
Abstract
The four Janus kinase (JAK) proteins and seven signal transducer and activator of transcription (STAT) transcription factors mediate intracellular signal transduction downstream of cytokine receptors, which are implicated in the pathology of autoimmune, allergic and inflammatory diseases. Development of targeted small-molecule therapies such as JAK inhibitors, which have varied selective inhibitory profiles, has enabled a paradigm shift in the treatment of diverse disorders. JAK inhibitors suppress intracellular signalling mediated by multiple cytokines involved in the pathological processes of rheumatoid arthritis and many other immune and inflammatory diseases, and therefore have the capacity to target multiple aspects of those diseases. In addition to rheumatoid arthritis, JAK inhibition has potential for treatment of autoimmune diseases including systemic lupus erythematosus, spondyloarthritis, inflammatory bowel disease and alopecia areata, in which stimulation of innate immunity activates adaptive immunity, leading to generation of autoreactive T cells and activation and differentiation of B cells. JAK inhibitors are also effective in the treatment of allergic disorders, such as atopic dermatitis, and can even be used for the COVID-19-related cytokine storm. Mechanism-based treatments targeting JAK–STAT pathways have the potential to provide positive outcomes by minimizing the use of glucocorticoids and/or non-specific immunosuppressants in the treatment of systemic immune-mediated inflammatory diseases.
Key points
-
Mechanism-based targeting of receptor-mediated signalling via Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathways in refractory systemic autoimmune diseases can potentially minimize glucocorticoid and non-specific immunosuppressant use.
-
JAK–STAT pathways are important for cellular interaction during rheumatoid arthritis pathological processes, causing synovial inflammation, autoantibody production, synovial proliferation and joint destruction, which are potential targets for JAK inhibition.
-
Inflammatory processes involving JAK–STAT signalling pathways are involved in the pathology of spondyloarthritis (including psoriatic arthritis), and are targets for JAK inhibition.
-
Innate immune system cytokines signal through JAK–STAT to adaptive immune mechanisms involving autoreactive T cells, B cell activation and autoantibody production, which are potential therapeutic targets in systemic lupus erythematosus.
-
Cytokines contribute to various pathophysiological mechanisms of organ-specific autoimmune diseases, and JAK inhibitors can target multiple aspects of inflammatory bowel diseases, alopecia, allergic disorders and cytokine storm.
-
Use of JAK inhibitors requires careful consideration of their multi-target effects, with adequate prior screening and regularly planned monitoring during treatment for infection, cardiovascular disorders, thrombosis and malignancy.
Similar content being viewed by others
Introduction
Cytokines have critical roles in the pathogenesis of immunological and inflammatory diseases and can be targeted therapeutically. Targeted, small-molecule therapies that inhibit Janus kinase (JAK) proteins (essential signalling mediators that act downstream of pro-inflammatory cytokines) have gained traction as efficacious options for the treatment of rheumatic and autoimmune diseases such as rheumatoid arthritis (RA), spondyloarthritis, psoriasis, atopic dermatitis and inflammatory bowel disease (IBD). RA is a systemic autoimmune disease that is characterized by persistent destructive synovitis and extra-articular manifestations, which can lead to severe disability and even mortality. Timely and appropriate treatment is essential to control joint damage, because rapid destruction occurs in the early phase of RA, resulting in joint deformity and irreversible functional impairment. The use of DMARDs, and particularly the development of biologic DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs), theoretically enables remission to be the goal of therapy in all patients. In addition, these drugs can prevent progression of joint damage and physical dysfunction in the long term1,2,3.
JAK inhibitors are an important class of tsDMARDs. The rationale underlying the use of these inhibitors is that JAKs have pivotal roles in particular pathological mechanisms, so that their targeted inhibition can result in effective disease control. Clinical results support this rationale, and JAK inhibitors have been approved for the treatment of RA and other systemic or organ-specific autoimmune diseases (Table 1). In this Review we describe the progress in JAK-targeting therapies for autoimmune rheumatic diseases, with a focus on the mechanisms of action, and discussion on a disease-by-disease basis.
What are JAK inhibitors?
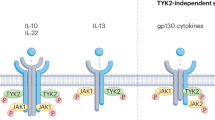
In contrast to bDMARDs, which are large molecules that must be administered parenterally, tsDMARDs are orally available small molecules that enter cellular cytoplasm and directly regulate intracellular signalling by inhibition of kinases or phosphodiesterases. Protein kinases are important regulators of cellular functions that constitute a diverse family, with 518 kinase-encoding genes identified by the Human Genome Project. JAKs belong to the tyrosine-kinase family4,5. Binding of a number of cytokines and growth factors to their receptors results in phosphorylation of receptor-associated JAKs. Phosphorylation activates the JAKs, and they, in turn, phosphorylate intracellular components of the receptors, which enables recruitment of transcription factors of the signal transducer and activator of transcription (STAT) family. Activated STAT proteins translocate to the nucleus and induce transcription. Intracellular signal transduction involves combinations of four JAK isoforms (JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)) and seven STAT family members. The usage of individual JAKs depends on their selective interactions with particular cytokine receptors. Evidence from genetic studies with mutated cell lines, animal models and humans established the essential role of JAKs in signalling by a subset of cytokines that use type I and type II cytokine receptors. More than 50 soluble factors, including IL-2, IL-3, IL-4, IL-5, IL-6 and IL-12, as well as interferons, endocrine factors (including growth hormone, prolactin and leptin) and colony-stimulating factors including erythropoietin, thrombopoietin and granulocyte–macrophage colony-stimulating factor (GM-CSF), exert their effects through specific combinations of JAKs6,7,8,9,10,11 (Fig. 1).

Extracellular binding by a number of cytokines and growth factors to their receptors results in intracellular phosphorylation of receptor-associated Janus kinases (JAKs). Activated JAKs in turn phosphorylate the intracellular components of the receptors, enabling recruitment of signal transducer and activator of transcription (STAT) transcription factors. Activated STATs accumulate in the nucleus and induce transcription. Intracellular signals are transduced through combinations of four JAK isoforms, JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), and seven STAT family members. The involvement of particular JAKs depends on their selective interactions with cytokine-receptor families. JAK inhibitors suppress the effects of cytokines by inhibiting STAT-mediated and other downstream signalling pathways. GM-CSF, granulocyte–macrophage colony-stimulating factor; LIF, leukaemia inhibitory factor; OSM, oncostatin M.
JAK inhibitors selectively interfere with the ATP-binding site of JAKs, resulting in suppression of downstream signalling pathways, which can have immunomodulatory effects in a wide range of pathological processes. Cytokines work in networks, with type I and type II cytokines inducing or being induced by TNF-family cytokines, and in mouse models, JAK inhibitors can inhibit production of TNF, which is a major component in the pathogenesis of RA12. Theoretically, the selectivity of each JAK inhibitor determines its effects on particular inflammatory responses, including those that promote RA9,10,11,12,13,14,15. Five JAK inhibitors (tofacitinib, baricitinib, peficitinib, upadacitinib and filgotinib) are currently approved by different agencies for the treatment of RA, and are categorized as tsDMARDs. In results from clinical trials in patients with RA, tsDMARDs (either as monotherapy or in combination with methotrexate) had multi-target, rapid and robust effects that were equivalent to or superior to those of bDMARDs16,17,18,19,20,21,22,23.
