
Abstract
Dysregulation of the immune system is a cardinal feature of Alzheimer disease (AD), and a considerable body of evidence indicates pathological alterations in central and peripheral immune responses that change over time. Considering AD as a systemic immune process raises important questions about how communication between the peripheral and central compartments occurs and whether this crosstalk represents a therapeutic target. We established a whitepaper workgroup to delineate the current status of the field and to outline a research prospectus for advancing our understanding of peripheral–central immune crosstalk in AD. To guide the prospectus, we begin with an overview of seminal clinical observations that suggest a role for peripheral immune dysregulation and peripheral–central immune communication in AD, followed by formative animal data that provide insights into possible mechanisms for these clinical findings. We then present a roadmap that defines important next steps needed to overcome conceptual and methodological challenges, opportunities for future interdisciplinary research, and suggestions for translating promising mechanistic studies into therapeutic interventions.
Key points
-
Alzheimer disease (AD) should be viewed as a systemic disease that involves dynamic processes in the peripheral and central immune compartments.
-
Clinical studies suggest that the peripheral and central immune systems are dysregulated in AD, are related to cognitive function and clinical status, and may change in a non-linear manner over time; burgeoning evidence also suggests that the roles of innate and adaptive immune processes differ depending on the pathological stage of AD.
-
Animal studies have provided insights into possible mechanisms for peripheral and central immune communication, including direct pathways that involve peripheral immune cell infiltration into the CNS, as well as indirect pathways that involve systemic inflammation-driven modulation of microglial function.
-
Longitudinal clinical studies to evaluate peripheral and central immune mechanisms are needed in combination with comprehensive assessment of the human exposome in demographically diverse cohorts.
-
Further mechanistic studies in animal models are critical to providing a better understanding of peripheral immune cell trafficking to the CNS, adaptive and innate immune system interfaces and astrocyte–microglia crosstalk.
-
Translational and interdisciplinary studies are needed to reconcile differences in immune pathways across species and to develop strategic immune-based therapies that are tailored to disease state and severity.
Similar content being viewed by others

Introduction
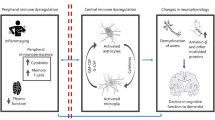
Dysregulation of the immune system is a cardinal feature of Alzheimer disease (AD). Considerable evidence indicates that pathological alterations occur in both central and peripheral immune responses and that these change over time. Immune dysfunction as a cause of AD pathogenesis and progression was previously thought to be restricted to the immune process in the CNS, but accumulating data indicate pivotal contributions of the peripheral immune system as well. Examining AD pathophysiology through the lens of a systemic immune process raises important questions about how communication occurs between the peripheral and central compartments, whether this communication functionally changes over time, and whether this crosstalk could be a therapeutic target1. Some data also indicate that functional alterations in immune cells and/or disruption of immune homeostasis result in a combination of protective and destructive downstream effects with complex implications — positive and negative — for AD according to disease stage.
In order to understand the myriad roles of peripheral–central immune dysregulation in AD and to develop effective therapeutic interventions that target the communication between these systems, we established a whitepaper workgroup as part of the Alzheimer’s Association Immunity and Neurodegeneration Professional Interest Area. Numerous comprehensive reviews of the role of the immune system in AD pathogenesis have been published previously2,3,4,5,6, so our aim was not to provide an exhaustive summary of prior studies nor to reach a consensus on the most robust immune biomarkers. Instead, the goal of this Roadmap is to delineate the current status of the field and to outline a research prospectus for advancing our understanding of the dynamic interplay between peripheral and central immune dysregulation in AD.
We begin with an overview of seminal clinical observations that suggest a role for peripheral immune dysregulation and peripheral–central immune crosstalk in AD, followed by formative animal data that provide insights into possible mechanisms that underlie these clinical findings. Although we discuss the role of central immune regulation in AD, we primarily consider central immune function from the perspective of its relationship with the periphery rather than in isolation. Finally, we present a roadmap that defines the next steps that are essential to overcoming conceptual and methodological challenges, highlights opportunities for interdisciplinary future research, and provides suggestions for translating promising mechanistic studies into therapeutic interventions.
