lacking the receptor for type I interferons, suggesting that the disease-causing mutations also have type I interferon-independent effects that contribute to disease21,22,23,27. Ongoing efforts in the field are aimed at further defining the molecular mechanisms of SAVI pathogenesis as well as the cell types involved in disease initiation, which might eventually lead to even better treatments for SAVI.
In 2015, researchers described another disease called COPA syndrome28. Patients with COPA syndrome have mutations in the COPA gene that encodes the α-COP component of the coatomer (a macromolecular complex involved in membrane trafficking), and deficiency of coatomer complex I (COPI) components causes activation of the STING pathway29. α-COP has a role in COPI vesicle biogenesis and retrograde transport of proteins from the Golgi to the endoplasmic reticulum (ER); hence, COPA mutations are thought to dysregulate the transit of proteins from the Golgi to the ER, or to other subcellular compartments30,31,32. In some ways, COPA syndrome clinically resembles SAVI, although the two diseases have some distinguishing features. For example, unlike SAVI, COPA syndrome does not typically cause severe peripheral vasculopathy or autoamputation of digits. Additionally, COPA syndrome can cause pulmonary haemorrhage as well as glomerulonephritis, and can elicit the formation of anti-neutrophil cytoplasmic antibodies (ANCA)33,34, similar to what occurs in patients with ANCA-associated vasculitis35. In 2020, multiple groups reported that COPA mutations lead to trapping of STING in the Golgi, which results in constitutive STING signalling30,31,32. Additionally, a SAVI-associated mutation in an adult also caused ANCA-associated vasculitis36, further suggesting potential phenotypic overlap resulting from mutations in the genes encoding STING and α-COP proteins. Thus, constitutive STING signalling is now implicated in the pathogenesis of multiple autoinflammatory and autoimmune diseases.
What might be most remarkable about these stories is that all of these rare diseases, which are generally quite heterogeneous in clinical phenotype, converge at the molecular level on a single pathway (Fig. 1). This convergence underscores the probable importance of the TREX1–cGAS–STING signalling pathway in the pathogenesis of various rheumatic diseases, in addition to its role in a variety of non-rheumatic diseases37,38. Even more mutations that dysregulate this pathway and the related signalling pathways will undoubtedly be uncovered in the future. More importantly, the discovery of these mutations, as well as of other rare mutations, will probably lead to novel therapies and therapeutic targets of pathways involved in more common types of autoimmune and autoinflammatory diseases.
Somatic mutations
One of the most innovative and paradigm-shifting discoveries at the interface of genetics and rheumatology has been the discovery of VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic)39. Prior to this discovery in 2020, patients with VEXAS were given a variety of clinical diagnoses on the basis of heterogeneous disease phenotypes. Whereas one patient with VEXAS might have been eligible to enrol in a study of classic polyarteritis nodosa, another might have been correctly diagnosed as having giant cell arteritis, and yet another with relapsing polychondritis, despite the fact that somatic mutations in a common gene have led to these distinct disease processes in each of these individuals. Researchers at the National Human Genome Research Institute and collaborators from other NIH Institutes studied the genome sequences from more than 2,500 individuals with undiagnosed inflammatory diseases39, focusing in particular on a set of ~800 genes implicated in ubiquitylation, a post-translational modification that regulates protein activation or protein degradation in cells39. By doing so, the researchers identified UBA1 as the culprit gene: all the patients with VEXAS had somatic mutations in UBA1 (ref. 39). The peripheral blood cells from these patients also showed decreased levels of ubiquitylation compared with healthy individuals, as well as increased activation of innate immune pathways in myeloid cells. Understanding the molecular basis of this disease might lead to the development of novel disease categories based primarily on the molecular features of the disease rather than the clinical phenotype, and enable more personalized therapy.