Tofacitinib was developed as a small-molecule drug that competitively binds to the ATP-binding site of JAK3. Tofacitinib was initially thought to selectively inhibit phosphorylation of JAK3, but it is now considered to inhibit JAK1, JAK2 and JAK3 to varying degrees in vitro and in vivo16,17,24. All of the currently approved JAK inhibitors are competitive antagonists. However, the in vitro assays used in preclinical studies to obtain selectivity data are not identical, and relating these data to in vivo efficacy and adverse events is not a simple matter. Selectivity to JAKs can be determined by the use of purified enzymes, or via a variety of cellular models using cytokine stimulation of cells, with assessment of STAT phosphorylation. To illustrate the difficulties with direct comparison of these methods, in vitro kinase assays demonstrate that tofacitinib is a potent inhibitor of JAK1 and JAK3, but that it is less active against JAK2 and TYK2, whereas baricitinib is a selective JAK1 and JAK2 inhibitor, and upadacitinib and filgotinib are selective JAK1 inhibitors8,9,10. However, in direct comparisons in cell-based assays, the ability of each JAK inhibitor to inhibit a specific cytokine-signalling pathway could not be readily inferred using preclinical selectivity data25,26,27,28. For example, baricitinib and tofacitinib similarly suppress the JAK–STAT-mediated differentiation of plasmablasts, T helper 1 (TH1) and T helper 17 (TH17) cells, as well as the capacity of dendritic cells to stimulate T cells25,26. In addition, the effects of JAK inhibitors on cytokine-receptor signalling are all generally similar when comparing the clinically effective doses for RA, suggesting that differentiation on the basis of pharmacological properties with individual JAKs comes with substantial caveats28. By contrast, in an industry-sponsored study of filgotinib, upadacitinib, tofacitinib and baricitinib, although JAK1-dependent pathways were the most potently affected by all four inhibitors, filgotinib demonstrated relatively little inhibition of the JAK2-dependent and JAK3-dependent pathways, compared with the other inhibitors27. The apparent selectivity of filgotinib might have benefits in terms of safety profiles, and there is preliminary evidence of a lower incidence of herpes zoster infection and venous thrombotic events with filgotinib from side-by-side tabulation of across-trial data28,29. However, any potential differences in safety profiles need to be confirmed with rigorously designed head-to-head studies and more real-world experience.
Although the downstream effects of JAK inhibition in vivo are not fully understood, JAK-dependent cytokine signalling in vivo is known to be influenced by individual variation in factors such as single-nucleotide polymorphisms (SNPs) affecting STAT isoforms, the penetration and distribution of drugs into tissues, the expression patterns of JAKs at sites of inflammation and the dynamic balance of T follicular helper (TFH) cells, T peripheral helper (TPH) cells, TH17 cells and regulatory T (Treg) cells7,8. Furthermore, specific aspects of particular experimental approaches could differentially affect results relating to the selectivity of inhibitors for JAK isoforms. In this Review, we discuss the potential of JAK inhibitors on a disease-by-disease basis.
JAK inhibition in RA
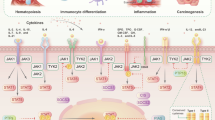
In genome-wide association studies (GWAS) of SNPs in patients with RA, among disease-susceptibility genes such as PTPN22, CTLA4 and STAT4, HLA-DRB1 had the strongest association. HLA-DRB1 alleles encode protein chains that include the shared epitope motif, and they are associated with production of anti-citrullinated protein antibodies. Although specific autoantigens have not been identified, the interaction of genetic and environmental factors, as well as citrullination of extracellular matrix molecules such as filaggrin and fibrinogen, induces autoimmunity in RA through epigenetic modification and conformational changes that disrupt immune tolerance to antigens. As a result, autoreactive T cells and B cells accumulate in synovial tissue, leading to angiogenesis, vasodilation and proliferation of synovial cells. In addition, differentiation of naive T cells to TH1 cells, TH17 cells, TFH cells and TPH cells, as well as activation of B cells, leads to the formation of lymphoid-follicle-like structures and germinal-centre-like structures, which induce the production of autoantibodies. Close cell–cell interaction results in excessive production of pro-inflammatory cytokines, leading to RA. Monocytes differentiate into immature dendritic cells in a process that is dependent on IL-4 and GM-CSF, and then can differentiate into dendritic-cell-derived osteoclasts in the presence of macrophage colony-stimulating factor (M-CSF) and RANKL (Fig. 2). Rheumatoid synovial fibroblasts also produce an excess of pro-inflammatory cytokines (mainly IL-6). The nature of these and other pathological processes in RA indicate that multiple cytokines, including IL-6, interferons and GM-CSF, are direct targets for JAK inhibitors, whereas the production of other cytokines, such as TNF, can be indirectly affected1,2,30,31 (Fig. 2).

The Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signalling pathways have pivotal roles in intracellular signalling in the pathogenesis of rheumatoid arthritis (RA), including synovial inflammation, autoantibody production, synovial proliferation and joint destruction, which are potential targets for JAK inhibition. Differentiation of naive T cells to T helper 1 (TH1), T helper 17 (TH17), T follicular helper (TFH) and T peripheral helper (TPH) cells, and differentiation of B cells to plasmablasts leads to production of autoantibodies. This close cell–cell interaction, including B cell differentiation to plasmablasts induced by TFH in lymphoid organs or TPH in peripheral inflamed tissue, results in high expression of pro-inflammatory cytokines. Monocytes differentiate into osteoclasts in a process dependent on macrophage colony-stimulating factor (M-CSF) and RANKL. Monocytes also differentiate into immature dendritic cells in the presence of IL-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF), and stimulation with M-CSF and RANKL further differentiates the cells to activated osteoclasts (dendritic cell-derived osteoclasts). Synovial fibroblasts produce an excess of pro-inflammatory cytokines, mainly IL-6. These pathological processes provide evidence that multiple cytokines, including IL-6, TNF, interferons and GM-CSF, are good targets for JAK inhibitors in RA. BAFF, B cell activating factor; MMP, matrix metalloproteinase; TGFβ, transforming growth factor-β.
An animal model of RA (SCID-HuRAg) was created by transplantation of synovium and cartilage from patients with RA into severe combined immunodeficiency mice. Continuous administration of tofacitinib to these mice using an osmotic minipump suppressed production of human IL-6, IL-8 and matrix metalloproteinase-3 (MMP-3) from the transplanted synovium, leading to a reduction of synovial inflammation and cartilage destruction compared with untreated SCID-HuRAg mice. In this model, tofacitinib also directly inhibited the production of IL-17 and IFNγ and the proliferation of CD4+ T cells, which in turn inhibited the production of MMP-3, IL-6 and IL-8 by synovial fibroblasts and CD14+ monocytes and suppressed cartilage destruction. These results demonstrated the important roles of JAK signalling for CD4+ T cells, TH1 cells and TH17 cells in synovial inflammation in RA32 (Fig. 2).
