
Abstract
The current understanding of neurological diseases is derived mostly from direct analysis of patients and from animal models of disease. However, most patient studies do not capture the earliest stages of disease development and offer limited opportunities for experimental intervention, so rarely yield complete mechanistic insights. The use of animal models relies on evolutionary conservation of pathways involved in disease and is limited by an inability to recreate human-specific processes. In vitro models that are derived from human pluripotent stem cells cultured in 3D have emerged as a new model system that could bridge the gap between patient studies and animal models. In this Review, we summarize how such organoid models can complement classical approaches to accelerate neurological research. We describe our current understanding of neurodevelopment and how this process differs between humans and other animals, making human-derived models of disease essential. We discuss different methodologies for producing organoids and how organoids can be and have been used to model neurological disorders, including microcephaly, Zika virus infection, Alzheimer disease and other neurodegenerative disorders, and neurodevelopmental diseases, such as Timothy syndrome, Angelman syndrome and tuberous sclerosis. We also discuss the current limitations of organoid models and outline how organoids can be used to revolutionize research into the human brain and neurological diseases.
Key points
-
Development of the human brain involves unique processes that are relevant to neurological disease but cannot be studied in animal models, so alternative model systems are required.
-
Organoids are 3D human cell culture models that originate from pluripotent stem cells and recapitulate the hallmarks of human neurodevelopment, enabling studies of human brain development in vitro.
-
Specific mutations can be introduced into organoids to study their effects on neurodevelopment; combined with high-throughput screening methods, this approach can determine the disease relevance of mutations in human tissue.
-
To study specific diseases, brain organoids can be generated from induced pluripotent stem cells from individual patients, thereby preserving the specific genetic background of the individual and generating an insightful model.
-
Through recapitulation of previously inaccessible periods of human brain development, brain organoids have enabled identification of novel mechanisms that underlie neurodevelopmental, neurodegenerative and infectious diseases.
-
Combining organoids, patient research and animal models enables us to take full advantage of each of these systems and will provide unprecedented insights into neurodevelopment and neurological diseases.
Similar content being viewed by others
Introduction
Neurological diseases are the leading cause of disability and the second leading cause of death worldwide1. Preventive strategies and interdisciplinary treatment regimens are improving outcomes of neurological conditions2, but advances in treatment require accurate understanding of disease aetiology and progression. This knowledge can be acquired by studying the disease in patients and by studying in vitro and animal disease models.
Studying disease in patients — for example, through liquid biopsies, surgery or post-mortem tissue collection — mostly provides snapshots of disease development. However, disease initiation is rarely captured in such studies. Noninvasive methods, such as neuroimaging, enable prospective examination of disease traits and progression but also cannot track early pathological processes at the cellular level. For ethical reasons, randomized trials cannot be conducted to study disease initiation. Consequently, capturing early pathophysiology in patients requires time-consuming and expensive longitudinal investigations that rely on so-called experiments of nature. For example, an epidemiological study published in 2022 that provided strong evidence that the Epstein–Barr virus is the leading cause of multiple sclerosis (MS)3 required a sample size of >10 million people who were monitored over decades3. However, even such studies cannot, in isolation, demonstrate causality.
Experiments in animal models can identify disease mechanisms and prove causality, as disease onset and progression can be controlled and monitored closely, enabling robust mechanistic research. This approach has been used to demonstrate causality in the example of Epstein–Barr virus in MS4. However, translation of animal research depends on the conservation of disease processes between rodents and humans. As our understanding of the human brain and its development evolves, an increasing number of disparities between species are being uncovered that bring into question the utility of animal models. Therefore, human in vitro models could bridge the gap between research in patients and model organisms (Fig. 1). With the rise of 3D in vitro models, known as organoids, complex disease processes can now be studied in a human context.

Studies in patients (left), such as sequencing, neuropathology or patient-derived xenograft models, provide a snapshot of disease at a given time point. Furthermore, these studies are usually not started until symptoms become apparent, meaning that the earliest pathogenic processes are not captured. Noninvasive and longitudinal studies to capture these early processes require large sample sizes and a lot of time. In animal studies (right), disease initiation can be controlled, so disease initiation, pathogenesis and progression can be studied throughout the disease course. Transfer of knowledge from animal studies to humans and vice versa relies on the assumption that disease mechanisms are conserved between humans and animal models, which is not always true. 3D human model systems such as organoids could be useful for bridging this gap, as they enable studies of early disease stages in human-derived tissue.
In this Review, we discuss advances in our understanding of human neurodevelopment, how these advances influence neurological disease modelling, and how organoids can improve such modelling. We contrast the benefits and limitations of organoids with those of animal models and 2D human models, and highlight recent advances that provide an extensive toolbox for neurological research. We introduce different approaches for in vitro 3D disease modelling and address how organoids have been used to study various conditions. Finally, we illustrate how organoids could be integrated into existing research frameworks of patient and animal studies.
Neurodevelopment across species
Conserved principles
The human brain differs notably from that of common laboratory animals, such as rodents, not only with respect to overall size, but also in the contributions of different cell types and the six-layered architecture and folding (gyrification) of the cortex. Nevertheless, major events in neurodevelopment follow principles that are conserved between rodents, primates and humans.
Regardless of the species, neurons in the brain originate from a pool of neuroepithelial cells. Neuroepithelial cells divide symmetrically at first, thereby increasing the progenitor pool, before transforming into radial glia cells (RGCs), which are the neural progenitor cells of the brain5 and generate excitatory and inhibitory neurons. The number of neurons produced by RGCs during neurogenesis differs vastly depending on the species6 but adheres to some general principles.
In the dorsal forebrain, two main progenitor populations — RGCs and intermediate progenitor cells (IPCs) — are established during development (reviewed elsewhere7) and produce neurons via two mechanisms: direct neurogenesis, in which one RGC produces one neuron via asymmetric division8; and indirect neurogenesis, in which RGCs first divide asymmetrically to generate an IPC, from which two neurons are produced via symmetric division9. Apical RGCs (aRGCs) divide in the ventricular zone, whereas IPCs divide in the subventricular zone. The ratio between the two modes of neurogenesis influences the number of neurons that a progenitor pool can generate, as intermediate populations such as IPCs strongly increase the neuronal output from a limited number of RGCs10. Accordingly, indirect neurogenesis accounts for most of the neuronal production in mammals8,9 and the proportion of neurons generated in this way is greatest in large gyrated brains, such as the human brain11. Newly generated neurons migrate along the basal processes of RGCs to populate the developing cortex in an inside–out manner — the deepest layers of the cortex are produced first and upper layer excitatory neurons are produced later12.
Whereas excitatory neurons are exclusively generated in the dorsal forebrain, cortical interneurons in rodents, primates and humans are thought to derive mainly from the ganglionic eminences (GEs) in the ventral forebrain13,14. The GEs also develop from a ventricular zone, but their organization differs from dorsal areas in that they include abundant non-epithelial progenitors14. Interneurons do not ascend along RGC fibres but undergo tangential migration (orthogonal to the RGC fibres) into the cortex, populating all layers and regions13. Subregions of the GEs — the caudal GE (CGE), medial GE (MGE) and lateral GE (LGE) — generate distinct interneuron subtypes15,16 that are largely conserved at the epigenetic level across species17.
The role of dorsal regions in generating cortical interneurons in different species is unclear. In mice, dorsal progenitors are known to generate interneurons that migrate to the olfactory bulb18, but whether cortical interneurons can be generated from dorsal progenitors has long been debated19. This debate continues, as studies published in 2022 provided evidence that dorsal progenitors generate interneurons in humans20 but not in mice21. Overall, the ventral areas differ greatly from the dorsal regions in terms of organization, timing and neuron specification, adding another layer of complexity to neurogenesis.
Neurodevelopment in the primate brain
Differences between species affect cell type composition and neurodevelopment. In comparison with other animals, the primate brain contains several distinct types of progenitors and neurons (Fig. 2), individual excitatory and inhibitory lineages are amplified, and other processes inside and outside the cerebrum differ. Specific genetic changes underlie these species differences.

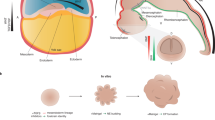
The human brain develops over a protracted period of time (centre), resulting in its complex structure. This development involves several processes that are unique to humans (parts a–e). a | Radial glial cell development. Apical radial glia cells (aRGCs, blue) are the neural stem cells that give rise to the human brain. aRGCs reside in the ventricular zone (VZ) and are connected to the ventricular surface via apical processes. At gestational week (GW) 14 (left), they pass through the cortical plate (CP) and connect to the pial surface via basal processes. Outer RGCs (oRGCs, brown) emerge in the subventricular zone (SVZ) and connect only to the pial surface via their basal process. Subsequently (at GW18, centre), the basal processes of aRGCs detach from the pial surface, and these cells become truncated radial glial cells (tRGCs). At this stage, the progenitor zone (right) is organized into a VZ that contains tRGCs, an inner SVZ (iSVZ) that contains intermediate progenitor cells (IPCs, dark yellow) and an outer SVZ (oSVZ) that contains oRGCs. Newly generated neurons (red) ascend along the basal processes of the radial glial cells towards the CP. b | Expansion of cortical layers II and III. Excitatory neurons are generated in an inside–out manner. Neurons migrate through the intermediate zone (IZ) towards the CP, which is delineated by the marginal zone (MZ) towards the pial surface. The first neurons to be generated are subplate neurons (dark blue), which form the subplate (SP). The deep SP and upper SP are formed sequentially. In humans, the SP expands greatly during development (compare GW13.5 with GW26–29) — during mid-gestation, the SP becomes larger than the CP and cortical layers I to VI combined. At later stages, an increased contribution of oRGCs to neurogenesis results in expansion of cortical layers II and III in the human brain (purple; compare GW26–29 with newborn). The SP also reduces in size and the prominent white matter (WM) emerges. c | Interneuron generation in the ventral forebrain within the ganglionic eminences. The human medial ganglionic eminence (MGE, left) contains doublecortin-positive cell-enriched nests (yellow) that contribute to neuronal production during the later stages of development. The MGE, lateral ganglionic eminence (LGE) and caudal ganglionic eminence (CGE) generate interneurons throughout neurogenesis (right) but the peak of neurogenesis in the CGE is later than in other regions and persists until the end of gestation. d | Interneuron migration to the cortex. In humans, this process persists until the first years of life, with large corridors of interneurons migrating in the so-called Arc into the forebrain (left). The proportion of interneurons in the human brain is larger than that in rodents — interneurons constitute up to 30% of all neurons in association cortices (centre), such as the prefrontal cortex (PFC), compared with around 15% in the human sensory cortices (V1) or mouse association (frontal cortex (FC)) or sensory cortices (V1)54. In addition, the contribution of CGE interneurons is greater in the human brain than in rodent brain. MGE and CGE interneurons differ in their final positioning in the cortex, with MGE interneurons (yellow) predominantly in deep and CGE interneurons (green) in the expanded upper layers (right). e | Cerebellum development. The rhombic lip (RL) generates granule cell progenitors that migrate to the external granule layer (green) and unipolar brush cells (UBCs, purple) that migrate into the cerebellar lobes. In the developing human cerebellum, the RL contains a VZ (blue) and an SVZ (red). The SVZ is established at approximately GW11, after which the RL is internalized by GW17 in humans; this internalization does not occur in other non-human primates. Part c, left panel adapted with permission from ref.53, UCSF. Part d, left panel adapted with permission from ref.282, Wiley. Part e adapted from ref.72, Springer Nature Limited.