State of the field
Clinical studies of immune dysregulation
The possibility that immune dysfunction is involved in AD was first recognized in Alois Alzheimer’s original 1907 article, in which he described morphological alterations in glia in autopsy samples from the brains of patients with AD. Colocalization of immune cells with amyloid plaques is now a well-recognized neuropathological feature of AD. More broadly, ageing is characterized by systemic changes to the innate and adaptive immune systems, including thymic involution, chronic expression of circulating pro-inflammatory markers (known as ‘inflammageing’), and senescent cellular phenotypes, which have a senescence-associated secretory profile and release several inflammatory mediators. Although immune system dysregulation was originally considered to be a ‘bystander’ in pathological ageing processes, genome-wide association studies (GWAS) and clinical research studies have implicated this dysfunction in AD pathogenesis and clinical progression.
In this section, we discuss evidence from human studies that addressed the complexity of the interplay between central and peripheral immune dysfunction and its association with salient clinical outcomes. We focus on this interplay to avoid the false dichotomy of central versus peripheral immune dysregulation. We primarily review the role of peripheral immune regulation in CNS-related outcomes and the tandem appraisal of the peripheral and central immune systems in ageing and AD.
Peripheral inflammation, cognitive decline and dementia
In vivo studies of immune dysregulation in humans have predominantly focused on circulating, peripheral inflammatory mediators. Increases in these markers have been associated with negative trajectories in the context of both ‘normal’ ageing7 and AD, although discordant results are noted throughout the literature8,9,10 and several large-scale studies have found no relationship between peripheral inflammation and AD11,12. Nevertheless, meta-analyses of observational and epidemiological studies have indicated that dysregulation of inflammatory markers is present in AD13,14 and that this dysregulation is associated with an increased risk of developing all-cause dementia15,16.
Early clinical studies of immune dysregulation focused on the role of peripheral inflammatory markers in predicting pathological outcomes in late life and indicated that alterations in these markers presage cognitive changes and/or symptomatic manifestations of disease. These formative epidemiological studies demonstrated that baseline levels of C-reactive protein17,18 and IL-6 (ref.18) and peripheral blood mononuclear cell production of tumour necrosis factor (TNF) and IL-1β19 in healthy adults are associated with future development of all-cause dementia or AD. Observations that higher levels of peripheral inflammatory markers in midlife to late life indicate an increased risk of dementia have been further supported by studies in which elevated levels of these markers were found to be associated with future cognitive decline more broadly (not just dementia)20,21,22 and to interact with vascular risk factors to negatively influence outcomes23. Moreover, a chronic pro-inflammatory state, defined by longitudinal elevations in the levels of peripheral inflammatory markers, has been associated with earlier disease onset of AD in APOE ε4 carriers24. Findings from the Atherosclerosis Risk in Communities (ARIC) study indicate that a greater extent of peripheral inflammation in midlife is associated with steeper cognitive decline over a 20-year period even after accounting for vascular risk factors and demographic variables25. Similar to the body of research linking midlife cardiovascular risk factors with late-life cognitive decline26, the implications of peripheral inflammation for brain health might differ according to the timing and duration of exposure.
Peripheral innate immunity and AD stages
Peripheral inflammatory markers in asymptomatic older adults have been associated with many clinical variables and biomarkers, including cognition27,28, brain structure24,29,30,31, functional brain connectivity32 and future amyloidosis33. However, resolving the temporal course and clinical significance of peripheral immune dysregulation in the context of symptomatic AD has been a considerable challenge. Attempts to use peripheral inflammatory markers to differentiate clinical stages of AD or to demarcate conversion from mild cognitive impairment (MCI) to dementia have yielded mixed results34,35,36,37,38,39. Although many peripheral inflammatory markers increase in tandem with clinical progression, there is no agreed-upon immune marker profile in the periphery for the staging of diagnostic severity. This is due, in part, to inconsistent evidence suggesting that some peripheral inflammatory markers peak in the early symptomatic stages of AD and decline in later stages (that is, late dementia)34,40,41. Studying absolute differences in cytokine levels might be less meaningful than delineating how these cytokines function and change at specific stages of clinical severity, as strong negative associations between peripheral inflammation and cognition have been observed even in the context of low absolute levels of inflammatory markers42.