Phase III clinical trials have demonstrated robust and rapid effects of JAK inhibitors compared with placebo in patients with RA who are methotrexate naive or who have an inadequate response to methotrexate or to bDMARDs15,16,17,18,19,20,21,22,23. Baricitinib 4 mg daily dosage, compared with TNF inhibitor adalimumab in a head-to-head phase III trial, achieved superiority in the primary outcome, the ACR20 response rate (20% improvement in the number of tender and the number of swollen joints, along with 20% improvement in three criteria among patient global assessment, physician global assessment, functional ability measure, visual analogue pain scale and erythrocyte sedimentation rate or C-reactive protein) at 12 weeks18. However, it should be noted that baricitinib 4 mg daily dosage for treatment of RA has approval in Europe, but not in the USA. Upadacitinib is more effective than adalimumab with regard to ACR20, ACR50 and ACR70 response rates, but only the ACR50 comparison demonstrates multiplicity-controlled statistical superiority20. Upadacitinib is superior to the selective T cell costimulatory modulator abatacept with regard to the mean change of DAS28-CRP (disease activity score in 28 joints using C-reactive protein concentrations) at 12 weeks (the primary outcome) and the remission rate22. Filgotinib 200 mg once-daily dosage (but not 100 mg once-daily dosage) is non-inferior to adalimumab21. In patients with RA who have an inadequate response to methotrexate, monotherapy with upadacitinib results in improvements in clinical and functional outcomes compared with continuation of methotrexate23. In the EULAR management guidelines for RA of 2019, the recommendation for JAK inhibitors was raised to the same level as for bDMARDs, that is, for use as second-line and third-line agents3. On the basis of the treat-to-target principle, JAK inhibitors should be used in combination with conventional synthetic DMARDs in patients with RA.
The JAK inhibitors upadacitinib and filgotinib are therapeutically effective in patients with difficult-to-treat RA, and exert their effects even in patients who have previously been treated with at least two bDMARDs33,34. Although there have been no direct comparative studies between JAK inhibitors in RA in general, results from a propensity score-based study indicate that baricitinib is more effective than tofacitinib35. We also showed in a network meta-analysis that peficitinib is comparable with baricitinib and tofacitinib in terms of efficacy36.
JAK inhibitors result in robust inhibition of bone erosion in RA. Relative to placebo, baricitinib inhibited joint inflammation and the progression of radiographic joint damage in patients with RA during phase III studies, and these effects were comparable with those observed with adalimumab37. These efficacies against joint destruction are supported by results from preclinical studies showing that baricitinib promotes mineralization of osteogenic cells and has osteoprotective effects38. Pathological bone erosion occurs when inflammatory granulation tissue, including proliferating and stratified synovial cells, grows until it contacts bone, at which point multinucleated osteoclasts destroy and resorb the bone and cause joint destruction. IL-6 and TNF induce proliferation of synoviocytes and expression of RANKL on synoviocytes and lymphocytes, thereby inducing the maturation and activation of osteoclasts. JAK inhibitors directly or indirectly inhibit osteoclast maturation by suppressing IFNβ-mediated signalling in osteoclasts and IL-6-mediated RANKL expression in synovial fibroblasts. Dendritic cell-derived osteoclasts stimulated by IL-4, GM-CSF, M-CSF and RANKL promote bone resorption as osteoclasts and T cell activation as antigen-presenting cells in the pathogenesis of chronic inflammatory and destructive synovitis, suggesting that dendritic cell-derived osteoclasts are also targets for JAK inhibitors in the suppression of bone erosion in RA39,40 (Fig. 2).
JAK inhibition in spondyloarthritis
Genetic, cellular and molecular mechanisms contribute to the pathogenesis of spondyloarthritis (SpA). In SpA, dysregulation of skin and gut barriers, caused by alteration of bacterial exposure and/or by genetic factors, is responsible for inflammation in the skin, gut and joints. Immune cells migrate from the peripheral blood to the inflamed joints. Invasion of immune cells such as dendritic cells, macrophages, innate lymphoid cells, mucosal-associated invariant T cells and mast cells into the tissue results in the production of numerous additional inflammatory mediators. Thus, various cytokines such as IFNγ, IL-6, IL-12, IL-17, IL-23 and TNF have important roles in pathogenesis.
IL-12 and IL-23 are produced in large quantities by all antigen-presenting cells, such as dendritic cells, monocytes and macrophages. IL-12 is important for the induction of TH1 cells, which produce IFNγ, TNF and other cytokines, and IL-23 is important for the induction of TH17 cells that produce, among others, IL-17A, IL-17F and IL-22 (Fig. 3). IL-17 is produced by TH17 cells, CD8 T cells, γδ T cells and type 3 innate lymphoid cells. These cytokines work in synergy to perpetuate persistent inflammation by interacting with a variety of cells, including chondrocytes, osteoblasts, osteoclasts and fibroblasts, leading to disease manifestations and complications41,42,43,44,45,46 (Fig. 3).

During pathological processes of spondyloarthritis, invasion of immune cells such as dendritic cells, T cells, type 3 innate lymphoid cells (ILC3) and neutrophils into the tissue results in the production of numerous additional inflammatory mediators. Thus, various cytokines, including IFNγ, IL-6, IL-12, IL-17, IL-23 and TNF, have important roles in pathogenesis, with involvement of multiple signalling pathways including the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathways among several types of immune and non-immune cells. Various cytokines work in synergy to perpetuate persistent inflammation by interacting with a variety of cells, including fibroblasts and monocytes/macrophages. Propagation of autoinflammation involves diverse cytokines, leading to disease symptoms and complications. Targeting these effector cytokines with JAK inhibitors can help to resolve arthritis and cartilage damage, as well as spine and joint damage in spondyloarthritis. M-CSF, macrophage colony-stimulating factor; MMP, matrix metalloproteinase; TGFβ, transforming growth factor-β; TH, T helper.
Approvals for JAK inhibitors beyond RA are being extended to various rheumatic and autoimmune diseases, including SpA. Results from phase III trials in ankylosing spondylitis (AS), the prototypic axial SpA (axSpA), indicate that the overall magnitude of response to tofacitinib is similar to that reported for TNF inhibitors47. Upadacitinib at 15 mg daily was assessed in the phase II/III placebo-controlled trial SELECT-AXIS 1, and more patients had an ASAS40 response (improvement of ≥40% and absolute improvement of ≥10 units in three or more of the domains: patient global assessment, pain assessment, function and inflammation) at week 14 in the upadacitinib group than in the placebo group48. Upadacitinib has been approved for treatment of AS by the European Medicines Agency. Overall, the efficacy of JAK inhibitors in AS seems to be comparable with that of TNF inhibitors, and the patterns of adverse events and changes in laboratory outcomes are similar to previous findings in other indications.
Among individuals with psoriasis, 30–40% have SpA, resulting in the designation psoriatic arthritis (PsA). However, only a subset of the heterogeneous PsA population develops axSpA, which is considered to differ from classic axSpA or non-radiographic axSpA by type of spinal involvement, disease characteristics and responses to therapy. PsA initially occurs as enthesitis associated with immune abnormalities, and subsequently the inflammation persists or spreads to synovitis. Because inflammation and new bone formation result in progressive and irreversible functional disability affecting peripheral joints and/or the spine, appropriate and timely treatment is a prerequisite for inhibition of damage progression. Other notable clinical manifestations of PsA include dactylitis, inflammation of the nails and entheses, eye lesions such as anterior uveitis, keratoconjunctivitis sicca and iritis, aortic regurgitation, interstitial lung disease and intestinal inflammation41,42,43,44,45,46,49.