Expansion of excitatory neurons
Neurodevelopment in primates and other gyrencephalic species relies heavily on an additional progenitor cell type that substantially increases neuron production: outer RGCs (oRGCs). oRGCs reside in the outer subventricular zone and, although their contribution to neuronal production in lissencephalic animals is negligible, they account for the majority of neurons in humans, non-human primates and some other species22,23 (Fig. 2a). In primates, the initial pool of oRGCs is expanded by a temporary shortening of the cell cycle, which results in a large population in the outer subventricular zone24. Rodents and other lissencephalic animals have very few oRGCs25,26,27, a difference that is due to several human-specific aspects of oRGC development, expansion and proliferation28,29,30,31,32,33,34,35,36.
The biology and development of oRGCs can be recapitulated in human cerebral organoids (hCOs)37. Studies of such organoids and primary tissues have revealed that the mTOR signalling pathway in oRGCs from humans differs even from that in chimpanzees and macaques35. In addition to increasing the number of neurons, oRGCs provide a scaffold for migrating newly generated neurons. In later stages of primate development, aRGCs convert to truncated radial glia (tRGCs), which form a non-continuous scaffold that primarily depends on the basal processes of oRGCs38 (Fig. 2a). Thus, oRGCs in the subventricular zone are critical for establishing the vast neuronal population of the human cortex.
In addition to the transient progenitor zones, the developing human brain contains several transient neuronal compartments that differ from those in other species. The subplate undergoes particularly dramatic dynamic changes. This region contains various neurons and glia and is involved in various processes, such as migration, maturation, axon pathfinding and circuit organization (reviewed elsewhere39). The subplate in humans expands dramatically during development in comparison with that in other species — it becomes more than fourfold thicker than the cortical plate, which is twofold thicker than in non-human primates40 (Fig. 2b).
The subplate is important in the organization of circuits that connect the two hemispheres and the cortex to the thalamus41. To establish the topological organization, corticothalamic and thalamocortical projections require the formation of transient circuits within the subplate. Spontaneous activity patterns in the subplate shape the connectivity of the cortical plate, and dysregulation of these processes has been implicated in psychiatric disease (reviewed elsewhere41). Similarly, defects of the subplate can cause structural malformations, such as agenesis of the corpus callosum42. The subplate is also crucial for migration of newly generated neurons — synaptic interactions of immature neurons with the subplate leads to their transition from multipolar to bipolar, which initiates fast migration along the RGC scaffold43. At later developmental stages, after guiding the axonal projections of the cortical plate, the subplate reduces in size and the expanded white matter that is characteristic of the human brain is formed. Thus, the subplate orchestrates the emergence and connectivity of diverse cell types in the human brain — its greater size than in other animals is necessary to support these processes.
The outer subventricular zone becomes densely populated around mid-gestation and continues to produce excitatory neurons. Consequently, the prominent outer subventricular zone in gyrencephalic species results in greater expansion of the upper cortical layers than in rodents11,44 (Fig. 2b). These neurons form intratelencephalic connections and have been linked with human-specific cognitive abilities45,46. Introduction of the human-specific gene ARHGAP11B into non-human primate fetuses increases the size of the outer subventricular zone and the numbers of neurons in the upper cortical layers while deep layers are unchanged30,32, thereby making the developmental process more similar to that in humans.
Overall, excitatory neurogenesis in the human brain involves various human-specific processes in progenitor proliferation, transient developmental structures and neuronal subtype distribution that lead to the complex connectivity of the human brain. Given that these developmental processes cannot be studied in rodents, human-derived organoids can be used to model these processes and their specialized cell types for investigation of their physiological roles and contributions to human pathology.
Generation of inhibitory neurons
In addition to differences in excitatory neuron development, the development of interneurons in the human brain differs from that in other animals (Fig. 2c,d). The protracted development of the human brain has a strong effect on the timeline of interneuron generation (reviewed elsewhere47). In mice, neurogenesis in the ventral GE regions occurs from around embryonic day 10.5 (E10.5) to E16 (refs.48,49), and the timing differs slightly between the MGE, LGE and CGE50. By contrast, ventral neurogenesis in humans peaks around mid-gestation14 and continues until the end of pregnancy — proliferation occurs in the MGE earlier than in the CGE51,52 (Fig. 2c). Differences in tissue architecture in the MGE are also important. In humans, this region contains doublecortin-positive cell-enriched nests, which are densely packed islands of immature neurons that contribute to neuron production during the second trimester, thereby increasing interneuron numbers in the human brain53 (Fig. 2c). Sustained proliferation of cells that express doublecortin suggests that, as in the dorsal forebrain, ventral areas rely on intermediate progenitor types to increase neuronal output14,53.
Interneuron identities are determined by complex genetic programmes that largely overlap in humans and mice13,17 but important differences have emerged. Some genes that are specific to interneuron subtypes in mice are less specific or not expressed in humans54; for example, the serotonin receptor subunit Htr3a, one of the main markers of CGE interneurons in mice, is not expressed by CGE interneurons in humans or other primates54,55. Conversely, in humans and other gyrencephalic animals, such as ferrets, the calcium-binding protein secretagogin is expressed by late-migrating CGE interneurons56, whereas this population is absent in rodents57,58.
Like the proliferation in the GEs, the migration of interneurons into the cortex occurs over an extended period in humans — including the postnatal period — in comparison with other animals47. Postnatal cortical interneuron migration has been observed in mice59 but to a lesser extent than in humans and other gyrencephalic animals56. In humans, interneurons migrate into the cortex from the MGE and CGE in a structure called the Arc (Fig. 2d) until well into the first year of life60. In addition, migratory streams that lead to the olfactory bulb in rodents are redirected to the cortex in humans and contribute to the diversity of cortical interneurons61.
This post-migratory cortical interneuron population differs between primates and rodents in several ways (Fig. 2d). GABAergic interneurons are more abundant in the human brain than in the mouse brain62, and even more so in areas that mediate higher cognitive function, such as association cortices; in these regions, interneurons account for up to ~30% of all neurons compared with <15% in mice54. Similarly, the distribution of interneuron subtypes differs. MGE-derived interneurons are found in deep layers across species and populate the mouse cortex uniformly, but CGE-derived interneurons are enriched in the upper layers in primates and humans (Fig. 2d), more so in areas associated with higher cognition, such as the prefrontal cortex, where they account for up to 50% of all interneurons54. In addition, interneurons in subcortical structures seem to differ, as demonstrated by the discovery of a primate-specific striatal interneuron type54.
Overall, the extended period of neurogenesis in humans influences not only progenitor organization and biology, but also the identity, composition and distribution of interneurons throughout the human brain. Use of organoids enables these unique aspects of development to be studied. For example, the migratory dynamics of interneurons over developmental time63,64,65,66 can be monitored, as can the relevance of specific cell types and their developmental trajectories to diseases such as Timothy syndrome63,67 and tuberous sclerosis68.
Human-specific cerebellar development
Human-specific developmental processes also occur in the cerebellum and are implicated in disease. The human cerebellum has a much greater surface area than that in other animals — ~750-fold greater than that in mice and tenfold greater than that in non-human primates69. The differences in size can be attributed to differences in cerebellar progenitor zones. In addition to the cerebellar ventricular zone that produces GABAergic neurons, the developing cerebellum contains a progenitor zone called the rhombic lip, the function of which differs between humans and mice. Rhombic lip progenitors generate excitatory neurons and expand into their own ventricular zone and subventricular zone in humans and non-human primates but not in mice. In a process unique to humans, the rhombic lip is also internalized after mid-gestation70,71 (Fig. 2e). This process is relevant to developmental disorders, as rhombic lip dysfunction has been associated with cerebellar vermis hypoplasia, also known as Dandy–Walker malformation71,72, and has been identified as the source of medulloblastoma group 3 and 4 — a severe childhood brain tumour73,74. Human cerebellar organoid models75 that include rhombic lip progenitors76 have recently been developed, enabling investigation of cerebellar development in humans.
The molecular basis of differences
Numerous studies have been conducted to advance our understanding of the molecular basis of the evolutionary differences that distinguish humans from other species77. Regulation of some conserved pathways is altered, resulting in changes in Robo28, mTOR35, PDGF34 and Notch signalling31, as well as morphological transitions that affect the organization of the ventricular zone and increases in progenitor numbers78. In addition, several human-specific gene duplications and variants have been identified that alter the functions of NOTCH2NL29,36, TBC1D3 (ref.33), ARHGAP11B30,32, PPP1R17 (ref.79) and human-specific enhancers, such as HARE5 (ref.80), and alter microRNA expression81,82,83, all of which are involved in progenitor expansion and regulation. Similarly, gene variants have been discovered that change neuronal maturation, spine morphology84,85 and connectivity86.
Differences between human and rodent brain development result not only from evolutionary gains in primates but also from secondary losses of acquired traits in rodents. A prominent example is cortical gyrification, which occurs in most amniotes87. Lissencephaly and reduced brain size in rodents is caused by secondary loss of miR-3607 (ref.88), which is expressed in evolutionarily more distant animals, such as ferrets, that exhibit gyrencephaly and similar neural progenitor cells to humans22,88,89. This example illustrates that diverse evolutionary mechanisms have led to the developmental differences that make us human. Organoids enable recapitulation of human developmental milestones and enable manipulation of developmental processes to understand how the complex human brain has emerged and what goes wrong in diseases of development78.
Brain organoids for modelling
Disease modelling with 3D human organoids has the potential to bridge the gap between conventional animal models and humans. Organoids have been developed for numerous organ systems (reviewed in detail elsewhere90,91) and are generally derived from pluripotent stem cells (PSCs), although some non-brain organoids can be generated from adult stem cells or fetus-derived cultures92,93. With respect to brain organoids, Sasai’s group pioneered the 3D in vitro culture of optic cup structures94,95 and cortical tissue96, and our group developed the cerebral organoid model and pioneered its use for disease modelling37. Organoids have a high degree of self-organization and contain progenitor cells and differentiated cell types. Numerous variations of these organoid models have been developed, and essentially every part of the human brain can now be recapitulated. Non-human brain organoid models exist, but we focus on hCOs.
Human brain modelling
Generation of hCOs starts with embryonic stem cells or induced PSCs (iPSCs). The stem cells aggregate into embryoid bodies, which are incubated in a medium that restricts fate to the neural lineage before being embedded into an extracellular matrix or transferred to a proliferative medium that supports progenitor expansion97. Upon formation of radially organized neural progenitors in ventricular zone-like rosettes, an organoid is established. During these initial weeks of culture, hCOs mostly comprise progenitor regions that expand symmetrically. The first neurons appear around the ventricular zone after ~20 days. Organoids can be cultivated for long periods; postnatal characteristics develop after >1 year98.
The cellular composition of hCOs is determined by the diversity of the initial progenitor pool. Unguided protocols involve no extracellular signalling molecules, resulting in a mixture of regional identities37. Guided protocols involve addition of morphogens to induce or restrict specific fates, leading to so-called restricted hCOs with more homogeneous cell populations99,100. These approaches can be used to generate different hCOs depending on the research question. Furthermore, fusion of differentially patterned hCOs into assembloids enables analysis of interactions between regions63,64, such as interneuron migration from ventral to dorsal hCOs63,64,65,66 or axonal projections101,102. Together, these possibilities result in a comprehensive toolbox for studying disease processes during human neurodevelopment (Fig. 3).