In addition to linking peripheral inflammatory markers with clinical AD severity, levels of peripheral inflammatory markers have also recently been compared with fluid and PET biomarkers of AD pathology to investigate their relationship with disease pathogenesis and stage. In this context, levels of peripheral inflammatory markers have generally been more strongly associated with cerebrospinal fluid (CSF) levels of phosphorylated tau43,44 and neurofilament light chain44 than with CSF levels of amyloid-β (Aβ). The majority of studies have been restricted to cross-sectional designs and have been limited in their appraisal of standardized (for example, amyloid–tau–neurodegeneration framework)45 pathological AD staging, both of which have hampered a full view of the temporal course. Of note, however, very recent data from a study on peripheral inflammation and AD-related neuroimaging biomarkers in asymptomatic older adults suggests the possibility that inflammation has protective effects at specific pathological stages46. Specifically, Yang and colleagues reported that higher levels of IL-12p70 were associated with slower rates of longitudinal cognitive decline in asymptomatic older adults, but only in individuals with higher amyloid burden on PET imaging; moreover, higher levels of IL-12p70 in the setting of higher amyloid burden were associated with less tau pathology and neurodegeneration46. This study raises questions about the dynamic role of peripheral inflammation in combating or compensating for accumulating amyloid pathology during early stages of the disease process, and highlights the need for prospective, longitudinal biomarker studies that are stratified by both clinical and pathological AD stages.
Although most human studies of immune dysfunction and AD stages have focused on plasma cytokine levels, phenotypic and functional assays of monocytes have also indicated a role for inflammatory dysregulation in AD dementia47,48. In the most recent study of this type, in vitro application of IL-33 to lipopolysaccharide-primed, Aβ42-stimulated monocytes reduced IL-1β production in monocytes from healthy elderly adults and adults with MCI but not in cells from people with AD dementia; moreover, it stimulated IL-10 production in monocytes from healthy older adults only. Collectively, these findings suggest that anti-inflammatory functional pathways may be altered in the MCI and dementia stages48. Other studies have also linked hyperreactive peripheral innate immune cells with clinical outcomes in patients with AD. Specifically, homeostasis of circulating neutrophils is altered, with a shift towards pro-inflammatory phenotypes and increased production of reactive oxygen species49. In this study, the ratio of harmful, hyperreactive senescent neutrophils to immunosuppressive neutrophils was higher in patients with AD dementia than in patients with MCI. Moreover, alterations in neutrophil homeostasis were more pronounced in patients who declined rapidly than in those who declined more slowly, strongly suggesting that neutrophil immunophenotypes not only reflect stage of disease but also presage cognitive decline49.
Systemic inflammatory events and clinical outcomes
Ageing is associated with an increased incidence of infections and surgical procedures, both of which introduce systemic immune alterations50,51,52. Systemic inflammatory health events range from common acute infections (for example, the common cold) and procedures (for example, orthopaedic surgery) to chronic infections (for example, herpes simplex virus and Chlamydia pneumoniae infection) and critical illness (for example, sepsis). So-called sickness behaviour53 — defined as a collection of behavioural changes that occur during infection and serve an evolutionarily adaptive role in the conservation of energy and resources — and cognitive decline54,55 have been associated with a range of such acute and chronic events, suggesting that direct pathways exist between the peripheral and central compartments. Even a single recent infection may alter peripheral immune markers and accelerate cognitive decline in elderly adults53,56,57.