Targeting effector cytokines with bDMARDs and JAK inhibitors can help to resolve enthesitis and subsequent arthritis, as well as spine and joint damage in PsA41,42,43,44,45,46. Tofacitinib is approved for PsA in multiple countries. In the phase III trial OPAL Broaden, tofacitinib had a comparable efficacy and safety profile to adalimumab in patients with PsA who had inadequate response to at least one conventional synthetic DMARD and were TNF inhibitor-naive50. In another landmark phase III trial, OPAL Beyond, tofacitinib was effective in patients with PsA who had previously had an inadequate response to TNF inhibitors51. Notably, 10 mg tofacitinib was not approved for PsA because of concerns regarding its safety–benefit ratio. Also, tofacitinib was not approved for patients with psoriasis but without PsA. The clinical development of baricitinib for PsA has been halted, possibly because of results in a phase II trial in patients with psoriasis, in which responses were only seen at the higher doses of 8 mg and 10 mg52. In a comparison of the efficacy and safety of upadacitinib with those of placebo or adalimumab in patients with PsA, the proportion of patients achieving ACR20 response at week 12 was greater with upadacitinib 15 mg or 30 mg than with placebo, and the 30 mg (but not 15 mg) dose of upadacitinib was superior to adalimumab. Adverse events were more frequent with upadacitinib than with placebo. In patients with active PsA and with inadequate response to bDMARDs, upadacitinib (15 mg or 30 mg) was more effective than placebo over 24 weeks for improvement of the signs and symptoms of PsA53,54. Brepocitinib, an inhibitor of TYK2 and JAK1, is effective for treatment of PsA, with a therapeutic response beginning as early as 4 weeks after commencement and maintained to 52 weeks55.
Deucravacitinib (BMS-986165) is a selective TYK2 inhibitor. Unlike currently approved inhibitors that all bind to the JAK catalytic domain, deucravacitinib targets the pseudokinase or regulatory domain, potentially resulting in higher selectivity56. Deucravacitinib was developed for multiple indications including psoriasis, PsA, systemic lupus erythematosus (SLE) and IBD57. Deucravacitinib is superior to both placebo and apremilast (an inhibitor of phosphodiesterase 4) in treating moderate to severe plaque psoriasis, according to results from a pivotal phase III trial58. Results from a phase II trial demonstrate that it also has favourable efficacy and safety in the treatment of active PsA59.
JAK inhibition in SLE
SLE is a multisystem autoimmune disease that is more common in women (particularly those of reproductive age) than in men, and that can affect the skin, joints, heart, kidneys, serosa, nerves and blood vessels, presenting with a variety of clinical symptoms. It is pathologically characterized by activation of autoreactive T cells and production of autoantibodies by B cells60,61,62,63. Glucocorticoids and conventional immunosuppressants are widely used treatments, but their targets are non-specific, and effective targeted therapies are needed.
Many SLE disease-susceptibility genes identified by GWAS are highly expressed in adaptive immune cells, including B cells, and B cell activation processes and overproduction of autoantibodies are notable pathological features of SLE64. B cells stimulated by TFH cells or autoreactive T helper cells undergo class switching, differentiating into autoantibody-producing cells in response to IL-21 and other cytokines. In addition, B cells produce various cytokines, such as IL-6 (refs65,66). Therefore, B cells have a central role in humoral immunity and autoimmune diseases, and therapies targeting B cells are expected to be effective for the treatment of SLE. However, many bDMARDs targeting B cells, such as the anti-CD20 antibody rituximab and the anti-CD22 antibody epratuzumab, have seemed promising, but have not yielded favourable results67.
GWAS have resulted in identification of the genes encoding IL-1 receptor-associated kinase 1, interferon regulatory factor 5, Toll-like receptor (TLR) 7, TYK2 and STAT4 as disease-susceptibility genes for SLE64. In addition, overproduction of interferons and the concomitant overexpression of interferon-induced genes (known as the ‘interferon signature’) is a canonical feature of SLE and other autoimmune diseases. Expression of these molecules is high in cells of the innate immune system, including dendritic cells65. TLRs are highly expressed in dendritic cells in patients with SLE, and their contribution to aberrant cell death, including neutrophil death through formation of neutrophil extracellular traps (NETs), induces the production of cytokines and chemokines, which has an important role in triggering subsequent loss of immune tolerance65 (Fig. 4). These cytokines, which are produced by the innate immune system, include soluble B cell activating factor (BAFF), type I interferons, type II interferon, type III interferons and IL-12 and/or IL-23, which in turn induce the differentiation and activation of T cells, and class switching and differentiation of B cells to autoantibody-producing cells in the adaptive immune system. Thus, these cytokines link the innate and adaptive immune systems and are of particular interest as targets for treatment66,67. Notably, in patients with SLE, serum levels of soluble BAFF and IFNα are positively associated with disease activity, which is also associated with critical organ disorders, such as lupus nephritis and neuropsychiatric SLE.

Genome-wide association analysis has identified disease-susceptibility genes for systemic lupus erythematosus (SLE), including genes encoding Toll-like receptor (TLR) 7 and interferon regulatory factor 5. When TLRs on dendritic cells bind to DNA and RNA released during apoptosis and NETosis, dendritic cells transduce signals and produce cytokines, including soluble B cell activating factor (BAFF), type I interferons, type II interferon, IL-12 and IL-23. Numerous cytokines and growth factors signal through Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathways to form a bridge between the innate and adaptive immune systems, resulting in T cell activation, B cell activation and autoantibody production. This signalling is of particular interest as a target for the treatment of SLE. Cytokines that link the innate and adaptive immune systems, such as type I interferons, IL-12 and IL-23, as well as those that activate T cell–B cell interaction, such as IL-21, IL-6 and IL-4, are potential targets of JAK inhibitors in SLE. BAFF-R, BAFF receptor; BCR, B cell receptor; FcR, Fc receptor; MHC, major histocompatibility complex; NET, neutrophil extracellular trap; TCR, T cell receptor.
Belimumab, an anti-BAFF antibody, was the first approved biologic for the treatment of SLE and is also approved for lupus nephritis68. Anifrolumab, a monoclonal antibody to type I interferon receptor, was recently approved for patients with moderate to severe SLE in the USA, Japan and the EU on the basis of results from two phase III trials, TULIP1 and TULIP2 (refs69,70). Many biologics are under development for SLE and lupus nephritis. However, because B cells are activated and antibody production function is enhanced in SLE, administration of large exogenous molecules such as biologics might actually result in the production of anti-drug antibodies.
The use of JAK inhibitors in SLE is currently being assessed (Fig. 4). Cytokines that bridge the innate and adaptive immune systems, such as type I interferons, IL-12 and IL-23, as well as those that activate T cell–B cell interaction, such as IL-21, IL-6 and IL-4, are likely targets of JAK inhibitors in SLE71. Results from a pilot phase Ib/IIa trial showed that the immunological response to tofacitinib in SLE is modulated by STAT4 risk allele rs7574865[T], which is associated with severe SLE manifestations72. In those with SLE who carry the STAT4 risk allele, tofacitinib is associated with low expression of interferon-response genes and reduction in proportions of low-density granulocytes and neutrophil NETosis, whereas in those without the STAT4 risk allele, tofacitinib is otherwise associated with low concentrations of activation and checkpoint markers, such as CD103, CXCR3, inducible costimulatory molecule (ICOS) and programmed cell death protein 1 (PD1), in multiple T cell subsets71,72. In a phase IIb clinical trial of baricitinib in patients with active SLE exhibiting skin and joint symptoms despite standard care, more patients in the baricitinib (4 mg) group achieved resolution of joint or skin symptoms at week 24 (according to SLE Disease Activity Index 2000 criteria) than in the placebo group. In addition to meeting this primary end point, the baricitinib treatment also achieved a response according to the SLE Responder Index criteria73. The phase III trials BRAVE I and II, in which the efficacy of baricitinib in SLE is under evaluation, are currently ongoing (NCT03616912 and NCT03616964). In addition, brepocitinib, an inhibitor of JAK1 and TYK2, is currently the subject of a phase II clinical trial for SLE (NCT03845517).