a | Organoids are generated from pluripotent stem cells, either embryonic or induced, that are grown in adherent 2D culture. These cells are aggregated in low-attachment plates to produce embryoid bodies, after which induction of neuroectoderm occurs. Organoid progenitors proliferate symmetrically in the first weeks, followed by neuron production and differentiation. During the initial culture period, organoids can be patterned to develop into representations of specific brain regions. Various protocols enable patterning for dorsal96,99,100,161,283,284,285 or ventral forebrain63,64,65, thalamus101, hypothalamus161, midbrain161,240,286, hindbrain287 and cerebellum75,76. b | Mature organoids recapitulate developmental hallmarks of the human brain, including ventricular zone (VZ) structures that contain apical radial glia, subventricular zone (SVZ) areas that contain intermediate progenitors and outer radial glia, and an emerging cortical plate (CP) that contains neurons. c | Restricted organoids can be fused to model interactions between distinct brain areas; for example, the tangential migration of interneurons from ventral to dorsal areas63,64,66,67, the striatal208 or thalamic projections101 to the cortex, hypothalamic projections to the pituitary gland288 or the connection of cortical neurons to muscle via the spinal cord289. d | To overcome difficulties such as restricted nutrient supply, sliced organoids — so-called air–liquid interface cerebral organoids — can be cultured.
The experimental timeline depends on whether the focus is on early progenitor biology78 and disease susceptibility103,104, neuronal maturation, migration and activity, or late non-neuronal populations105,106, and can last for more than 1 year. Organoids are constantly accessible during these long experiments, enabling flexible application of compounds66,107, modification of culture conditions107,108 and/or genetic perturbations109.
Approaches to disease
Organoids can be used to study disease processes in two main ways. The first is to study the effects of known risk factors for diseases. Widespread use of diagnostic genomic sequencing and the rise of genome-wide association studies have identified a plethora of genetic variants that are associated with diseases, including structural brain defects, such as microcephaly110, and neurodevelopmental disorders, such as autism spectrum disorders (ASD)111. Organoids can be used to investigate the causal relationships between these risk variants and defined cellular phenotypes. Furthermore, generating organoids from patient-derived iPSCs enables replication of the patient-specific genomic background.
The second use of organoids to study disease is to investigate the mechanisms that underlie development of diseases with a known cause. For example, many monogenetic diseases have been identified, including early infantile epileptic encephalopathies112,113, neurocutaneous syndromes114, lysosomal storage disorders115 and neurodegenerative disorders116, but the link between developmental processes and phenotypes often remains poorly understood. Organoids can be used to dissect the mechanisms of pathogenic mutations in human tissue (Fig. 4). Similarly, organoids can be used to screen potential therapeutic agents.

a | Prenatal Zika virus infection causes microcephaly (left), characterized by a drastic reduction in brain size and head circumference. Studies in organoids have revealed the mechanisms involved (right). In healthy organoids (top), radial glia cells (RGCs) in the ventricular zone (VZ) are radially organized and have tight apical junctions (AJ; blue ovals represent nuclei of RGCs for which the cell body is not shown for clarity). In organoids infected with Zika virus, AJs between RGCs are destroyed and centrosome errors occur, leading to disruption of the VZ. Apoptosis of RGCs accounts for the reduced neuronal output and the size defect. b | Organoids can be generated from patients with familial Alzheimer disease (AD), sporadic AD or Down syndrome. AD pathology — including amyloid-β (Aβ) plaques, tau tangles and enlarged early endosomes — develops in these organoids and neuronal apoptosis is increased. This pathology develops more quickly in organoids derived from people with familial AD or Down syndrome (2 months) than in organoids derived from people with sporadic AD (6 months). These organoid models are starting to incorporate interactions of neurons with microglia and the blood–brain barrier (BBB), which could provide further insights into disease mechanisms. c | Frontotemporal dementia with tau pathology (FTD-tau) is caused by mutations in the MAPT gene, which encodes tau. In organoids derived from people with FTD-tau, dysfunction of the autophagy–lysosomal pathway (ALP) occurs early and the excitatory lineage splicing regulator ELAVL4 co-localizes with tau in stress granules, resulting in splicing dysfunction, aberrant development of the excitatory lineage and consequent neuronal dysfunction, excitotoxicity and apoptosis. d | Timothy syndrome is a neurodevelopmental disease caused by mutations in the CaV1.2. Work on assembloids of ventral and dorsal organoids derived from patients with Timothy syndrome revealed inefficient saltatory migration movements of interneurons. Increased Ca2+ influx via the mutant CaV1.2 channel altered the cytoskeleton to reduce the length of saltatory movements and remodelled GABA receptors to increase the frequency of saltatory movements. e | In Angelman syndrome, loss of the UBE3A gene that encodes a ubiquitin protein ligase leads to accumulation of big potassium channels in neurons (top). 2D and organoid experiments have shown that neurons in Angelman syndrome have increased fast components of the after hyperpolarization (fAHP, middle). In organoids derived from patients with Angelman syndrome, synchronicity of calcium events was greater than in organoids from healthy people (bottom). f | Organoids derived from people with tuberous sclerosis complex (TSC) recapitulated the tuber and tumour phenotypes and demonstrated that abnormalities of caudal late interneuron progenitor (CLIP) cells underlie the disease. CLIP cells are vulnerable to heterozygous TSC2 mutations, which leads to their over-proliferation that initiates formation of tubers (dysmorphic interneurons (IN) and giant cells) or tumours. Part f adapted with permission from ref.68, Kelli Holoski.
Assessing disease traits with organoids
Major structural defects such as microcephaly were the first developmental disorders to be studied in organoids37. The cardinal symptom of primary microcephaly (referred to as microcephaly primary hereditary (MCPH)) is a small head circumference117. The condition is caused by various genes, the functions of which converge on several common pathways, such as DNA replication and repair and centrosome biology110. Our understanding of how associated gene alterations cause MCPH has relied heavily on mouse models, which have revealed changes to important aspects of basic neurodevelopment (reviewed elsewhere118). For example, mice with mutations in Mcph1, the first gene to be associated with MCPH119, have a smaller cerebral cortex than wild-type mice120,121, and the model revealed a premature switch from symmetric to asymmetric cell division, which reduces the initial aRGC pool size and underlies the phenotype121.
Some genes associated with MCPH, including CDK5RAP2 and ASPM, have been linked to the evolutionary expansion of the brain in primates122,123, indicating that not all MCPH-related genes are entirely conserved between humans and rodents. Cdk5rap2-knockout mice do have a smaller brain but the phenotype varies between mouse strains124. By contrast, CDK5RAP2 mutations in hCOs recapitulate MCPH-like effects and have demonstrated that the size of the progenitor pool is reduced owing to premature neurogenic divisions37. Similarly, ASPM mutation, which is the most common cause of MCPH, results in drastic brain size reductions in humans125 but causes only a moderate126,127,128 and variable129 phenotype in mice. This disparity could result from differences in expression of ASPM in the subventricular zone between mice and humans, making the rodents less susceptible to the mutation118. Accordingly, ferrets, which have a prominent subventricular zone similar to that in humans, develop microcephaly upon ASPM mutation130. Features of microcephaly also developed in hCOs that were generated from iPSCs from patients with ASPM mutations131. These findings demonstrate that when mutations cause disease through processes that are not conserved between humans and rodents, alternatives to mouse models are needed.
In a study published in 2020, organoids were used to assess the effects of 172 MCPH-associated genes109. The methodology developed for this study enables loss-of-function analysis for multiple genes in parallel. Instead of developing one organoid model per gene, mosaic organoids were created in which one gene per cell was mutated using the CRISPR–Cas9 system. Each individual starter cell was also uniquely barcoded so that the number of daughter cells generated could be measured. This enabled measurement of the effect of each MCPH-associated gene on proliferation in the organoid. This approach demonstrated that 32 genes caused a reduction in proliferation, consistent with the reduced proliferation during brain development seen in microcephaly, and their effects were independently validated in homogeneous, single-gene knockout organoids by investigating lineage development and organoid size. Phenotypic patterns differed between variants, and the pathways affected included some that have previously been associated with microcephaly and some novel pathways. One novel pathway identified regulates brain size in association with endoplasmic reticulum stress and caused microcephaly through dysregulation of extracellular matrix proteins109.
This study showed that the effects of genes associated with a defined developmental phenotype can be efficiently screened through use of hCOs. This method can be used to address a wide range of underlying mechanisms and pathways. It can also establish causality between risk genes and disease phenotypes, which is critical not only for advancing diagnosis and treatment but also for future mechanistic studies.
Low-risk and unknown variants in organoids
ASD is highly heritable but has complex genetic causes (reviewed elsewhere132). Other than in ASD-related monogenetic syndromes, high-risk de novo mutations have only a small role in the disease133; instead, an individual’s risk is usually determined by a combination of multiple common and new variants. Over 100 genes have been associated with ASD, many of which converge on pathways that regulate transcription or synaptogenesis130. In this context, the use of organoids generated from patient-derived iPSCs provides a major advantage over animal models and the study of single risk gene knockouts in organoids134, as it enables investigation of the effects of numerous combined, low-risk mutations, some of which might not be known, while accounting for genetic background.
One such study has been done to investigate morphological and transcriptomic abnormalities in organoids generated from iPSCs from four patients with ASD and their neurotypical relatives135. Each patient had severe idiopathic ASD with macrocephaly without a known causal genetic mutation. However, the organoid study revealed an imbalance of excitatory and inhibitory neurons and synapses in all four. This imbalance has previously been proposed as a mechanism of ASD136 and is supported by computational137 and transcranial magnetic stimulation studies138 in patients.
A similar approach was used with organoids derived from patients with ASD and healthy relatives to identify and compare neurodevelopmental pathophysiological processes in ASD with and without macrocephaly. The disease mechanism differed between patients with and without macrocephaly but converged within each cohort despite distinct individual genetic backgrounds139. In ASD with macrocephaly, progenitor proliferation and excitatory cortical plate neurons were increased, whereas increases in the early-generated preplate neurons (the opposite pattern) were seen in ASD without macrocephaly. Nevertheless, some alterations were shared between the two ASD cohorts, mostly in the regulation of oRGCs. These studies demonstrate that the use of organoids to model the combination of risk variants within individuals can identify convergent pathways involved in pathogenesis. The findings have revealed that the pathways involved in ASD are important in processes that are unique to or amplified in humans, demonstrating the value of human model systems.
In addition to studying patient-specific phenotypes, understanding the role of risk genes in healthy neurodevelopment is crucial for identifying the cell types that are affected in disease. Transcriptomic analysis of brains from patients with ASD have revealed that processes in the mid-fetal period underlie ASD pathogenesis140,141, thereby narrowing the set of genes that could be involved to those that are expressed during this window. Large-scale efforts have generated databases of somatic mutations142, gene expression143 and epigenomics144,145 in brain development146 and disease147, and these resources are invaluable for determining the temporal window during which risk genes are expressed. Use of high-throughput single-cell RNA (scRNA) sequencing techniques with tissue from people with ASD has identified cell type-specific effects on upper layer projection neurons148. hCOs can be used to investigate neurodevelopment in vitro and complete the developmental timeline of cell-type specific processes149. Numerous studies have been done to examine single-cell gene expression and chromatin accessibility in hCOs35,99,139,145,149,150,151,152,153,154. Importantly, organoids enable very dense sampling over time to understand the temporal dynamics of risk gene expression as well as cell type specificity. Identification of sensitive periods for different cell types can facilitate the development of aetiological theories155. Coupling of high-throughput screening technologies with in vitro single-cell profiling in organoids promises to provide even greater insight into the effects of risk variants.