In a seminal study published in 2009, recent inflammatory events were associated with increased levels of peripheral immune markers and a twofold increase in the rate of cognitive decline over a 6-month period58. With regard to longer-term outcomes, a recent study showed that a history of infections requiring hospital treatment was associated with future development of dementia, even when analyses were limited to individuals in whom dementia was diagnosed more than 10 years after the infection59. Of note, such infections were more strongly associated with vascular dementia than with AD dementia, suggesting that infection-related mechanisms involve both inflammatory and vascular pathways59.
Other studies have suggested that systemic inflammatory events can influence the neuroinflammatory response in AD in more complex ways; for example, terminal systemic infections have been associated with an immunosuppressive, rather than an inflammatory, environment in the brain of patients with AD, characterized by upregulation of anti-inflammatory genes (IL4R and CHI3L1) and decreased expression of pro-inflammatory proteins60. However, the role of acute and chronic systemic events in exacerbating or unmasking AD-specific degenerative processes remains relatively under-studied, and little is known about how the period of exposure or the role of concomitant AD pathology affects immune phenotypes in response to these events.
Correlations between peripheral and central compartments
Methodological constraints have limited widespread in vivo assessment of CNS immune dysfunction in humans. Nevertheless, a growing body of evidence indicates that a CNS inflammatory response, typically characterized by upregulation of CSF pro-inflammatory markers, occurs early in AD. Clinical and pathological associations with this inflammatory response are mixed and marker dependent, emphasizing the fact that granular assessment of specific pathways at each stage of disease severity is critical. For example, several studies have found that higher CSF levels of C–C motif chemokine ligand 2 predict faster clinical decline in AD61,62. However, other studies have suggested that increased levels of soluble triggering receptor expressed on myeloid cells 2 (TREM2)63 and confluence of several pro-inflammatory and anti-inflammatory factors in the CSF are predictive of slower clinical decline64. Using the National Institute on Aging–Alzheimer’s Association 2011 criteria65 for stratifying participants on the basis of preclinical biomarker stage, one study found that CSF levels of inflammation were lower in stage 1 (biomarker evidence of amyloid positivity only) than in stage 0 (no biomarker abnormality) but markedly higher in stage 2 (biomarker evidence of amyloid positivity and elevated total tau levels). This unexpected result of a nadir in the inflammatory process prior to significant tau accumulation highlights that the pattern of CSF levels of immune proteins is likely to be non-linear and can differ with the presence or absence of Aβ and tau pathology66.
PET imaging studies have also raised numerous questions about the longitudinal dynamics of central innate immune activation and the balance between beneficial and detrimental roles of resident innate immune cells in responding to AD pathology. A meta-analysis of studies in which translocator protein (TSPO) PET was used to estimate microglia and astrocyte activation showed that neuroinflammatory processes increase in tandem with disease progression67. Other PET studies have suggested that CNS immune responses fluctuate with multiple peaks or differ in their dynamics according to pathological stage67. A longitudinal PET study has also suggested that the functions of distinct neuroinflammatory signatures depend on patient-specific factors rather than strictly on disease stage68.
Few clinical studies have been done to directly investigate communication between peripheral and central innate immunity in AD. The breadth of evidence for associations between peripheral inflammation, acute systemic health events and CNS-related outcomes strongly suggests that communication between the two systems occurs, but the directionality and timing of these pathways and the potential for their therapeutic modification are poorly understood. Simultaneous measurements of and comparisons between CNS and peripheral inflammation are sparse, although some findings suggest that levels of most analytes correlate only modestly between the compartments44,69. Importantly, biomarkers of AD-related pathology are associated with unique inflammatory signatures in the blood and the CSF43,44,69, and indices of peripheral and CNS immune dysregulation contribute to models of AD classification43,69. The distinct inflammatory profiles suggest joint and/or independent contributions of the peripheral and central immune systems to AD, further highlighting that the peripheral inflammatory milieu is unlikely to be simply a downstream epiphenomenon of CNS dysfunction.