JAK inhibition in other diseases
Although ulcerative colitis and Crohn’s disease differ in their clinical signs and pathological features, these IBDs share gut microbial abnormalities that are involved in immune disorders. Biologic agents that target TNF, IL-12, IL-23 and gut-selective integrins have beneficial effects in the treatment of IBD, but these agents are not effective for all patients74.
According to results from GWAS, IBD is associated with SNPs in JAK2, STAT3, TYK2 and IL23R75. Crohn’s disease shares about 30% of its genetic polymorphisms with ulcerative colitis, including the variants in IL23R. Several cytokines, including IL-5, IL-6, IL-7, IL-12, IL-13, IL-15, IL-17, IL-18, IL-21, IL-22, IL-23, IL-27, IL-32, IL-33 and IFNγ, have important roles in the pathogenesis of IBD. Among them, IFNγ, IL-6 and IL-7 are more involved in Crohn’s disease, which is associated predominantly with TH1 cell and TH17 cell immune responses, whereas patients with ulcerative colitis have elevated IL-5, IL-13, IL-15 and IL-33, consistent with a TH2 cell-based response. These cytokines function through the JAK–STAT pathway and involve all members of the JAK family, so JAK inhibitors have potential for the treatment of IBD76,77,78,79.
Tofacitinib is approved for the treatment of adults with moderately to severely active ulcerative colitis. In three phase III studies, patients with moderate to severe ulcerative colitis who had not responded to conventional therapy or biologics were treated with tofacitinib (10 mg twice a day) and had a higher rate of clinical remission, clinical response and mucosal healing at week 8 than the placebo group. In addition, the groups of patients who received tofacitinib 5 mg or 10 mg in two divided doses as maintenance therapy for ulcerative colitis had higher frequencies of remission at week 54 than the placebo group80. However, clinical trials of tofacitinib for Crohn’s disease have been disappointing, with no differences in response or remission at various doses compared with placebo81. By contrast, the selective JAK1 inhibitors filgotinib and upadacitinib increased remission rates in patients with moderate to severe Crohn’s disease in phase II trials, and larger phase III trials for ulcerative colitis are currently underway (NCT03653026 and NCT02914522). Phase II clinical trials with an inhibitor of TYK2 and JAK1, brepocitinib (PF-06700841), for ulcerative colitis (NCT02958865) and Crohn’s disease (NCT03395184) are complete. In addition, several clinical trials of retretinib, a JAK3 inhibitor, are ongoing for Crohn’s disease (NCT03395184), ulcerative colitis (NCT02958865) and RA (NCT02969044).
Beyond arthritis and IBD, JAK inhibitors are being studied in other autoimmune, inflammatory and allergic diseases including non-infectious uveitis, giant cell arteritis, systemic sclerosis, Sjögren syndrome and dermatomyositis (Table 1). In patients with atopic dermatitis, both baricitinib and upadacitinib effectively achieve rapid improvement of clinical activity compared with placebo82,83. Baricitinib has been approved for treatment of atopic dermatitis in Europe84. Upadacitinib has superior efficacy in atopic dermatitis to dupilumab (a monoclonal antibody targeting IL-4 and IL-13), but it is also associated with higher rates of serious infection, including one death owing to influenza83. Evidence for therapeutic efficacy of JAK inhibitors has also been demonstrated in conditions such as alopecia areata, vitiligo and palmoplantar pustulosis85,86. In a phase II trial for treatment of alopecia areata, ritlecitinib (a JAK3 inhibitor) and brepocitinib showed marked efficacy and good tolerability after 24 weeks of treatment87.
An unanticipated role for JAK inhibitors is their use in treatment of COVID-19, to attenuate the dysregulated production and action of pro-inflammatory cytokines, including IL-2, IL-6, IL-12, IFNγ and GM-CSF, in the COVID-19-associated cytokine storm. Extreme elevation of cytokine concentrations is associated with pulmonary and endothelial disease, myocardial damage and mortality88. Baricitinib differs from other JAK inhibitors in that it also inhibits AP2-associated protein kinase 1, a pivotal regulator of clathrin-dependent endocytosis, and thus could inhibit viral entry into target cells89. In clinical trials, the combination of baricitinib plus remdesivir was superior to remdesivir monotherapy for both improvement in oxygenation and reduction in select inflammatory markers in patients with COVID-19 pneumonia receiving supplemental oxygen, high-flow oxygen or non-invasive ventilation88,90. The Adaptive COVID-19 Treatment Trial head-to-head comparison of baricitinib and dexamethasone for treatment of severe COVID-19 was terminated prematurely because early results met pre-defined futility criteria, indicating that it was unlikely that continuation of the study would demonstrate a difference between the two treatment arms91. Another industry-sponsored trial involving addition of baricitinib to the combination of remdesivir and dexamethasone for treatment of severe COVID-19 did not meet its primary end point of a composite outcome of progression to high-flow oxygen, non-invasive ventilation, invasive mechanical ventilation or death, but a reduction in death by 38.2% was observed in those receiving baricitinib92. Thus, baricitinib received Emergency Use Authorization for treatment of severe COVID-19 in concomitant use with remdesivir by the FDA in November 2020, and then as monotherapy in July 2021 (ref.93). Baricitinib was also approved in Japan94 and identified as one of the promising candidate therapeutics in Europe95. Clinical trials involving several JAK inhibitors in the treatment of COVID-19 are ongoing, and should provide valuable information on the usefulness of these agents. In addition to current indications, the question arises as to whether JAK inhibitors could have roles in the treatment of sepsis and acute respiratory distress syndrome.
Safety concerns with JAK inhibition
Until warnings from the FDA were published in 2021 (ref.96), the consensus was that the short-term and long-term safety of JAK inhibitors were comparable with those of bDMARDs. As potent immunosuppressive agents, the incidence rates of infections, including opportunistic infections, are comparable with those for bDMARDs, with the exception of the rate of herpes zoster infections, which is slightly higher for JAK inhibitors97,98. Analyses from randomized controlled trials of tofacitinib and baricitinib have suggested a possible dose-dependent pattern of infection risk99,100. Studies on the long-term safety of tofacitinib with follow-up of up to 9.5 years identified no changes over time in incidence rates of infection, opportunistic infection, serious infection, malignancy, thrombosis or cardiovascular disorders101. In an integrated safety analysis of five phase III trials, upadacitinib had comparable short-term and long-term safety with methotrexate and adalimumab, except for a higher risk of herpes zoster and of creatine phosphokinase elevation with upadacitinib than with adalimumab98,102. JAK inhibitors are also associated with potentially serious effects, including malignancy, major adverse cardiovascular events (MACEs) and venous thromboembolic events103. The ORAL-Surveillance study (NCT02092467) compared the safety of tofacitinib and TNF inhibitors. The results of the study have not yet been published, but the preliminary data are available on the sponsor’s website104 and in the trial register (NCT02092467). The initial preliminary result in 2019 demonstrated an association with the risk of venous thromboembolism and death in patients taking tofacitinib 10 mg twice-daily dosage, but not 5 mg twice-daily dosage, prompting an FDA warning in relation to high-dose tofacitnib105. However, later results show a higher incidence of MACEs and malignancies excluding non-melanoma skin cancer in patients with RA treated with either 5 mg or 10 mg twice-daily dosage of tofacitinib than in patients treated with a TNF inhibitor96. In response to this study, the FDA released an updated boxed warning in September 2021 regarding the increased risk of death, MACEs, malignancies and thrombosis with JAK inhibitors compared with TNF inhibitors96. It also limits all approved uses to certain patients who have not responded to or cannot tolerate one or more TNF blockers. Although this study only compared tofacitinib with adalimumab, the FDA was concerned about a JAK-inhibitor class effect, and the warning was extended to two other JAK inhibitors approved in the USA for treatment of inflammatory diseases, baricitinib and upadacitinib. Whether the use of inhibitors with different JAK subtype selectivity or the use of JAK inhibitors in different diseases would improve cardiovascular and carcinogenic risk clearly warrants further investigation. Additionally, in clinical scenarios where TNF inhibitors have failed or not been appropriate, the choice between other biologics and JAK inhibitors is unclear.