Disease mechanisms in organoids
In addition to studying the relevance of risk genes to known phenotypes, organoids can be used to investigate novel disease mechanisms (Table 1). Access to and the ability to manipulate human-specific cell types during development can provide substantial insights; in the following sections, we discuss examples of such insights into virus-related brain diseases, neurodegenerative disease and several genetic neurodevelopmental syndromes.
Virus-associated microcephaly
Brain organoids are an excellent tool for evaluating vulnerability to viral diseases during neurodevelopment, demonstrated by the rapid emergence of insightful models of Zika virus-associated microcephaly during the 2015 outbreak in Latin America156,157. Maternal Zika virus infection was quickly linked to an increase in primary microcephaly, supported by the detection of virus in the amniotic fluid158 and the brain of a microcephalic fetus159. Mouse and 2D and 3D human models were soon established to study how Zika virus infection causes microcephaly103,160,161,162,163,164,165,166,167,168.
Neural progenitors in 2D culture were easily infected and their growth was reduced163, and the concept that Zika virus targets neural progenitors was later confirmed by infection of fetus-derived neural progenitor cells but not neurons168. However, microcephaly-like phenotypes require a 3D architecture and could only be recapitulated by infecting organoids103,160,161 (Fig. 4a). The virus-induced phenotype was specific to Zika virus; infection with Dengue virus103, another member of the flavivirus family, and lymphocytic choriomeningitis virus165 did not reproduce the effect. Progenitors in the ventricular zone and subventricular zone were affected in these models, supporting the hypothesis that transient amplified populations of human neurodevelopment are involved in the disease161,165,166,168. Organoids have been used to study the entry of Zika virus into neural progenitors and the mechanisms by which infection leads to microcephaly.
Use of organoids also helped to reveal how Zika virus affects RGCs, which proliferate less, produce fewer neurons and undergo apoptosis after infection103,160,161, resulting in destruction of the ventricular zone architecture and the RGC scaffold168. Studies of organoid and mouse models demonstrated that the Zika virus protein NS2A is responsible, as it disrupts the apical junction of RGCs, explaining the tissue architecture disturbance167. Furthermore, studies in organoids and fetal brain slices also suggested that centrosome damage occurs in Zika virus-associated microcephaly165,168, which is a common mechanism in MCPH110. These studies demonstrate how organoids can be used to replicate complex tissue phenotypes that occur as a result of viral infection and to provide mechanistic insight into the effects of infection.
Use of organoids also overcame deficiencies in mouse and 2D models of Zika virus infection. In humans, but not in mice, Zika virus suppresses type I interferon (IFN) responses, meaning that ablation of IFN signalling is needed to assure infection in most mouse models169. Furthermore, in human models, Zika virus attenuation of type I IFN is considerably stronger in organoids than in 2D culture, which could explain the discrepancies in infection rates between these models and shows the value of the 3D system104. Organoids can also shed light on differences between neurotropic viruses. For example, infection of organoids with Zika virus or herpes simplex virus (HSV-1) revealed that both reduced type I IFN signalling but that distinct interferons mediated the IFN response in Zika virus and HSV-1 infection104.
Thus, hCOs can be used to study the cellular mechanisms, dynamics, and outcomes of CNS infections, further demonstrated by studies of SARS-CoV-2 (refs.170,171,172) and cytomegalovirus infection173 in the past 2 years. 3D models recreate the in vivo infection more faithfully than 2D models, and enable investigation of the mechanisms in a tissue-like context. Furthermore, the cellular diversity and scalability of organoids enable evaluation of several viruses in different cell types at the same time.
Alzheimer disease
Alzheimer disease (AD) is the most common neurodegenerative disorder. The neuropathological hallmarks are amyloid-β (Aβ) plaques and neurofibrillary tangles that contain hyperphosphorylated tau174. Sporadic AD is most common, but familial AD can occur, usually as a result of mutations in or duplications of APP, which encodes amyloid precursor protein (APP), or mutations in PSEN1 or PSEN2, which encode the core proteins of secretases that cleave APP to release Aβ peptides. In addition, people with Down syndrome, that is caused by an additional copy of chromosome 21, are at high risk of AD because APP is located on chromosome 21. The neuropathology of AD has been studied extensively in mouse models, but many of these models do not capture all aspects of AD or require mutations in several genes to recapitulate the full phenotype175,176. Human-derived models are, therefore, essential for a full understanding of the pathophysiology.
The first human-derived models of AD pathogenesis were neurons that had been differentiated in 2D from iPSCs of people with sporadic AD, familial AD177 or Down syndrome178,178. In these neurons, pathogenic Aβ, tau and endosome abnormalities were increased. From this system, a 3D model was developed, in which the development of Aβ and tau pathology was faster179. In subsequent studies, organoids derived from iPSCs of people with familial AD spontaneously developed pathology without overexpression180,181 (Fig. 4b). Inhibition of the secretases reversed pathology in 2D models177,178 and 3D models179,180,182 in a time-dependent manner. The 3D models of familial AD have, therefore, enabled more accurate modelling of Aβ and tau pathology, and the use of iPSCs from people with familial AD and Down syndrome enables generation of patient-specific models177,178,179,180,181,183.
Beyond familial AD, several risk genes have been identified in sporadic AD, and organoids can be used to study the role of these genes in AD pathophysiology. Many of the risk genes identified are expressed in microglia184,185, and the importance of microglia in AD pathology is supported by evidence of neuroinflammation in a 3D culture model that combined neurons, astrocytes and microglia186. The strongest risk factor for sporadic AD is the APOE*ε4 allele, and in 2D and organoid models that included microglia, Aβ deposits were cleared less efficiently by microglia that expressed APOE*ε4 than by those that expressed APOE*ε3 (ref.187). Interestingly, expression of APOE*ε4 in cerebral organoids resulted in development of AD pathology at a later stage (6 months)187 than did APP duplication or mutation of PSEN1 or PSEN2 (2 months)180, suggesting that different pathogenesis time lines can be replicated.
The brain vasculature is another non-neuronal player in AD pathogenesis, and single-cell profiling of vasculature from people with AD has shown that AD risk genes are abundantly expressed in brain endothelial cells in humans but not in mice188. Vascular cells can be incorporated into organoids, enabling investigation of some blood–brain barrier properties in the context of Aβ pathology189. However, the lack of functional vascularization in organoids remains a major limitation of current studies (Fig. 4b). Nevertheless, existing models — especially 3D tissue models — have provided important insights into AD pathology.
Other neurodegenerative diseases
Although tau tangles are a hallmark of AD, mutations in MAPT, which encodes tau, can cause frontotemporal dementia (FTD-tau)190. This disease is heterogeneous, but dysregulation of glutamatergic signalling from excitatory neurons is thought to cause the symptoms191. 2D models of neurons derived from iPSCs of people with a MAPT mutation have identified impaired cytoskeletal remodelling of the axon initial segment, which results in activity changes192. In addition, dysregulation of the autophagy–lysosomal pathway has been identified in FTD-tau193. However, 2D models could not disentangle the developmental regulation of these processes. In a study published in 2021, a comprehensive brain organoid model for FTD was developed by use of several patient iPSC lines and isogenic controls194, and this model revealed a selective loss of excitatory neurons in the late stages of development. This loss was initiated by disruption of the autophagy–lysosomal pathway, which promoted tau aggregation. Furthermore, expression of the splicing regulator ELAVL4 was increased and co-localized with tau in stress granules, resulting in splicing dysregulation and impaired excitatory neuron function. This dysregulation of excitatory neuron development resulted in excitotoxicity and apoptosis in the late stages of development (Fig. 4c). Thus, the extended duration of organoid development enabled the different steps in the development of FTD-tau pathology to be identified.
Organoids have also been used to model other neurodegenerative disorders, including Parkinson disease195,196,197 and Creutzfeldt–Jakob disease198 (Table 1). Despite the fact that symptomatic neurodegenerative diseases generally occur in older people, organoids can be used to study the early pathogenic processes. More complex systems that include microglia and vasculature will enable more accurate modelling of the neuroinflammatory aspects of these diseases. Final disease stages are unlikely to be replicated in organoid models, but their use to uncover early disease processes could lead to new approaches for drug development182,199.
Neurodevelopmental syndromes
In contrast to neurodegenerative disorders, most neurodevelopmental syndromes are initiated during prenatal development. For many of these syndromes, single, causal gene mutations have been defined; the combination of these mutations and iPSC and organoid modelling allows powerful mechanistic studies, as organoids can recapitulate the emergence of phenotypes during neurodevelopment in a human system. Fascinating work of this kind has been done in diverse diseases, such as neurocutaneous syndromes (neurofibromatosis200,201 and tuberous sclerosis complex (TSC)68,202), DiGeorge syndrome203, genetic lissencephaly (Miller–Dieker syndrome204,205 and Seckel syndrome206), fragile X syndrome207, Phelan–McDermid syndrome208, Timothy syndrome63,67 and Angelman syndrome209 (Table 1). In the following sections, we focus on three disorders in which organoid studies have led to new aetiological theories: Timothy syndrome, Angelman syndrome and TSC.
Timothy syndrome
Timothy syndrome is caused by an autosomal dominant mutation in CACNA1C, which encodes the α-subunit of the L-type calcium channel CaV1.2 (ref.210). In mice, high expression of the mutant channel is lethal, but low expression211 leads to ASD-like characteristics, although the model did not replicate severe cardiac disease and other symptoms associated with Timothy syndrome. This limitation of the mouse model prompted investigation of CACNA1C mutations in a human context.
In a 2D model of neurons derived from iPSCs of patients with Timothy syndrome, CaV1.2 mutation resulted in a loss of channel inactivation and increased intracellular calcium212. Furthermore, work in 2D human models and in mice showed that the mutation leads to an activity-dependent but calcium-independent retraction of dendrites213. In addition to their functional and morphological roles, L-type calcium channels have been implicated in regulation of tangential migration214, and assembloids of ventral and dorsal organoids have been used to study this migration in Timothy syndrome. Interneuron migration follows a saltatory movement pattern that is preserved in these assembloids, enabling modelling of migration in disease63,64,66. In Timothy syndrome, this assembloid model revealed a cell-autonomous decrease in the length of individual saltations and an increase in the frequency of saltations63. Use of these assembloids also demonstrated that the migratory defects are mediated by two different pathways: saltation length is controlled by L-type calcium channel-dependent regulation of the actin skeleton via calcium, whereas saltation frequency is modulated by GABA receptor signalling67 (Fig. 4d). Previous use of organoids to study the effects of neurotransmitter signalling on interneuron migration has identified similar effects of GABA receptor inhibition66, emphasizing the robustness of the model across laboratories.
This work demonstrates that disease-relevant phenotypes of interneuron migration can be studied by using patient iPSCs to generate assembloids. The accessibility and high-throughput nature of organoid cultures enables application of drugs and changes in the external environment (for example, by changing media composition), ultimately providing a platform to generate mechanistic insights into a range of pathways.