Adaptive immunity in AD
Burgeoning human data demonstrate that cellular adaptive immunity and associated neuroimmune interactions have an instrumental role in AD pathophysiology. Several studies have demonstrated that homeostasis of peripheral T cell immunity is dysregulated in AD, suggesting that altered distributions and activation statuses of lymphocyte functional subsets in the periphery have complex implications in disease pathogenesis. In line with this idea, peripheral alterations in naive and memory subsets of CD4+ T cells have been documented in patients with mild AD dementia compared with healthy older adults — the proportion of naive cells was decreased and that of effector memory and terminally differentiated effector memory (TEMRA) CD4+ cells was increased70,71. Accordingly, other studies have demonstrated that peripheral CD4+ T cells from patients with AD include a high proportion of circulating IFNγ-secreting T helper (TH) 1 cells and are skewed towards TH17 and TH9 phenotypes72,73. Some clinical evidence indicates that immunomodulation mediated by regulatory T (Treg) cells — CD4+ T cells that suppress excessive immune responses — is also altered in AD. Although still at an early stage of inquiry, studies suggest that the overall proportion of peripheral Treg cells is reduced70,74 or that Treg cell subsets and Treg cell-mediated suppression are increased in MCI (but not in mild or severe AD dementia)75,76.
In addition to T cell activation in the peripheral compartment, patients with AD have a much higher proportion of activated CD8+ T cells in the CSF than healthy older adults, and this abnormality correlates with AD-related cognitive and neuroanatomical outcomes77. The potential for adaptive immune crosstalk between the peripheral and central compartments is illustrated by a recent comprehensive study that examined adaptive immune changes in multiple groups of patients with AD. The study provided evidence that AD is associated with an adaptive immune signature in the blood (the presence of CD3+CD8+CD27− TEMRA cells), which is linked with increased effector functions and negatively correlates with a measure of global cognitive function78. Moreover, single-cell T cell receptor sequencing and repertoire analyses have revealed clonal expansion of cytotoxic pro-inflammatory CD8+ TEMRA cells in the CSF of patients with MCI or AD, which was also associated with higher proportions of granzyme A+CD8+ T cells in AD-affected than in control hippocampi. CD8+ T cells were also observed adjacent to Aβ plaques and/or associated with microtubule-associated protein 2-positive neuronal processes78, and other studies have found a correlation between T cell infiltration and phosphorylated tau load in the brain of patients with AD79. Levels of extravascular CD3+ T cells have been shown to correlate with tau but not Aβ pathology in the brain of patients with AD, suggesting that T cell extravasation into the brain is driven by tau-related neurodegenerative changes rather than by cerebral amyloidosis80.
Collectively, these data suggest that peripheral homeostasis of T cells is partially altered in patients with AD, resulting in dysregulated distribution, subsets and functional differentiation of T cells associated with neurodegeneration. Current data suggest that peripheral CD4+ and CD8+ T cells are more highly differentiated and pro-inflammatory than normal in AD, that Treg cell-mediated immunomodulation is altered, and that antigen-experienced T cells that patrol the intrathecal space of AD-affected brains undergo clonal expansion. Additional studies are needed to further establish the antigen specificity and functional significance of these T cells and whether T cell infiltration from the periphery is a feature specific to AD or common to several neurodegenerative conditions with neuroinflammatory features, such as Lewy body dementia and Parkinson disease.
Gut microbiota and peripheral–central immune crosstalk
Differences in the composition and diversity of gut microbiota have been observed in patients with AD compared with asymptomatic older adults, indicating an additional channel through which the peripheral milieu could influence the CNS81. Studies in humans have identified a decrease in the abundance of Firmicutes bacteria and an increase in the abundance of Bacteroidetes bacteria in the microbiome of people with AD relative to controls, and these alterations have been associated with high CSF levels of p-tau and a high p-tau to Aβ42 ratio82. Directly relevant to immune dysregulation, one study showed that the abundance of a pro-inflammatory microbiota taxon (Escherichia and Shigella) was higher and the abundance of an anti-inflammatory microbiota taxon (Eubacterium rectale) was lower among participants who were Aβ-positive and cognitively impaired than among participants who were Aβ-negative83. These alterations in gut microbiota were associated with systemic inflammation in the blood — the abundance of Escherichia and Shigella correlated positively with levels of IL-1β, NACHT, LRR and PYD domains-containing protein 3 (NLRP3) and C–X–C motif chemokine 2 (CXCL2)83.