The VEXAS study raises another intriguing concept: the possibility that many rheumatic diseases might be caused by rare somatic or inherited mutations. For example, some inherited mutations might cause disease with incomplete penetrance, and might therefore be difficult to detect or prove as causal. One intriguing hypothesis is that a variety of somatic mutations can function as triggers for common diseases, similar to how somatic mutations can cause cancer. A major barrier to testing this hypothesis is that identification of the correct tissue for sequencing is challenging. Disease-causing somatic mutations might occur in only a small percentage of somatic cells, which means that the correct cell type would need to be studied and sequenced carefully. Furthermore, disease-causing somatic mutations might be so infrequent in a specific cell population that the mutation would fall below the limit of reliable detection in next-generation sequencing. These challenges distinguish studies of rheumatic diseases from studies of mutations in cancer, as tumour DNA can easily be separated and sequenced in comparison with genomic DNA from healthy control tissue from the same individual40. Thus, deep sequencing approaches that sequence a specific gene many hundreds or thousands of times, combined with the identification of the correct target tissue, will be necessary to facilitate identification of certain somatic mutations, followed by mechanistic studies to determine their importance in the aetiology of rheumatic diseases.
Route to personalized medicine
A molecular diagnosis creates opportunities for individualized treatment approaches. For some autoinflammatory diseases (for example, TNF receptor-associated periodic syndrome, neonatal-onset multisystem inflammatory disease, Muckle–Wells syndrome and familial cold autoinflammatory syndrome), therapies that target specific cytokines (such as IL-1β and TNF) can effectively suppress disease41. Studies of these diseases have also led to the discovery of the NLRP3 inflammasome, which is now recognized as a central mediator of innate inflammatory responses42. Indeed, inhibitors of NLRP3 are now in clinical development43. However, other autoinflammatory and related diseases are still difficult to treat, despite the availability of a wide range of DMARDs. For example, many patients with VEXAS still succumb to disease despite immunosuppressive therapy39, and lung disease can still progress in patients with SAVI despite treatment with JAK inhibitors24,25,26. For other rare rheumatic diseases, including RVCL, immunomodulatory therapies have been entirely ineffective8.
A major barrier to the development of therapies is that for many autoinflammatory syndromes, the number of patients is too small to conduct a well-powered study. Animal models can be helpful for the study of these diseases, not only for testing hypotheses relating to pathogenic mechanisms, but also for the development and testing of novel therapies. For example, a variety of personalized therapies have been considered for the treatment of RVCL, including proteolysis-targeting chimeras (PROTACs), which are molecules that can bind to a mutant protein of interest and target it for ubiquitylation and subsequent degradation by the proteasome44. Other researchers have speculated that CRISPR/Cas9-mediated or TALEN-mediated genome editing might be a reasonable approach for the treatment of RVCL. For such strategies, rigorous proving of the molecular, cellular and immunological mechanisms is important. For example, the cell compartment responsible for promoting various autoinflammatory diseases and vasculopathies, including SAVI, COPA syndrome and RVCL, is still not fully understood. Thus, to attempt gene therapy to correct a disease-causing mutation, the appropriate target cell must first be identified, and the best option for such preliminary studies is an animal model. Indeed, data from bone marrow chimaera experiments in a mouse model of SAVI point to a critical role for mutant STING-expressing radioresistant parenchymal and/or stromal cells in the recruitment and activation of pathogenic lymphocytes in SAVI-associated lung disease45, as well as a role for type II interferon (IFNγ) receptor signalling45,46. In addition to clinical phenotyping and cell culture experiments to study disease-causing mutations, the path towards the development of effective personalized therapies should ideally include extensive and rigorous studies of disease-causing mutations in animals, such as in mice that express human versions of the disease-causing mutant proteins16,21,22,47. Such animal models of human autoinflammatory disease should enable a better understanding of the underlying mechanisms, and permit the testing of siRNA, small molecular inhibitors, gene-editing and PROTAC therapies before proceeding onto phase I trials in humans.