Angelman syndrome
Angelman syndrome is a rare genetic disorder characterized by intellectual deficits and epilepsy, among other symptoms215. It is caused by loss of function of the imprinted UBE3A gene, which encodes E3 ubiquitin protein ligase that ubiquitinates proteins for subsequent degradation. The loss of function can be caused by deletions or large mutations of the maternal allele, paternal uniparental disomy or imprinting defects, and leads to defects in ion channel processing. The imprinting that results in maternal-only expression of UBE3A is unique to the brain216 and linked to the neuron-specific expression of a neighbouring gene217.
Several mouse models have been generated to advance our understanding of the disease mechanisms, and model animals have motor dysfunction and susceptibility to seizures218. An underlying excitatory–inhibitory imbalance was identified219, with specific defects in interneurons220,221. In neurons derived from iPSCs of people with Angelman syndrome, parental imprinting is established during in vitro development, paving the way for human models222. In one such model, maturation of mutated 2D differentiated neurons was delayed223. Thus, mice and 2D models enabled several aspects of the disease to be studied, but how mutations in UBE3A cause these phenotypes remained unclear. Accumulation of UBE3A substrates was proposed as the mechanism, although a single responsible protein had not been identified224.
Subsequent work in which organoids were used in combination with 2D and mouse models of Angelman syndrome has identified the responsible protein209. Comparison of 2D-differentiated neurons that derived from healthy people and people with Angelman syndrome revealed an increase in the magnitude and duration of the fast component of after-hyperpolarization of the action potential (Fig. 4e). This difference was caused by elevated expression of voltage-dependent big potassium (BK) channels in neurons from patients with Angelman syndrome. BK channels were shown to be a substrate of UBE3A, and deletion or point mutation of UBE3A decreased degradation of these channels. Studies in organoids reproduced these findings and enabled calcium imaging, which revealed increased synchronicity of neuronal activity in Angelman syndrome. Inhibition of BK channels with paxilline reversed all these phenotypes in organoids. Furthermore, the findings in human models led to the demonstration that increased BK channel currents are conserved in Angelman syndrome mouse models, and that paxilline treatment could ameliorate seizure susceptibility in these mice. Overall, this study demonstrates how 2D and organoid models can be used to uncover mechanistic targets of disease and how these findings can be validated in mouse models if mechanisms are conserved.
Tuberous sclerosis
TSC is caused by mutations in TSC1 or TSC2, which encode the proteins hamartin and tuberin that together regulate cell growth. The disease is characterized by tumours and lesions in multiple organs225. In the brain, tumours develop near the lateral ventricle wall as subependymal nodules that can progress into subependymal giant cell astrocytomas (SEGAs), and tubers — local disorganized regions with dysplastic cells that are thought to cause epilepsy — develop in the cortex. Tumours and tubers develop during prenatal development226. Use of organoids has demonstrated that human-specific processes underlie the pathogenesis of TSC68.
Heterozygous mouse models of TSC lack morphological abnormalities227 but homozygous deletion of Tsc1 or Tsc2 is lethal. By contrast, inducible knockouts of Tsc1 or Tsc2 in mice can replicate several characteristics of the disease, such as hyperexcitability228, nodular tumours and dysmorphic cells229,230,231,232. This discrepancy in phenotypes led to the assumption that TSC is caused by loss of heterozygosity (LOH) — that is, loss of the healthy allele — in a two-hit process225. Furthermore, in a human spheroid model of TSC that was induced to form only dorsal forebrain tissue, LOH was also required for excitatory neuron dysregulation, similar to the findings in mice202. However, although LOH is common in SEGAs, it is rare in cortical tubers233. Thus, how pathogenesis could involve heterozygous cells remained unclear209. One possibility was that disease initiation in heterozygous cells involves cell types that are not present in the dorsal forebrain.
In a study published in 2022, we generated unguided organoids from multiple patients with TSC and isogenic controls68 (Fig. 4f). In line with previous findings, heterozygous organoids remained unchanged during early stages of development. However, at later stages, tumour lesions developed from heterozygous interneuron progenitors, revealing the disease-initiating population. When grown in different culture conditions, organoids derived from patients with TSC could also recapitulate cortical tuber phenotypes68. Subsequent scRNA profiling and comparison with sequencing data from fetal tissue indicated specific dysregulation of CGE interneuron progenitors known as caudal late interneuron progenitors, or CLIP cells, during mid-gestation as the underlying cause of the phenotypes. CLIP cells generate tumours and give rise to dysmorphic interneurons and so-called giant cells that initiate tuber pathology. Both lesions arise from heterozygous cells, as CLIP cells are vulnerable to heterozygous TSC2 mutations. Only in tumour lesions is the second allele lost through copy-neutral LOH as the disease progresses. Excitatory dysmorphic neurons emerge later than dysmorphic interneurons, indicating that they are secondary effects of the presence of dysmorphic interneurons68.
Thus, the TSC organoid model explains the differences in mutational profiles between tumours and tubers but also establishes that both derive from CLIP cells. This vulnerable disease-initiating population could only be studied with a human-based model that recreates the protracted development of the human brain with its unique cell types.
Current limitations of organoid models
The value of organoid models for investigating human brain development and disease has been demonstrated in numerous studies. Nevertheless, several limitations of these models must be considered. Organoids grow quickly during lengthy periods of culture and their size exceeds the limit for passive diffusion of oxygen, causing formation of necrotic tissue in the core. Whether this compromises the overall utility of the model151 or merely affects individual cells remains under debate98,234. Meta-analysis of scRNA data235 and multi-omics data154 suggest that in vitro conditions create an artificial stressed state in a defined set of cells while the remaining tissue remains unaffected. To improve nutrient supply, vascularized organoids have been developed with co-culture236,237, induction189 and transplantation238 methods, and cultured organoid slices have also been used102,239. However, these techniques currently lack the scalability to replace regular organoid cultures and require improvements.
Besides improving culture conditions, a major focus in recent years has been to create increasingly precise organoid models of specific brain areas. Protocols have been developed for models of the forebrain, midbrain197,240, spinal cord241, cerebellum75,76 and other brain regions242. These approaches are likely to improve the reproducibility and robustness of models by minimizing the inherent variability of the progenitor pool. However, disease modelling with restricted organoids can only recreate phenotypes in these constrained regions, necessitating careful experimental planning.
A major advantage of human in vitro models is that they replicate human-specific cell types. Currently, however, brain organoids are largely made up of neural tissue and lack non-neuronal cell types. One such cell type is oligodendrocytes, which myelinate axons in the CNS. These cells differentiate in distinct developmental stages from RGCs in the human brain243 and have complex roles in additional processes, such as interneuron migration244. Oligodendrocytes can be induced and matured in organoids, but specific culture conditions are required105,106,245. Consequently, these cells are rare in standard protocols. They can form spontaneously in organoids246, although controlled mixing procedures provide greater control over the ratio of cell types237,247,248,249. Methods for generating holistic 3D cultures that have the same high throughput as traditional methods are required to build more precise models and to study processes such as demyelination and neuroinflammation. Such approaches could also increase the lifetime and maturity of in vitro cultures.
Organoids are electrically active and can even reproduce complex functional networks and disease phenotypes209,250,251,252, so have been used to study activity-related processes. However, one of the most important limitations of organoids is that the tissue architecture and organization that influence activity patterns in vivo are missing. This limitation is illustrated by comparing organoids with normal cortical architecture. The markers expressed by neurons that are generated in organoids recapitulate the serial specification of layer identity in excitatory neurons and the distinct fates of upper-layer and deep-layer neurons are established, but their spatial organization does not recapitulate that in the brain. Separation of layer markers was improved in one study published in 2020 (ref.239) but the fine-tuned architecture of the six-layered human cortex is still far from being recreated. Similarly, although distinct brain regions can be produced and fused in vitro, they do not replicate the intricate arrangement of the human brain and therefore remain a reductionist model. Overall, organoids are a powerful tool for neurological research when used appropriately, but require rigorous evaluation and validation with in vivo material to realize their full potential.
Organoids in neurological research
Human 3D organoid models have contributed to advances in our understanding of human brain development and neurological diseases. Considering the benefits and limitations of these model systems, organoids will undoubtedly become an important tool for neurological research, especially when paired with other approaches (Fig. 5). Close collaboration with clinical experts is required to ensure that the versatility possible with gene editing in organoids and iPSC-derived models is used to capture medically relevant information. For example, organoids can be used to explore the effects of disease-associated gene variants in the context of specific cellular phenotypes. Ultimately, this application could directly benefit patient care by providing information about the disease risk associated with specific traits. Thus, existing and future novel organoid disease models could be used to assess and inform the relevance of new variants.

Close collaboration with medical specialists is required to increase the accuracy of organoid models (step 1). Patient research is the foundation for developing organoid models (step 2). Identification of disease-associated genes enables these genes to be screened in organoids to inform disease risk and provide insights into disease mechanisms (step 3). Identification of causative genes enables development of accurate screening platforms. When these genetic mechanisms are conserved in rodents, genetic mouse models can be developed to perform in vivo experiments (step 4). However, induced pluripotent stem cells (iPSCs) from patients can also be used to establish patient-derived organoids (step 5). These models can be used for drug testing (step 6) that leads to improved therapies. Cells from these organoid models can also be transplanted (step 7) into animals for in vivo evaluation of human cell types (step 8). All of these organoid-based models can provide insights into disease mechanisms (step 9). The increased understanding of disease and improvements in therapies that result feed back into patient care and patient research.
Accurate disease models are needed to identify patient-relevant phenotypes and pathways, and patient-specific organoid models can facilitate development of such models. As in the studies of MCPH discussed above, use of such models to understand the effects of single mutations37 can inform strategies to screen for other risk genes109. This approach will be important for establishing powerful new screening models. In the case of conserved developmental processes, the impact of a mutation can be reliably investigated in vivo in genetically engineered mouse models, but combining insights from human organoid models and mouse models enables more reliable and robust modelling of conserved processes.
Transplantation of organoid-derived cells into rodent brains is another excellent tool for determining the in vivo effects and clinical importance of specific human cell types. Organoids can be used to study human-specific cell types during development, and orthotopic transplantation could enable studies of in vivo processes and intricate cell type interactions. The combination of patient-derived organoids, risk gene assessment and additional models, such as transplantation models, will be powerful for studying disease mechanisms. However, development of accurate aetiological theories and improvements in treatment necessitate crosstalk with patient research. Investigation of disease in patients with noninvasive approaches, sequencing, neuropathological studies and patient-derived in vitro or xenograft models is invaluable for elucidating acute disease processes. These methods can provide insights into the disease process in people, although mostly catch snapshots of disease progression. By contrast, organoids are excellent for studying disease initiation and early pathophysiology. Only by combining these approaches will we be able to ensure that organoid research focuses on disease-relevant developmental processes and enrich our understanding of neurological disease.
Conclusion
Brain organoids enable the generation and perturbation of human tissue in vitro, providing an unprecedented opportunity to decipher neurological diseases. Such models are becoming increasingly relevant as our understanding of the unique processes that contribute to brain development in humans increases. Through analyses that would not be possible in animal models or in patients, human in vitro models have provided insights into previously inaccessible stages of development, leading to groundbreaking studies of neurodevelopmental, neurodegenerative and infectious disorders that have showcased the versatility of organoid models. We anticipate that the development of more complex organoid systems in the coming years, together with patient research and animal models, will further advance our understanding of disease development and ultimately help to improve patient treatment.
References
GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 459–480 (2019).