Overall, accumulating data suggest that alterations in gut microbiota are associated with AD clinical status, AD-related biomarkers and inflammatory state. Strikingly, emerging evidence suggests that healthy ageing is driven by so-called microbial drift towards person-specific microbiota composition84. Multimodal longitudinal studies are needed to further characterize the intersections between peripheral immunity, central immunity and gut microbiota changes, but compelling data indicate that the microbiome is an important therapeutic target for modulating immune functions and AD progression.
Mechanistic insight from animal studies
Collectively, clinical studies of immune regulation in AD in humans suggest that peripheral and central immune systems are dysregulated and dynamically change in a non-linear manner over time, innate and adaptive immune processes are associated with presentation and course of the disease, and crosstalk between the central and peripheral systems is likely. However, how this communication occurs and at what stages it is beneficial or deleterious are unclear. Possible routes of communication between the peripheral and central compartments include circumventricular organs, direct transport across the blood–brain barrier (BBB) and stimulation of vagal afferents (reviewed in detail elsewhere85,86,87). Current technology limits in vivo study of the mechanisms behind the clinical findings in humans, but animal studies of immune dysregulation provide critical insight into possible mechanisms of central–peripheral immune crosstalk and its temporal dynamics in AD. Of note, in the discussion of animal models, we have used the common nomenclature ‘APP/PS1’ as an umbrella term for several types of double-transgenic APP and PS1 strains.
Peripheral inflammatory markers, neuroinflammation and cognition
The ways in which peripheral inflammatory factors affect neuroinflammation remains a key mechanistic question, and animal studies have provided important insights. In one study of the APP23 mouse model of Aβ pathology, treatment with different regimens of peripheral lipopolysaccharide injection to alter blood levels of pro-inflammatory cytokines had opposing effects on the development of Aβ pathology. In mice treated with regimens to increase peripheral pro-inflammatory profiles, microglia exhibited a form of immune memory called immune training, which exacerbates neuroinflammatory responses, cerebral β-amyloidosis and neuronal death. In mice treated with regimens to induce peripheral immune tolerance, microglia exhibited immune tolerance, which dampened inflammatory responses, increased microglial Aβ uptake and improved neuronal survival88. These data suggest that microglia retain a long-lasting memory of peripheral inflammation that can influence the progression of Aβ pathology in mouse models. Systemic inflammation also reduced microglial clearance of Aβ in a study of the APP/PS1 mouse model89. Knockout of the NLRP3 inflammasome blocked these microglial changes, suggesting that inhibition of NLRP3 pathways is critical to minimize the pathological effects of peripheral inflammation.
More broadly, other studies have shown that blood-borne factors in the systemic milieu can inhibit adult neurogenesis and cognitive function in an age-dependent fashion in mice. For example, alterations in plasma levels of selected chemokines, including eotaxin, modulate adult neurogenesis and affect learning and memory90. Although this study did not specifically address AD, the findings suggest direct communication between the periphery and CNS, and underscore the potential for peripheral systemic factors to affect AD pathogenesis.
Peripheral innate immune cell infiltration
Evidence from studies of aged animals and murine models of AD-like pathology has established that brain neuroinflammation comprises not only activation of brain-resident immune-competent cells but also infiltration of activated peripheral immune cells. Peripheral macrophages are among the major innate immune cells that can infiltrate the CNS in neuroinflammatory conditions. However, the extent of such infiltration in AD and the role of such infiltrating cells remain controversial. Some evidence suggests that peripheral myeloid cells can infiltrate brain tissue and mitigate Aβ deposition and cognitive impairments in AD mouse models91,92,93,94. However, replacement of brain-resident myeloid cells with peripheral monocytes did not modify Aβ load in two mouse models of cerebral β-amyloidosis (APP23 and APP/PS1 mice)95,96. These data raise questions about the respective roles of peripheral monocytes and microglia in clearing Aβ burden.