Some of the adverse events associated with JAK inhibitors are predicted by mechanisms related to the blockade of cytokines that use JAK–STAT for signalling, which could explain the risk of serious and/or opportunistic infections such as herpes zoster106. However, the occurrence of thromboembolism, although relatively rare, is an unexpected and unexplained event104,106. Whether this event involves activation of the coagulation–fibrinolysis system or of platelets and endothelial cells is not yet known. Thus, although the use of JAK inhibitors is convenient because of their oral administration, it should be carefully considered12. Adequate screening should be performed for factors such as infection, cardiovascular disorders, thrombosis and malignancy. JAK inhibitors should be administered by physicians who are able to provide systemic management of adverse events. Contraindications to the use of JAK inhibitors are related to pharmacokinetic and pharmacodynamic profiles and adverse events, and include: severe active infection (acute or chronic), including latent tuberculosis and opportunistic infections with the apparent exception of COVID-19; active malignancy; severe organ damage (including severe hepatic or renal disease); pregnancy and lactation; and history of venous thromboembolism. The safety and efficacy of JAK inhibitors in children have been assessed in some indications. Tofacitinib is currently approved for treatment of polyarticular JIA in the USA107, and is being studied in systemic JIA (NCT03000439). Ruxolitinib (an inhibitor of JAK1 and JAK2) is approved for treatment of both acute and chronic graft-versus-host disease in patients >12 years old108. In general, JAK inhibitors are not recommended for use in combination with bDMARDs or potent immunosuppressants such as cyclosporine and tacrolimus, because these combinations might overly suppress the immune system and unacceptably increase the risk of infection and lymphoma. Finally, appropriately and regularly planned monitoring during treatment should be performed for known risks including infection, cardiovascular disorders, thrombosis and malignancy. Long-term safety studies regarding the development of infection and malignancy (such as lymphoma) need to be conducted.
Conclusions
JAK inhibitors exert immunomodulatory effects on a wide range of highly heterogeneous diseases by inhibiting STAT-mediated signalling pathways of numerous cytokines. Thus, mechanism-based therapies targeting several cytokines and their signalling have brought a paradigm shift in the treatment strategy for refractory systemic autoimmune diseases. The success of JAK inhibitors has facilitated research on intracellular signal transduction in immune cells and its relevance to pathological processes, as well as the development of inhibitors of targets including spleen tyrosine kinase, Bruton’s tyrosine kinase and IL-1 receptor-associated kinase 4, which are undergoing clinical trials109,110. Notably, some JAK inhibitors also have activity against Tec family tyrosine kinases (ritlecitinib) and spleen tyrosine kinase (gusacitinib)111,112. However, the top research priority in this field should be to improve therapeutic strategies, including strategies to maintain a balanced efficacy and safety profile, as well as thorough implementation of screening at treatment initiation, and monitoring during treatment. Furthermore, mechanism-based targeted therapies such as JAK inhibitors could ultimately enable either complete withdrawal or avoidance of glucocorticoid use in some autoimmune diseases. In many of these conditions, intensive and appropriate induction therapies are prerequisites for the achievement of disease remission and to sustain remission without damage to organs including joints and spine. After sustained remission, drug-free remission and even cure in the later stages of treatment might become possible, following appropriate and rigorous clinical trials. However, factors that act to inhibit the transition from remission to cure could exist, not only in the immune system but also in mesenchymal, intestinal, nerve and metabolic systems113. JAK inhibitors target multiple cytokines, growth factors and endocrine factors, so could have the potential to regulate any active factor inhibiting the transition to cure. Elucidation of such factors and approaches to regulate them could be an important strategy in addressing the challenges and unmet needs in the management of autoimmune diseases.
References
Smolen, J. S. et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 8, 18001 (2018). This is a comprehensive, very informative and highly educative review on rheumatoid arthritis.
Tanaka, Y. Rheumatoid arthritis. Inflamm. Regen. 40, 1–8 (2020).
Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 79, 685–699 (2020).
Macchi, P. et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 377, 65–68 (1995).
Johnston, J. A. et al. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature 370, 151–153 (1994).
Tanaka, Y. et al. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann. Rheum. Dis. 71, i70–i74 (2012).
O’Shea, J. J. et al. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 368, 161–170 (2012). This is one of the most highly cited review papers on the basic physiology and molecular and cellular biology of JAKs in the immune system.
O’Shea, J. J., Kontzias, A., Yamaoka, K., Tanaka, Y. & Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 72, ii111–ii115 (2013).
Tanaka, Y. The JAK inhibitors: do they bring a paradigm shift for the management of rheumatic diseases? Rheumatology 58, i1–i3 (2019).
Choy, E. H. Clinical significance of Janus kinase inhibitor selectivity. Rheumatology 58, 953–962 (2019).
Gadina, M. et al. Translating JAKs to Jakinibs. J. Immunol. 204, 2011–2020 (2020).
GhoreschI, K. et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243 (2011).
Nash, P. et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann. Rheum. Dis. 80, 71–87 (2021). This is a recent consensus statement for management and treatment with JAK inhibitors by specialists in the field of rheumatology.
Schwartz, D. M. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2016).
Villarino, A. V. et al. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18, 374–384 (2017).
Tanaka, Y. et al. Tofacitinib study investigators. Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res. 63, 1150–1158 (2011).
van der Heijde, D. et al. ORAL Scan Investigators. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 65, 559–570 (2013).
Taylor, P. C. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Tanaka, Y. et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double-blind, placebo-controlled phase III trial (RAJ3). Ann. Rheum. Dis. 78, 1320–1332 (2019).
Fleischmann, R. et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 71, 1788–1800 (2019).
Combe, B. et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann. Rheum. Dis. 80, 848–858 (2021).
Rubbert-Roth, A. et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N. Engl. J. Med. 383, 1511–1521 (2020).
Smolen, J. S. et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet 393, 2303–2311 (2019).
Changelian, P. S. et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878 (2003).
Kubo, S. et al. Janus kinase inhibitor baricitinib modulates human innate and adaptive immune system. Front. Immunol. 28, 1510–1521 (2018).
Kubo, S. et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann. Rheum. Dis. 73, 2192–2198 (2014).