Feigin, V. L. et al. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol. 19, 255–265 (2019).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Lanz, T. V. et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 603, 321–327 (2022).
Rakic, P. Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735 (2009).
Otani, T., Marchetto, M. C., Gage, F. H., Simons, B. D. & Livesey, F. J. 2D and 3D stem cell models of primate cortical development identify species-specific differences in progenitor behavior contributing to brain size. Cell Stem Cell 18, 467–480 (2016).
Cárdenas, A. & Borrell, V. Molecular and cellular evolution of corticogenesis in amniotes. Cell Mol. Life Sci. 77, 1435–1460 (2020).
Noctor, S. C., Martínez-Cerdeño, V., Ivic, L. & Kriegstein, A. R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136–144 (2004).
Haubensak, W., Attardo, A., Denk, W. & Huttner, W. B. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl Acad. Sci. USA 101, 3196–3201 (2004).
Kowalczyk, T. et al. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb. Cortex 19, 2439–2450 (2009).
Kriegstein, A., Noctor, S. & Martínez-Cerdeño, V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci. 7, 883–890 (2006).
Rakic, P. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science 183, 425–427 (1974).
Ma, T. et al. Subcortical origins of human and monkey neocortical interneurons. Nat. Neurosci. 16, 1588–1597 (2013).
Hansen, D. V. et al. Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat. Neurosci. 16, 1576–1587 (2013).
Fogarty, M. et al. Spatial genetic patterning of the embryonic neuroepithelium generates GABAergic interneuron diversity in the adult cortex. J. Neurosci. 27, 10935–10946 (2007).
Xu, Q. Origins of cortical interneuron subtypes. J. Neurosci. 24, 2612–2622 (2004).
Luo, C. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017).
Kohwi, M. et al. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. J. Neurosci. 27, 6878–6891 (2007).
Letinic, K., Zoncu, R. & Rakic, P. Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649 (2002).
Delgado, R. N. et al. Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 601, 397–403 (2022).
Bandler, R. C. et al. Single-cell delineation of lineage and genetic identity in the mouse brain. Nature 601, 404–409 (2022).
Fietz, S. A. et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 13, 690–699 (2010).
Hansen, D. V., Lui, J. H., Parker, P. R. & Kriegstein, A. R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561 (2010).
Betizeau, M. et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 80, 442–457 (2013).
Kalebic, N. et al. Neocortical expansion due to increased proliferation of basal progenitors is linked to changes in their morphology. Cell Stem Cell 24, 535–550.e9 (2019).
Shitamukai, A., Konno, D. & Matsuzaki, F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695 (2011).
Wang, X., Tsai, J.-W., LaMonica, B. & Kriegstein, A. R. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–561 (2011).
Cárdenas, A. et al. Evolution of cortical neurogenesis in amniotes controlled by robo signaling levels. Cell 174, 590–606.e21 (2018).
Fiddes, I. T. et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356–1369.e22 (2018).
Florio, M. et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347, 1465–1470 (2015).
Han, S. et al. Proneural genes define ground-state rules to regulate neurogenic patterning and cortical folding. Neuron 109, 2847–2863.e11 (2021).
Heide, M. et al. Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset. Science 369, 546–550 (2020).
Ju, X.-C. et al. The hominoid-specific gene TBC1D3 promotes generation of basal neural progenitors and induces cortical folding in mice. eLife 5, e18197 (2016).
Lui, J. H. et al. Radial glia require PDGFD–PDGFRβ signalling in human but not mouse neocortex. Nature 515, 264–268 (2014).
Pollen, A. A. et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell 176, 743–756.e17 (2019).
Suzuki, I. K. et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell 173, 1370–1384.e16 (2018).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Nowakowski, T. J., Pollen, A. A., Sandoval-Espinosa, C. & Kriegstein, A. R. Transformation of the radial glia scaffold demarcates two stages of human cerebral cortex development. Neuron 91, 1219–1227 (2016).
Kostović, I. The enigmatic fetal subplate compartment forms an early tangential cortical nexus and provides the framework for construction of cortical connectivity. Prog. Neurobiol. 194, 101883 (2020).
Kostovic, I. & Rakic, P. Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J. Comp. Neurol. 297, 441–470 (1990).
Molnár, Z., Luhmann, H. J. & Kanold, P. O. Transient cortical circuits match spontaneous and sensory-driven activity during development. Science 370, eabb2153 (2020).
Doyle, D. Z. et al. Chromatin remodeler Arid1a regulates subplate neuron identity and wiring of cortical connectivity. Proc. Natl Acad. Sci. 118, e2100686118 (2021).
Ohtaka-Maruyama, C. et al. Synaptic transmission from subplate neurons controls radial migration of neocortical neurons. Science 360, 313–317 (2018).
Jabaudon, D. Fate and freedom in developing neocortical circuits. Nat. Commun. 8, 16042 (2017).
Marín-Padilla, M. The mammalian neocortex new pyramidal neuron: a new conception. Front. Neuroanat. 7, 51 (2014).
Silbereis, J. C., Pochareddy, S., Zhu, Y., Li, M. & Sestan, N. The cellular and molecular landscapes of the developing human central nervous system. Neuron 89, 248–268 (2016).
Kim, J.-Y. & Paredes, M. F. Implications of extended inhibitory neuron development. Int. J. Mol. Sci. 22, 5113 (2021).
Anthony, T. E., Klein, C., Fishell, G. & Heintz, N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron 41, 881–890 (2004).
Xu, Q., Tam, M. & Anderson, S. A. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 506, 16–29 (2008).
Laclef, C. & Métin, C. Conserved rules in embryonic development of cortical interneurons. Semin. Cell Dev. Biol. 76, 86–100 (2018).
Arshad, A. et al. Extended production of cortical interneurons into the third trimester of human gestation. Cereb. Cortex 26, 2242–2256 (2016).
Malik, S. et al. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J. Neurosci. 33, 411–423 (2013).
Paredes, M. F. et al. Nests of dividing neuroblasts sustain interneuron production for the developing human brain. Science 375, eabk2346 (2022).
Krienen, F. M. et al. Innovations present in the primate interneuron repertoire. Nature 586, 262–269 (2020).
Hodge, R. D. et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019).
Ellis, J. K. et al. Ferret brain possesses young interneuron collections equivalent to human postnatal migratory streams. J. Comp. Neurol. 527, 2843–2859 (2019).
Raju, C. S. et al. Secretagogin is expressed by developing neocortical GABAergic neurons in humans but not mice and increases neurite arbor size and complexity. Cereb. Cortex 28, 1946–1958 (2018).
Shi, Y. et al. Mouse and human share conserved transcriptional programs for interneuron development. Science 374, eabj6641 (2021).
Inta, D. et al. Neurogenesis and widespread forebrain migration of distinct GABAergic neurons from the postnatal subventricular zone. Proc. Natl Acad. Sci. USA 105, 20994–20999 (2008).
Paredes, M. F. et al. Extensive migration of young neurons into the infant human frontal lobe. Science 354, aaf7073 (2016).
Schmitz, M. T. et al. The development and evolution of inhibitory neurons in primate cerebrum. Nature 603, 871–877 (2022).
Loomba, S. et al. Connectomic comparison of mouse and human cortex. Science 377, eabo0924 (2022).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). In this study, spheroids of different brain regions were combined to form assembloids, which were used to model Timothy syndrome.
Bagley, J. A., Reumann, D., Bian, S., Levi-Strauss, J. & Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 14, 743–751 (2017). In this study, organoids of different brain regions were fused to investigate interneuron migration between brain regions.
Xiang, Y. et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398.e7 (2017).
Bajaj, S. et al. Neurotransmitter signaling regulates distinct phases of multimodal human interneuron migration. EMBO J. 40, e108714 (2021).
Birey, F. et al. Dissecting the molecular basis of human interneuron migration in forebrain assembloids from Timothy syndrome. Cell Stem Cell 29, 248–264.e7 (2022). In this study, interneuron migration was studied in the Timothy syndrome disease model.
Eichmüller, O. L. et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 375, eabf5546 (2022). Generation of a brain organoid model of tuberous sclerosis in this study changed the prevailing view of the disease by identifying a specific cell population in the caudal GE as the source of disease pathology.
van Essen, D. C. Surface-based atlases of cerebellar cortex in the human, macaque, and mouse. Ann. NY Acad. Sci. 978, 468–479 (2002).
Aldinger, K. A. et al. Spatial and cell-type transcriptional landscape of human cerebellar development. Nat. Neurosci. 24, 1163–1175 (2021).
Haldipur, P. et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science 366, 454–460 (2019).
Haldipur, P. et al. Evidence of disrupted rhombic lip development in the pathogenesis of Dandy–Walker malformation. Acta Neuropathol. 142, 761–776 (2021).
Hendrikse, L. D. et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature 609, 1021–1028 (2022).
Smith, K. S. et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature 609, 1012–1020 (2022).
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K. & Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550 (2015).
Nayler, S., Agarwal, D., Curion, F., Bowden, R. & Becker, E. B. E. High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep. 11, 12959 (2021).
Heide, M. & Huttner, W. B. Human-specific genes, cortical progenitor cells, and microcephaly. Cells 10, 1209 (2021).
Benito-Kwiecinski, S. et al. An early cell shape transition drives evolutionary expansion of the human forebrain. Cell 184, 2084–2102.e19 (2021).
Girskis, K. M. et al. Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 109, 3239–3251.e7 (2021).
Boyd, J. L. et al. Human–chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr. Biol. 25, 772–779 (2015).
Arcila, M. L. et al. Novel primate miRNAs coevolved with ancient target genes in germinal zone-specific expression patterns. Neuron 81, 1255–1262 (2014).
Nowakowski, T. J. et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat. Neurosci. 21, 1784–1792 (2018).
Tomasello, U. et al. miR-137 and miR-122, two outer subventricular zone non-coding RNAs, regulate basal progenitor expansion and neuronal differentiation. Cell Rep. 38, 110381 (2022).
Charrier, C. et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 149, 923–935 (2012).
Dennis, M. Y. et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149, 912–922 (2012).
Schmidt, E. R. E. et al. A human-specific modifier of cortical connectivity and circuit function. Nature 599, 640–644 (2021).
Kelava, I., Lewitus, E. & Huttner, W. B. The secondary loss of gyrencephaly as an example of evolutionary phenotypical reversal. Front. Neuroanat. 7, 16 (2013).
Chinnappa, K. et al. Secondary loss of miR-3607 reduced cortical progenitor amplification during rodent evolution. Sci. Adv. 8, eabj4010 (2022).
Gertz, C. C., Lui, J. H., LaMonica, B. E., Wang, X. & Kriegstein, A. R. Diverse behaviors of outer radial glia in developing ferret and human cortex. J. Neurosci. 34, 2559–2570 (2014).
Kim, J., Koo, B.-K. & Knoblich, J. A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584 (2020).
Corsini, N. S. & Knoblich, J. A. Human organoids: new strategies and methods for analyzing human development and disease. Cell 185, 2756–2769 (2022).
Hendriks, D., Artegiani, B., Hu, H., de Sousa Lopes, S. C. & Clevers, H. Establishment of human fetal hepatocyte organoids and CRISPR–Cas9-based gene knockin and knockout in organoid cultures from human liver. Nat. Protoc. 16, 182–217 (2021).
Bonfanti, P. et al. Ex vivo expansion and differentiation of human and mouse fetal pancreatic progenitors are modulated by epidermal growth factor. Stem Cell Dev. 24, 1766–1778 (2015).