Considerable efforts are ongoing to develop small molecules that target cGAS or STING. Insights into the structural biology of the cGAS–STING pathway have enabled the development of selective small-molecule inhibitors with the potential to block cGAS–STING signalling48,49. Approaches that target the DNA binding or catalytic activity of cGAS, to reduce the generation of cGAMP, hold promise48. Molecules that compete with cGAMP for binding to STING are also promising approaches49. Inhibitors of STING are now available, such as H-151, that block STING activation by preventing the palmitoylation of STING, a critical event that coordinates the translocation of STING from the ER to the Golgi and enables subsequent downstream signalling. PROTAC molecules designed to instigate STING degradation50 might also have potential for the treatment of AGS as well as SAVI or COPA syndrome in humans. Such therapeutics could directly target the TREX1–cGAS–STING pathway and have the potential to interfere with all downstream STING events regardless of the effector pathways promoting disease. For example, STING can regulate T cell activation and the interferon response, both of which are relevant in the pathogenesis of numerous common rheumatic diseases.
Conclusions
The molecular mechanisms underlying most common autoimmune and autoinflammatory diseases are still not well understood, and a clinical diagnosis is almost always based primarily on a constellation of symptoms. Furthermore, the treatment of a rheumatic disease is typically based on the clinical diagnosis. However, in some cases, patients diagnosed with common rheumatic syndromes do not respond to treatments that are considered standard of care for their disease, suggesting the existence of molecular and immunological heterogeneity amongst patients categorized under the same diagnosis umbrella.
Next-generation sequencing in combination with bioinformatics is providing useful insight into various diseases and has enabled the identification of molecular commonalities in seemingly unrelated clinical syndromes. Diseases with similar symptoms were previously assumed to share a similar molecular mechanism, but emerging insights from genome sequencing challenge this assumption and are shifting how we view these diseases. Furthermore, the current study of rheumatic diseases is based on clinical classification, which might explain, at least in part, why molecular and genetic heterogeneity has presented major challenges for rheumatic disease research. Thus, rather than broadly categorizing diseases as autoimmune or autoinflammatory, a molecular systems-based view of disease is likely to become an important part of the future of clinical rheumatology. As a next step, large genotype–phenotype databases should generate a wealth of information that might lead to a molecular diagnosis and personalized therapy for both rare and common autoinflammatory and autoimmune conditions.
References
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
The Human Genome Project (genome.gov). https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost (2022).
Tan, E. M. et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 25, 1271–1277 (1982).
Hochberg, M. C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 40, 1725 (1997).
Slatko, B. E., Gardner, A. F. & Ausubel, F. M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 122, e59 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Grand, M. G. et al. Cerebroretinal vasculopathy. A new hereditary syndrome. Ophthalmology 95, 649–659 (1988).
Stam, A. H. et al. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain 139, 2909–2922 (2016).
Kothari, P. H. et al. TREX1 is expressed by microglia in normal human brain and increases in regions affected by ischemia. Brain Pathol. 28, 806–821 (2018).
Richards, A. et al. C-terminal truncations in human 3’-5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 39, 1068–1070 (2007).
de Jesus, A. A. et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J. Clin. Invest. 130, 1669–1682 (2020).
Crow, Y. J. et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 (2006).
Rice, G. et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 81, 713–725 (2007).
Mazur, D. J. & Perrino, F. W. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3’→5’ exonucleases. J. Biol. Chem. 274, 19655–19660 (1999).
Gall, A. et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 (2012).
Hasan, M. et al. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 43, 463–474 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02723448 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04611880 (2022).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014).
Crowl, J. T., Gray, E. E., Pestal, K., Volkman, H. E. & Stetson, D. B. Intracellular nucleic acid detection in autoimmunity. Annu. Rev. Immunol. 35, 313–336 (2017).
Warner, J. D. et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279–3292 (2017).