Traves, P. G. et al. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann. Rheum. Dis. 80, 865–875 (2021).
McInnes, I. B. et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res. Ther. 21, 183 (2020).
Tanaka, Y., Kavanaugh, A., Wicklund, J. & McInnes, I. B. Filgotinib, a novel JAK1-preferential inhibitor for the treatment of rheumatoid arthritis: an overview from clinical trials. Mod. Rheumatol. https://doi.org/10.1080/14397595.2021.1902617 (2021).
Smolen, J. S. et al. Rheumatoid arthritis. Lancet 388, 2023–2038 (2016).
McInnes, I. B. & Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 389, 2328–2337 (2017).
Maeshima, K. et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheumatol. 64, 1790–1798 (2012).
Genovese, M. C. et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet 391, 2513–2524 (2018).
Genovese, M. C. et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA 322, 315–325 (2019).
Miyazaki, Y. et al. Efficacy and safety of tofacitinib versus baricitinib in patients with rheumatoid arthritis in real clinical practice: analyses with propensity score-based inverse probability of treatment weighting. Ann. Rheum. Dis. 80, 1130–1136 (2021).
Tanaka, Y. et al. Comparative efficacy and safety of peficitinib versus tofacitinib and baricitinib for treatment of rheumatoid arthritis: a systematic review and network meta-analysis. Rheumatol. Ther. 8, 729–750 (2021).
Emery, P. et al. Baricitinib inhibits structural joint damage progression in patients with rheumatoid arthritis-a comprehensive review. Arthritis Res. Ther. 23, 3 (2021).
Murakami, K. et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS One 12, e0181126 (2017).
Narisawa, M. et al. Human dendritic cell-derived osteoclasts with high bone resorption capacity and T cell stimulation ability. Bone 142, 115616 (2021).
Tanaka, Y. Managing osteoporosis and joint damage in patients with rheumatoid arthritis: an overview. J. Clin. Med. 10, 1241 (2021).
McGonagle, D. M. et al. Pathophysiology, assessment and treatment of psoriatic dactylitis. Nat. Rev. Rheumatol. 15, 113–122 (2019).
Bravo, A. & Kavanaugh, A. Bedside to bench: defining the immunopathogenesis of psoriatic arthritis. Nat. Rev. Rheumatol. 15, 645–656 (2019).
Van den Bosch, F. & Coates, L. Clinical management of psoriatic arthritis. Lancet 391, 2285–2294 (2018).
Ritchlin, C. T. et al. Psoriatic arthritis. N. Engl. J. Med. 376, 957–970 (2017).
Schett, G. et al. Enthesitis: from pathophysiology to treatment. Nat. Rev. Rheumatol. 13, 731–741 (2017).
Sieper, J. et al. The IL-23-IL-17 pathway as a therapeutic target in axial spondyloarthritis. Nat. Rev. Rheumatol. 15, 747–757 (2019).
Deodhar, A. et al. Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 80, 1004–1013 (2021).
van der Heijde, D. et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet 394, 2108–2117 (2019).
Keeling, S. & Maksymowych, W. P. JAK inhibitors, psoriatic arthritis, and axial spondyloarthritis: a critical review of clinical trials. Expert Rev. Clin. Immunol. 17, 701–715 (2021).
Mease, P. et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N. Engl. J. Med. 377, 1537–1550 (2017).
Gladman, D. et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N. Engl. J. Med. 377, 1525–1536 (2017).
Papp, K. A. et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 174, 1266–1276 (2016).
Mease, P. J. et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 80, 312–320 (2020).
McInnes, I. B. et al. Trial of upadacitinib and adalimumab for psoriatic arthritis. N. Engl. J. Med. 384, 1227–1239 (2021).
Nogueira, M. et al. JAK inhibitors for treatment of psoriasis: focus on selective TYK2 inhibitors. Drugs 80, 341–352 (2020).
Burke, J. R. et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 11, eaaw1736 (2019).
Danese, S. & Peyrin-Biroulet, L. Selective tyrosine kinase 2 inhibition for treatment of inflammatory bowel disease: new hope on the rise. Inflamm. Bowel Dis. 27, 2023–2030 (2021).
Armstrong, A. et al. Efficacy and safety of deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, compared with placebo and apremilast in moderate to severe plaque psoriasis: results from the phase 3 POETYK PSO-1 study [abstract]. Ann. Rheum. Dis. 80, 795 (2021).
Mease, P. J. et al. Efficacy and safety of deucravacitinib (BMS-986165), an oral, selective tyrosine kinase 2 inhibitor, in patients with active psoriatic arthritis: results from a phase 2, randomized, double-blind, placebo-controlled trial [abstract]. Arthritis Rheumatol. 72 (Suppl. 10), L03 (2020).
Hahn, B. H. Systemic lupus erythematosus. In Harrison’s Principles of Internal Medicine, 20th edn (ed. Jameson, J. R. et al.) pp 2515–2526 (McGraw-Hill, 2018). This is a comprehensive and highly educative textbook on SLE.
Fanouriakis, A. et al. Update οn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum. Dis. 80, 14–25 (2021).
Dörner, T. & Lipsky, P. E. Beyond pan-B-cell-directed therapy — new avenues and insights into the pathogenesis of SLE. Nat. Rev. Rheumatol. 12, 645–657 (2016).
Murphy, G. & Isenberg, D. A. New therapies for systemic lupus erythematosus — past imperfect, future tense. Nat. Rev. Rheumatol. 15, 403–412 (2019).
Liu, Z. & Davidson, A. Taming lupus — a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 18, 871–882 (2012).
Gupta, S. & Kaplan, M. L. Bite of the wolf: innate immune responses propagate autoimmunity in lupus. J. Clin. Invest. 131, e144918 (2021).
Tanaka, Y. State-of-the-art treatment of systemic lupus erythematosus. Int. J. Rheum. Dis. 23, 465–471 (2020).
Tanaka, Y. et al. Lymphocyte phenotype and its application to precision medicine in systemic autoimmune diseases. Semin. Arthritis Rheum. 48, 1146–1150 (2019).
Navarra, S. et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 377, 721–731 (2011).
Furie, R. et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 1, e208–e219 (2019).
Morand, E. F. et al. Trial of anifrolumab in active systemic lupus erythematosus. N. Engl. J. Med. 382, 211–221 (2020).
Hagberg, N. et al. The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-γ production in T cells from patients with SLE. Ann. Rheum. Dis. 77, 1070–1077 (2018).
Hasni, S. et al. A phase 1B/2A trial of tofacitinib, an oral Janus kinase inhibitor, in systemic lupus erythematosus [abstract]. Lupus Sci. Med. https://doi.org/10.1136/lupus-2019-lsm.183 (2019).
Wallace, D. J. et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 392, 222–231 (2019).
Schreiner, P. et al. Mechanism-based treatment strategies for IBD: cytokines, cell adhesion molecules, JAK inhibitors, gut flora, and more. Inflamm. Intest. Dis. 4, 79–96 (2019).
de Lange, K. M. & Barrett, J. C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 64, 91–100 (2015).
Salas, A. et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 323–337 (2020).
Danese, S. et al. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut 68, 1893–1899 (2019).
Neurath, M. F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 269–278 (2017).
Harris, C. & Cummings, J. R. F. JAK1 inhibition and inflammatory bowel disease. Rheumatology 60, ii45–ii51 (2021).
Sandborn, W. J. et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 376, 1723–1736 (2017).