Eiraku, M. et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011). This study is the first to describe in vitro generation of self-organized neuronal tissue by generating optic-cup cultures from mouse embryonic stem cells.
Nakano, T. et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785 (2012). In this study, human embryonic stem cells were used for the first time to create optic-cup structures in vitro.
Kadoshima, T. et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl Acad. Sci. USA 110, 20284–20289 (2013). This study demonstrates self-organization of cortical tissue by generating neuronal tissue in vitro from human embryonic stem cells.
Lancaster, M. A. & Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014). In this study, 3D cultures derived from human pluripotent stem cells were used for the first time to model disease and recapitulate human-specific features of brain development.
Gordon, A. et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 24, 331–342 (2021).
Velasco, S. et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527 (2019).
Yoon, S.-J. et al. Reliability of human cortical organoid generation. Nat. Methods 16, 75–78 (2019).
Xiang, Y. et al. hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell 24, 487–497.e7 (2019).
Giandomenico, S. L. et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 22, 669–679 (2019).
Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 (2016). This study shows that Zika virus can infect human cortical progenitors in cerebral organoids and cause microcephaly-like phenotypes.
Krenn, V. et al. Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly. Cell Stem Cell 28, 1362–1379.e7 (2021).
Marton, R. M. et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491 (2019).
Madhavan, M. et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706 (2018).
Caporale, N. et al. From cohorts to molecules: adverse impacts of endocrine disrupting mixtures. Science 375, eabe8244 (2022).
Pașca, A. M. et al. Human 3D cellular model of hypoxic brain injury of prematurity. Nat. Med. 25, 784–791 (2019).
Esk, C. et al. A human tissue screen identifies a regulator of ER secretion as a brain-size determinant. Science 370, 935–941 (2020). In this study, massive parallel lineage tracing in organoids allowed high-throughput screening to simultaneously evaluate 172 microcephaly risk genes and uncovered endoplasmic reticulum secretion as a pathway involved in microcephaly.
Jayaraman, D., Bae, B.-I. & Walsh, C. A. The genetics of primary microcephaly. Annu. Rev. Genomics Hum. Genet. 19, 177–200 (2018).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Milh, M., Riccardi, F. & Denis, J. Genetics of neonatal onset epilepsies: an overview. Rev. Neurol. 176, 2–9 (2020).
Spoto, G. et al. Neonatal seizures: an overview of genetic causes and treatment options. Brain Sci. 11, 1295 (2021).
Gürsoy, S. & Erçal, D. Genetic evaluation of common neurocutaneous syndromes. Pediatr. Neurol. 89, 3–10 (2018).
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 4, 27 (2018).
Gan, L., Cookson, M. R., Petrucelli, L. & Spada, A. R. L. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 21, 1300–1309 (2018).
Gilmore, E. C. & Walsh, C. A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2, 461–478 (2013).
Pinson, A., Namba, T. & Huttner, W. B. Malformations of human neocortex in development–their progenitor cell basis and experimental model systems. Front. Cell Neurosci. 13, 305 (2019).
Jackson, A. P. et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 71, 136–142 (2002).
Zhou, Z.-W. et al. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 12, 645–655 (2013).
Gruber, R. et al. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1–Cdc25 pathway. Nat. Cell Biol. 13, 1325–1334 (2011).
Montgomery, S. H. & Mundy, N. I. Microcephaly genes evolved adaptively throughout the evolution of eutherian mammals. BMC Evol. Biol. 14, 120 (2014).
Bae, B., Jayaraman, D. & Walsh, C. A. Genetic changes shaping the human brain. Dev. Cell 32, 423–434 (2015).
Lizarraga, S. B. et al. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137, 1907–1917 (2010).
Desir, J., Cassart, M., David, P., Bogaert, P. V. & Abramowicz, M. Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero-posterior gradient pre- and post-natally. Am. J. Med. Genet. A 146A, 1439–1443 (2008).
Pulvers, J. N. et al. Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc. Natl Acad. Sci. USA 107, 16595–16600 (2010).
Fujimori, A. et al. Disruption of Aspm causes microcephaly with abnormal neuronal differentiation. Brain Dev. 36, 661–669 (2014).
Jayaraman, D. et al. Microcephaly proteins Wdr62 and Aspm define a mother centriole complex regulating centriole biogenesis, apical complex, and cell fate. Neuron 92, 813–828 (2016).
Capecchi, M. R. & Pozner, A. ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination. Nat. Commun. 6, 8763 (2015).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020).
Li, R. et al. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 8, 823–833 (2017).
Lord, C. et al. Autism spectrum disorder. Nat. Rev. Dis. Primers 6, 5 (2020).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014).
Paulsen, B. et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 602, 268–273 (2022). In this study, organoid models with different ASD-related mutations were generated and revealed convergent developmental changes.
Mariani, J. et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390 (2015). Use of induced pluripotent stem cells of patients with ASD in this study identified convergent mechanisms in idiopathic ASD.
Rippon, G., Brock, J., Brown, C. & Boucher, J. Disordered connectivity in the autistic brain: challenges for the “new psychophysiology”. Int. J. Psychophysiol. 63, 164–172 (2007).
Rosenberg, A., Patterson, J. S. & Angelaki, D. E. A computational perspective on autism. Proc. Natl Acad. Sci. USA 112, 9158–9165 (2015).
Masuda, F. et al. Motor cortex excitability and inhibitory imbalance in autism spectrum disorder assessed with transcranial magnetic stimulation: a systematic review. Transl. Psychiat 9, 110 (2019).
Alexandre, J. et al. ASD modelling in organoids reveals imbalance of excitatory cortical neuron subtypes during early neurogenesis. Preprint at bioRxiv https://doi.org/10.1101/2022.03.19.484988 (2022).
Willsey, A. J. et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013).
Li, M. et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018).
McConnell, M. J. et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: the Brain Somatic Mosaicism Network. Science 356, eaal1641 (2017).
Stranger, B. E. et al. Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat. Genet. 49, 1664–1670 (2017).
Akbarian, S. et al. The PsychENCODE project. Nat. Neurosci. 18, 1707–1712 (2015).
Ziffra, R. S. et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021).
de la Torre-Ubieta, L. et al. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304.e18 (2018).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Amiri, A. et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362, eaat6720 (2018).
Kanton, S. et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019).
Bhaduri, A. et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020).
Trevino, A. E. et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367, eaay1645 (2020).
Fleck, J. S. et al. Inferring and perturbing cell fate regulomes in human cerebral organoids. Preprint at bioRxiv https://doi.org/10.1101/2021.08.24.457460 (2021).
Uzquiano, A. et al. Single-cell multiomics atlas of organoid development uncovers longitudinal molecular programs of cellular diversification of the human cerebral cortex. Preprint at bioRxiv https://doi.org/10.1101/2022.03.17.484798 (2022).
Panagiotakos, G. & Pasca, S. P. A matter of space and time: emerging roles of disease-associated proteins in neural development. Neuron 110, 195–208 (2021).
Campos, G. S., Bandeira, A. C. & Sardi, S. I. Zika virus outbreak, Bahia, Brazil. Emerg. Infect. Dis. 21, 1885–1886 (2015).
Zanluca, C. et al. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 110, 569–572 (2015).
Calvet, G. et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect. Dis. 16, 653–660 (2016).
Mlakar, J. et al. Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016). In this study, cerebral organoids were used together with other model systems to investigate the effects of Zika virus infection.
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016). In this study, a mini-bioreactor culture system was developed to grow brain organoids and used to show that Zika virus infection can cause microcephaly phenotypes in organoids.
Dang, J. et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19, 258–265 (2016).
Tang, H. et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18, 587–590 (2016).
Hanners, N. W. et al. Western Zika virus in human fetal neural progenitors persists long term with partial cytopathic and limited immunogenic effects. Cell Rep. 15, 2315–2322 (2016).
Gabriel, E. et al. Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20, 397–406.e5 (2017).
Watanabe, M. et al. Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 21, 517–532 (2017).
Yoon, K.-J. et al. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell 21, 349–358.e6 (2017).
Onorati, M. et al. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 16, 2576–2592 (2016).
Grant, A. et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 (2016).
Jacob, F. et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell 27, 937–950.e9 (2020).
Pellegrini, L. et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell 27, 951–961.e5 (2020).
Ramani, A. et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 39, e106230 (2020).
Sun, G. et al. Modeling human cytomegalovirus-induced microcephaly in human iPSC-derived brain organoids. Cell Rep. Med. 1, 100002 (2020).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991).
Ashe, K. H. & Zahs, K. R. Probing the biology of Alzheimer’s disease in mice. Neuron 66, 631–645 (2010).
Wray, S. Modelling neurodegenerative disease using brain organoids. Semin. Cell Dev. Biol. 111, 60–66 (2020).
Israel, M. A. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220 (2012).
Shi, Y. et al. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl Med. 4, 124ra29 (2012).
Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014).
Raja, W. K. et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS ONE 11, e0161969 (2016).
Gonzalez, C. et al. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatr. 23, 2363–2374 (2018).
Jorfi, M., D’Avanzo, C., Tanzi, R. E., Kim, D. Y. & Irimia, D. Human neurospheroid arrays for in vitro studies of Alzheimer’s disease. Sci. Rep. 8, 2450 (2018).
Arber, C. et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatr. 25, 2919–2931 (2020).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Morabito, S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet. 53, 1143–1155 (2021).
Park, J. et al. A 3D human tri-culture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018). In this study, neurons, astrocytes and microglia were combined to study AD.
Lin, Y.-T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7 (2018).
Yang, A. C. et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022).
Cakir, B. et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175 (2019).
Woollacott, I. O. C. & Rohrer, J. D. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J. Neurochem. 138, 6–31 (2016).
Benussi, A. et al. Toward a glutamate hypothesis of frontotemporal dementia. Front. Neurosci. 13, 304 (2019).
Sohn, P. D. et al. Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470.e5 (2019).
Bain, H. D. C. et al. The role of lysosomes and autophagosomes in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 45, 244–261 (2019).
Bowles, K. R. et al. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184, 4547–4563.e17 (2021). In this study, a brain organoid model of frontotemporal dementia was generated and used to uncovered the steps that lead to glutamatergic neuron loss.
Kim, H. et al. Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids. Stem Cell Rep. 12, 518–531 (2019).
Smits, L. M. et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 5, 5 (2019).
Jo, J. et al. Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and α-synuclein mutations. Ann. Neurol. 90, 490–505 (2021).
Groveman, B. R. et al. Human cerebral organoids as a therapeutic drug screening model for Creutzfeldt–Jakob disease. Sci. Rep. 11, 5165 (2021).
Park, J.-C. et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 12, 280 (2021).
Anastasaki, C. et al. Human iPSC-derived neurons and cerebral organoids establish differential effects of germline NF1 gene mutations. Stem Cell Rep. 14, 541–550 (2020).
Wegscheid, M. L. et al. Patient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis. Cell Rep. 36, 109315 (2021).
Blair, J. D., Hockemeyer, D. & Bateup, H. S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 24, 1568–1578 (2018).
Khan, T. A. et al. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med. 26, 1888–1898 (2020).
Iefremova, V. et al. An organoid-based model of cortical development identifies non-cell-autonomous defects in Wnt signaling contributing to Miller–Dieker syndrome. Cell Rep. 19, 50–59 (2017).