Motwani, M. et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc. Natl Acad. Sci. USA 116, 7941–7950 (2019).
Bouis, D. et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J. Allergy Clin. Immunol. 143, 712–725.e715 (2019).
Sanchez, G. A. M. et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Invest. 128, 3041–3052 (2018).
Frémond, M. L. et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients. J. Allergy Clin. Immunol. Pract. 9, 803–818.e811 (2021).
Volpi, S. et al. Efficacy and adverse events during Janus kinase inhibitor treatment of SAVI syndrome. J. Clin. Immunol. 39, 476–485 (2019).
Luksch, H. et al. STING-associated lung disease in mice relies on T cells but not type I interferon. J. Allergy Clin. Immunol. 144, 254–266.e8 (2019).
Watkin, L. B. et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet. 47, 654–660 (2015).
Steiner, A. et al. Deficiency in coatomer complex I causes aberrant activation of STING signalling. Nat. Commun. 13, 2321 (2022).
Deng, Z. et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J. Exp. Med. 217, e20201045 (2020).
Lepelley, A. et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J. Exp. Med. 217, e20200600 (2020).
Mukai, K. et al. Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat. Commun. 12, 61 (2021).
Tsui, J. L. et al. Analysis of pulmonary features and treatment approaches in the COPA syndrome. ERJ Open Res. 4, 00017 (2018).
Cabrera-Pérez, J. S. et al. A Zebra at the rodeo: dyspnea, hematuria, and a family history of arthritis. Arthritis Care Res. 74, 165–170 (2020).
Kitching, A. R. et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 6, 71 (2020).
Staels, F. et al. Adult-onset ANCA-associated vasculitis in SAVI: extension of the phenotypic spectrum, case report and review of the literature. Front. Immunol. 11, 575219 (2020).
Chu, T. T. et al. Tonic prime-boost of STING signalling mediates Niemann–Pick disease type C. Nature 596, 570–575 (2021).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Beck, D. B. et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 383, 2628–2638 (2020).
Welch, J. S. et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278 (2012).
Jesus, A. A. & Goldbach-Mansky, R. IL-1 blockade in autoinflammatory syndromes. Annu. Rev. Med. 65, 223–244 (2014).
Stehlik, C. & Reed, J. C. The PYRIN connection: novel players in innate immunity and inflammation. J. Exp. Med. 200, 551–558 (2004).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04972188 (2021).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Gao, K. M., Motwani, M., Tedder, T., Marshak-Rothstein, A. & Fitzgerald, K. A. Radioresistant cells initiate lymphocyte-dependent lung inflammation and IFNγ-dependent mortality in STING gain-of-function mice. Proc. Natl Acad. Sci. USA 119, e2202327119 (2022).
Stinson, W. A. et al. The IFN-γ receptor promotes immune dysregulation and disease in STING gain-of-function mice. JCI Insight 7, e155250 (2022).
Sakai, T. et al. DNase-active TREX1 frame-shift mutants induce serologic autoimmunity in mice. J. Autoimmun. 81, 13–23 (2017).
Lama, L. et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun. 10, 2261 (2019).
Hong, Z. et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc. Natl Acad. Sci. USA 118, e2105465118 (2021).
Liu, J. et al. Novel CRBN-recruiting proteolysis-targeting chimeras as degraders of stimulator of interferon genes with in vivo anti-inflammatory efficacy. J. Med. Chem. 65, 6593–6611 (2022).
Acknowledgements
The authors would like to thank E. Roberson for helpful discussions about genome sequencing technology.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Rheumatology thanks S. Masters and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Miner, J.J., Fitzgerald, K.A. A path towards personalized medicine for autoinflammatory and related diseases. Nat Rev Rheumatol 19, 182–189 (2023). https://doi.org/10.1038/s41584-022-00904-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-022-00904-2
This article is cited by
-
A nosology of immune diseases from deep immunophenotyping
Nature Reviews Rheumatology (2024)