In addition to peripheral macrophages, natural killer cells infiltrate the brain in the APP/PS1 mouse model of Aβ pathology97 and neutrophils have been observed in the brains of patients with AD and three mouse models of Aβ pathology (APP/PS1, 5×FAD and 3×Tg-AD mice)98,99. Studies in the 3×Tg-AD mouse model have shown that neutrophil depletion or inhibition of neutrophil trafficking reduces Aβ neuropathology and improves memory98. Other studies have implicated neutrophils in the reduced cerebral blood flow observed in AD — in APP/PS1 and 5×FAD mice, the number of cortical capillaries with stalled blood flow was higher than in wild-type animals, largely as a result of neutrophil adherence in capillary segments, which blocked blood flow100. Administration of antibodies against the neutrophil marker Ly6G rapidly reduced the number of stalled capillaries, immediately increasing cerebral blood flow and rapidly improving performance in spatial and working memory tasks100. These studies suggest that neutrophils contribute to AD pathogenesis and cognitive impairment, and hence constitute a potential new therapeutic target that originates in the periphery98.
With respect to the mechanisms by which peripheral innate immune cells infiltrate the CNS, the BBB dysfunction that is observed in AD has been proposed as the basis for a direct and/or indirect route. Alterations in BBB function that could promote such infiltration include increased permeability as a result of deterioration of the molecular and cellular components of the endothelial, pericyte and/or capillary walls, leading to aberrant promotion of migration of activated leukocytes from the periphery into the brain. For a detailed description of this process the reader is referred to a review by Sweeney et al.101.
Innate immune mechanisms of AD pathogenesis
In the context of neurodegenerative diseases, several studies have investigated the pathomechanisms by which systemic inflammation could cause long-term cognitive decline and contribute to neurodegeneration102. Systemic inflammation affects neuronal populations that are compromised in AD, such as cholinergic neurons and their projections103, and such damage could directly affect memory function and cognitive performance. However, systemic inflammation also compromises the key functions of microglia, such as the phagocytosis and degradation of Aβ deposits88,89, which could fuel secondary harmful events. In this way, the influence of peripheral immunological processes on AD pathogenesis could be indirect, via crosstalk with the central immune system.
In the CNS, cerebral deposition of Aβ peptides initiates immune cascades in microglia and astroglia and this immune stimulation initially activates a phagocytic clearance response. However, as inflammation becomes chronic, deleterious effects of chronically activated microglia take over and contribute to disease progression, neuronal dysfunction and damage104. In APP/PS1 transgenic mice, blockade of the microglial NLRP3 inflammasome, a central and upstream innate immune pathway, prevented inflammatory polarization of microglia and the development of AD pathology and spatial memory dysfunction105. NLRP3-dependent IL-1β production and peripheral induction of several other cytokines, including TNF, impairs neuronal function and causes direct neuronal damage and synaptic deficits106,107,108,109. Inflammatory signals can synergistically interact with and potentiate further neurodegenerative mechanisms, such as excitotoxicity110.
Adding further complexity, chronic inflammatory activation of microglia can lead to pyroptosis, which results in the release of apoptosis-associated speck-like protein containing a CARD (ASC) complexes, also known as ASC specks. In the periphery, release of ASC specks ensures a rapid and efficient response of the innate immune system111, but in the brain could serve as a seed for Aβ deposition and thereby directly cause spread of pathology and progression112. Furthermore, some evidence suggests that microglial immune activation also influences neuronal tau pathology. Specifically, in tau transgenic mice, senescent microglia and astroglia have an inflammatory senescence-associated secretory profile, and ablation of these senescent cells prevents neuronal tau pathology113. Genetic knockout of the NLRP3 inflammasome also prevents Aβ-induced spread of tau pathology114. Together, these findings suggest that inflammatory signals contribute to, and possibly cause, tau pathology and subsequent neuronal death.