Bershteyn, M. et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 20, 435–449.e4 (2017).
Gabriel, E. et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 35, 803–819 (2016).
Kang, Y. et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 24, 1377–1391 (2021).
Miura, Y. et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 38, 1421–1430 (2020).
Sun, A. X. et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 366, 1486–1492 (2019). In this study, cerebral organoids were used to model Angelman syndrome and uncovered dysregulation of large potassium channels as the underlying mechanism of hyperactivity phenotypes.
Splawski, I. et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004).
Bader, P. L. et al. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc. Natl Acad. Sci. USA 108, 15432–15437 (2011).
Paşca, S. P. et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662 (2011).
Krey, J. F. et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209 (2013).
Bortone, D. & Polleux, F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71 (2009).
Buiting, K., Williams, C. & Horsthemke, B. Angelman syndrome–insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593 (2016).
Vu, T. H. & Hoffman, A. R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 17, 12–13 (1997).
Rougeulle, C., Cardoso, C., Fontés, M., Colleaux, L. & Lalande, M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet. 19, 15–16 (1998).
Jiang, Y. et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811 (1998).
Wallace, M. L., Burette, A. C., Weinberg, R. J. & Philpot, B. D. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800 (2012).
Wallace, M. L., Woerden, G. M., van, Elgersma, Y., Smith, S. L. & Philpot, B. D. Ube3a loss increases excitability and blunts orientation tuning in the visual cortex of Angelman syndrome model mice. J. Neurophysiol. 118, 634–646 (2017).
Judson, M. C. et al. GABAergic neuron-specific loss of Ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 90, 56–69 (2016).
Chamberlain, S. J. et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl Acad. Sci. USA 107, 17668–17673 (2010).
Fink, J. J. et al. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 8, 15038 (2017).
Sell, G. L. & Margolis, S. S. From UBE3A to Angelman syndrome: a substrate perspective. Front. Neurosci. 9, 322 (2015).
Henske, E. P., Jozwiak, S., Kingswood, J. C., Sampson, J. R. & Thiele, E. A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2, 16035 (2016).
Gelot, A. B. & Represa, A. Progression of fetal brain lesions in tuberous sclerosis complex. Front. Neurosci. 14, 899 (2020).
Lozovaya, N. et al. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat. Commun. 5, 4563 (2014).
Bateup, H. S. et al. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522 (2013).
Meikle, L. et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558 (2007).
Feliciano, D. M., Quon, J. L., Su, T., Taylor, M. M. & Bordey, A. Postnatal neurogenesis generates heterotopias, olfactory micronodules and cortical infiltration following single-cell Tsc1 deletion. Hum. Mol. Genet. 21, 799–810 (2012).
Feliciano, D. M., Su, T., Lopez, J., Platel, J. C. & Bordey, A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest. 121, 1596–1607 (2011).
Goto, J. et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc. Natl Acad. Sci. USA 108, E1070–E1079 (2011).
Martin, K. R. et al. The genomic landscape of tuberous sclerosis complex. Nat. Commun. 8, 15816 (2017).
Qian, X., Song, H. & Ming, G. Brain organoids: advances, applications and challenges. Development 146, dev166074 (2019).
Vértesy, Á. et al. Gruffi: an algorithm for computational removal of stressed cells from brain organoid transcriptomic datasets. EMBO J. 41, e111118 (2022).
Shi, Y. et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 18, e3000705 (2020).
Wörsdörfer, P. et al. Generation of complex human organoid models including vascular networks by incorporation of mesodermal progenitor cells. Sci. Rep. 9, 15663 (2019).
Mansour, A. A. et al. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441 (2018).
Qian, X. et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 26, 766–781.e9 (2020).
Fiorenzano, A. et al. Single-cell transcriptomics captures features of human midbrain development and dopamine neuron diversity in brain organoids. Nat. Commun. 12, 7302 (2021).
Hor, J. H. et al. Cell cycle inhibitors protect motor neurons in an organoid model of spinal muscular atrophy. Cell Death Dis. 9, 1100 (2018).
Pellegrini, L. et al. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 369, eaaz5626 (2020). In this study, a platform was developed for generating choroid plexus organoids that recapitulate the selective permeability of the human choroid plexus.
Huang, W. et al. Origins and proliferative states of human oligodendrocyte precursor cells. Cell 182, 594–608.e11 (2020).
Lepiemme, F. et al. Oligodendrocyte precursors guide interneuron migration by unidirectional contact repulsion. Science 376, eabn6204 (2022).
Kim, H. et al. Pluripotent stem cell-derived cerebral organoids reveal human oligodendrogenesis with dorsal and ventral origins. Stem Cell Rep. 12, 890–905 (2019).
Ormel, P. R. et al. Microglia innately develop within cerebral organoids. Nat. Commun. 9, 4167 (2018).
Xu, R. et al. Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modeling brain development and pathology. Stem Cell Rep. 16, 1923–1937 (2021).
Popova, G. et al. Human microglia states are conserved across experimental models and regulate neural stem cell responses in chimeric organoids. Cell Stem Cell 28, 2153–2166.e6 (2021).
Sabate-Soler, S. et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 70, 1267–1288 (2022).
Trujillo, C. A. et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell 25, 558–569.e7 (2019).
Samarasinghe, R. A. et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 24, 1488–1500 (2021). In this study, organoids and calcium imaging were used to investigate epileptiform activity in the context of a Rett syndrome model.
Fair, S. R. et al. Electrophysiological maturation of cerebral organoids correlates with dynamic morphological and cellular development. Stem Cell Rep. 15, 855–868 (2020).
Zhang, W. et al. Modeling microcephaly with cerebral organoids reveals a WDR62–CEP170–KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 10, 2612 (2019).
Wang, L. et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat. Commun. 11, 4038 (2020).
Li, Y. et al. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell 20, 385–396.e3 (2017).
Allende, M. L. et al. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 59, 550–563 (2018).
Xu, Y.-P. et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res. 29, 265–273 (2019).
Qiao, H. et al. Herpes simplex virus type 1 infection leads to neurodevelopmental disorder-associated neuropathological changes. PLoS Pathog. 16, e1008899 (2020).
Thomas, C. A. et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 21, 319–331.e8 (2017).
Pérez-Brangulí, F. et al. Human SPG11 cerebral organoids reveal cortical neurogenesis impairment. Hum. Mol. Genet. 28, 961–971 (2019).
Conforti, P. et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl Acad. Sci. USA 115, E762–E771 (2018).
Groveman, B. R. et al. Sporadic Creutzfeldt–Jakob disease prion infection of human cerebral organoids. Acta Neuropathol. Commun. 7, 90 (2019).
Szebényi, K. et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 24, 1542–1554 (2021).
Dooves, S., Velthoven, A. J. H., van, Suciati, L. G. & Heine, V. M. Neuron–glia interactions in tuberous sclerosis complex affect the synaptic balance in 2D and organoid cultures. Cells 10, 134 (2021).
Gomes, A. R. et al. Modeling Rett syndrome with human patient-specific forebrain organoids. Front. Cell Dev. Biol. 8, 610427 (2020).
Trujillo, C. A. et al. Pharmacological reversal of synaptic and network pathology in human MECP2-KO neurons and cortical organoids. EMBO Mol. Med. 13, e12523 (2021).
Mellios, N. et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatr. 23, 1051–1065 (2018).
Schafer, S. T. et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 22, 243–255 (2019).
Jong, J. Ode et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 12, 4087 (2021).
Srikanth, P. et al. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl. Psychiat 8, 77 (2018).
Ye, F. et al. DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron 96, 1041–1054.e5 (2017).
Xu, R. et al. OLIG2 drives abnormal neurodevelopmental phenotypes in human iPSC-based organoid and chimeric mouse models of Down syndrome. Cell Stem Cell 24, 908–926.e8 (2019).
Klaus, J. et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 25, 561–568 (2019).
Steinberg, D. J. et al. Modeling genetic epileptic encephalopathies using brain organoids. EMBO Mol. Med. 13, e13610 (2021).
Papes, F. et al. Transcription factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content. Nat. Commun. 13, 2387 (2022).
Morelli, K. H. et al. MECP2-related pathways are dysregulated in a cortical organoid model of myotonic dystrophy. Sci. Transl Med. 14, eabn2375 (2022).
Linkous, A. et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 26, 3203–3211.e5 (2019).
Jacob, F. et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e22 (2020).
Bian, S. et al. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 15, 631–639 (2018).
Ogawa, J., Pao, G. M., Shokhirev, M. N. & Verma, I. M. Glioblastoma model using human cerebral organoids. Cell Rep. 23, 1220–1229 (2018).
Krieger, T. G. et al. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro Oncol. 22, 1138–1149 (2020).
Rezazadeh, A. et al. Periventricular nodular heterotopia in 22q11.2 deletion and frontal lobe migration. Ann. Clin. Transl. Neurol. 5, 1314–1322 (2018).
Sakaguchi, H. et al. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 6, 8896 (2015).
Lancaster, M. A. et al. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 35, 659–666 (2017).
Paşca, A. M. et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015).
Jo, J. et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19, 248–257 (2016).
Eura, N. et al. Brainstem organoids from human pluripotent stem cells. Front. Neurosci. 14, 538 (2020).
Kasai, T. et al. Hypothalamic contribution to pituitary functions is recapitulated in vitro using 3D-cultured human iPS cells. Cell Rep. 30, 18–24.e5 (2020).
Andersen, J. et al. Generation of functional human 3D cortico-motor assembloids. Cell 183, 1913–1929.e26 (2020). This study combined cortical, spinal cord and muscle organoids to generate corticomotor assembloids.
Acknowledgements
The authors thank all members of the laboratory of J.A.K. for discussions that led to the ideas expressed in this Review. The authors apologize to their colleagues whose work could not be cited owing to the limited space. Work in the laboratory of J.A.K. is supported by the Special Research Programme (SFB) of the Austrian Science Fund (FWF, F7804-B), the FWF Stand Alone Grants P 35680 and P 35369, the Austrian Federal Ministry of Education, Science and Research, the City of Vienna, and a European Research Council (ERC) Advanced Grant under the European Union’s Horizon 2020 programmes (no. 695642 and no. 874769).
Author information
Authors and Affiliations
Contributions
O.L.E. researched data for the article. Both authors made substantial contributions to discussion of the content, wrote the article and reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
J.A.K. is on the supervisory and scientific advisory board of a:head bio AG (aheadbio.com) and is an inventor on several patents relating to cerebral organoids. O.L.E. declares no competing interests.
Peer review
Peer review information
Nature Reviews Neurology thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Eichmüller, O.L., Knoblich, J.A. Human cerebral organoids — a new tool for clinical neurology research. Nat Rev Neurol 18, 661–680 (2022). https://doi.org/10.1038/s41582-022-00723-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-022-00723-9
This article is cited by
-
Patient-derived organoids in human cancer: a platform for fundamental research and precision medicine
Molecular Biomedicine (2024)
-
APOE3ch alleviates Aβ and tau pathology and neurodegeneration in the human APPNL-G-F cerebral organoid model of Alzheimer’s disease
Cell Research (2024)
-
Apically localized PANX1 impacts neuroepithelial expansion in human cerebral organoids
Cell Death Discovery (2024)
-
Genetics of human brain development
Nature Reviews Genetics (2024)
-
Shaping the Neurovascular Unit Exploiting Human Brain Organoids
Molecular Neurobiology (2024)