Panés, J. et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut 66, 1049–1059 (2017).
Simpson, E. L. et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br. J. Dermatol. 183, 242–255 (2020).
Guttman-Yassky, E. et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 145, 877–884 (2020).
European Medicines Agency. New oral treatment for moderate to severe atopic dermatitis https://www.ema.europa.eu/en/news/new-oral-treatment-moderate-severe-atopic-dermatitis (2020).
Pourang, A. & Mesinkovska, N. A. New and emerging therapies for alopecia areata. Drugs 80, 635–646 (2020).
Zheng, C. & Tosti, A. Alopecia areata: new treatment options including Janus kinase inhibitors. Dermatol. Clin. 39, 407–415 (2021).
King, B. et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. Am. Acad. Dermatol. 85, 379–387 (2021).
Nissen, C. B. et al. The role of antirheumatics in patients with COVID-19. Lancet Rheumatol. 3, e447–e459 (2021).
Stebbing, J. et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol. Med. 12, e12697 (2020).
Kalil, A. C. et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med. 384, 795–807 (2021).
National Institutes of Health. NIH closes enrollment in trial comparing COVID-19 treatment regimens. Head-to-head trial unlikely to show difference between regimens https://www.nih.gov/news-events/news-releases/nih-closes-enrollment-trial-comparing-covid-19-treatment-regimens (2021).
Marconi, V. C. et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. https://doi.org/10.1016/S2213-2600(21)00331-3 (2021).
U.S. Food & Drug Administration. Baricitinib LOA FINAL 07.28.21 https://www.fda.gov/media/143822/download (2021).
Pharmaceuticals and Medical Devices Agency. PMDA’s efforts to combat COVID-19 https://www.pmda.go.jp/english/about-pmda/0002.html (2021).
European Commission. COVID-19 Therapeutics Strategy: Commission identifies five promising candidate therapeutics https://ec.europa.eu/commission/presscorner/detail/en/ip_21_3299 (2021).
U. S. Food & Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions, https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death (2021).
Pawar, A., Desai, R. J., Gautam, N. & Kim, S. C. Risk of admission to hospital for serious infection after initiating tofacitinib versus biologic DMARDs in patients with rheumatoid arthritis: a multidatabase cohort study. Lancet Rheumatol. 2, E84–E94 (2020).
Cohen, S. B. et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann. Rheum. Dis. 80, 304–311 (2020).
Wollenhaupt, J. et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res. Ther. 21, 89 (2019).
Wang, F. et al. Efficacy and safety of tofacitinib, baricitinib, and upadacitinib for rheumatoid arthritis: a systematic review and meta-analysis. Mayo Clin. Proc. 95, 1404–1419 (2020).
Cohen, S. B. et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open 6, e001395 (2020).
Conaghan, P. G. et al. Upadacitinib in rheumatoid arthritis: a benefit-risk assessment across a phase III program. Drug Saf. 44, 515–530 (2021).
Bechman, K. et al. A systematic review and meta-analysis of infection risk with small molecule JAK inhibitors in rheumatoid arthritis. Rheumatology 58, 1755–1766 (2019).
Pfizer. Pfizer shares co-primary endpoint results from post-marketing required safety study of Xeljanz® (tofacitinib) in subjects with rheumatoid arthritis (RA) https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-co-primary-endpoint-results-post-marketing (2021).
U.S. Food & Drug Administration. FDA approves Boxed Warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR), https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and (2019).
Winthrop, K. L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 13, 234–243 (2017).
Pfizer. U.S. FDA approves Pfizer’s Xeljanz® (tofacitinib) for the treatment of active polyarticular course juvenile idiopathic arthritis, https://investors.pfizer.com/investor-news/press-release-details/2020/U.S.-FDA-Approves-Pfizers-XELJANZ-tofacitinib-for-the-Treatment-of-Active-Polyarticular-Course-Juvenile-Idiopathic-Arthritis/default.aspx (2020).
U.S. Food & Drug Administration. FDA approves ruxolitinib for chronic graft-versus-host disease, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-acute-graft-versus-host-disease (2021).
Tanaka, Y. et al. Safety and efficacy of fostamatinib in rheumatoid arthritis patients with an inadequate response to methotrexate in phase II OSKIRA-ASIA-1 and OSKIRA-ASIA-1X study. Rheumatology 60, 2884–2895 (2021).
Cohen, S. et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, phase II trial (ANDES Study). Arthritis Rheumatol. 72, 1435–1446 (2020).
Robinson, M. F. et al. Efficacy and safety of PF-06651600 (ritlecitinib), a novel JAK3/TEC inhibitor, in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 72, 1621–1631 (2020).
Pavel, A. B. et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 1011–1024 (2019).
Schett, G., Tanaka, Y. & Isaacs, J. D. Why remission is not enough: underlying disease mechanisms in RA that prevent cure. Nat. Rev. Rheumatol. 17, 135–144 (2021). This review documents future perspectives on pathological relevance and treatment to achieve “cure” of rheumatoid arthritis.
Acknowledgements
The authors thank all the medical staff in all participating institutions for helpful discussions and advice.
Author information
Authors and Affiliations
Contributions
Y.T. researched data for the article. Y.T., J.J.O. and S.N. made substantial contributions to discussion of the content. Y.T. wrote the article. All authors contributed to review/editing of the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
Y.T. has received speaking fees and/or honoraria from AbbVie, Amgen, Astellas, AstraZeneca, Boehringer Ingelheim, Bristol-Myers, Chugai, Eisai, Eli Lilly, Gilead, Mitsubishi-Tanabe and YL Biologics, and received research grants from Abbvie, Asahi-Kasei, Boehringer Ingelheim, Chugai, Corrona, Daiichi-Sankyo, Eisai, Kowa, Mitsubishi-Tanabe and Takeda, and consultant fees from AbbVie, Ayumi, Daiichi-Sankyo, Eli Lilly, GlaxoSmithKline, Sanofi and Taisho. S.N. has received speaking fees and/or honoraria from Asahi-kasei, Astellas, Boehringer Ingelheim, Bristol-Myers, GlaxoSmithKline, Pfizer and Sanofi, and has received research grants from Mitsubishi-Tanabe and Novartis. J.J.O. declares receipt of US patent royalties related to JAK inhibitors and NIH Cooperative Research and Development Agreement with Pfizer. Y.M.L. declares no competing interests.
Additional information
Peer review information
Nature Reviews Rheumatology thanks M. Dougados, E. Choy and V. Strand for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tanaka, Y., Luo, Y., O’Shea, J.J. et al. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol 18, 133–145 (2022). https://doi.org/10.1038/s41584-021-00726-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-021-00726-8
This article is cited by
-
Drug survival and change of disease activity using a second janus kinase inhibitor in patients with difficult-to-treat rheumatoid arthritis who failed to a janus kinase inhibitor and subsequent biologics
Advances in Rheumatology (2024)
-
Recent advances and evolving concepts in Still’s disease
Nature Reviews Rheumatology (2024)
-
Emerging therapeutic targets in systemic sclerosis
Journal of Molecular Medicine (2024)
-
Does baricitinib reduce disease activity in patients with systemic lupus erythematosus? A systematic review and meta-analysis of randomized controlled trials
Clinical Rheumatology (2024)
-
Macrophage re-programming by JAK inhibitors relies on MAFB
Cellular and Molecular Life Sciences (2024)