These central inflammatory mechanisms could all be influenced by systemic inflammation. The widespread dissemination of newly developed protocols to study the behaviour of microglia around Aβ in vivo115 is likely to shed light on these processes and lead to the development of more effective mechanism-based therapies to prevent proteinopathies.
Adaptive immune mechanisms of AD pathogenesis
Clinical studies have indicated a role for the adaptive immune system in AD, but animal studies have provided additional mechanistic insights into the complexity of adaptive immune modulation of microglial responses and Aβ pathology. For example, in one study in Rag-5×FAD mice that were deficient in T cells, B cells and natural killer cells, adaptive immunity seemed to restrain Aβ pathology, possibly by modulating microglial function116. Conversely, results of a study in a RAG-deficient APP/PS1 mouse model, which lack functional T cells and B cells, suggested that adaptive immune processes modulate and dampen microglial responses to misfolded Aβ peptides in the brain, ultimately worsening Aβ pathology117. These contradictory findings suggest that the adaptive immune system has a multifaceted and incompletely understood role in the modification of microglial function, although the differential deficiency in natural killer cells and differences in the mouse models could be relevant to the discordant results.
The increased infiltration of T cells observed in the brain parenchyma of patients with AD79,80,118,119,120 has led to further exploration in mouse models of AD. Most functional studies have focused on Aβ pathology and also suggest a complex picture involving detrimental and beneficial effects of different T cell subtypes. One study in the APP/PS1 mouse model of Aβ pathology showed that Aβ-specific CD8+ T cells do not trigger detrimental autoimmune neuroinflammation121. In APP-Tg J20 mice, Aβ-reactive CD4+ T cells promoted Aβ clearance but also promoted meningoencephalitis122. A study in APP/PS1 mice suggested that Aβ-specific, IFNγ-producing TH1 CD4+ T cells increase Aβ deposition and microglial activation and negatively affect spatial learning123, whereas two other studies have shown that Aβ-specific CD4+ TH2 cells reverse cognitive decline and synaptic loss without infiltrating the brain parenchyma124,125. These studies highlight the fact that Aβ-reactive CD4+ T cells in AD have complex functional effects, with multiple outcomes that depend on the relative magnitude and functionality of different types of T cell responses in addition to the profile of the local neuroinflammatory microenvironment.
Studies in mouse models of Aβ pathology suggest that Treg cells have a critical role in controlling the magnitude of Aβ-specific CD4+ T cell responses in physiological and pathological settings126. In line with this observation, depletion of peripheral Treg cells at early disease stages in APP/PS1 mice accelerated the onset of cognitive deficits and reduced plaque-associated microglia, but did not substantially reduce Aβ deposition. Conversely, selective amplification of Treg cells through treatment with low-dose IL-2 restored cognitive functions and increased plaque-associated microglia. These studies suggest that Treg cells delay progression of Aβ-related cognitive deficits early in the disease course, at least partially by modulating activated microglia127. By contrast, however, peripheral Treg cells can worsen established Aβ amyloid pathology by reducing the parenchymal recruitment of leukocytes through the choroid plexus128.
Much less is known about the interplay between T cell responses, central innate neuroinflammation and tau pathology. A selective association of CD8+ T cell infiltration with tau deposition has been observed in the hippocampus of the THY-tau22 mouse model of AD-like tau pathology129. Early, total T cell depletion prevented cognitive deficits and reduced innate neuroinflammation in these mice, suggesting that tau pathology drives the development of deleterious T cell responses that promote detrimental central innate neuroinflammatory responses129.
An important but unanswered question is the extent to which changes in adaptive immunity reflect normal ageing or progression of disease. In mouse studies, systematic comparisons of AD-like mice and age-matched healthy littermates show that any observed changes in immune function relate to disease progression rather than normal ageing. Further investigations are needed to address whether such immune changes are a cause or a consequence of disease, but several studies of T cell-targeted interventions at very early, presymptomatic stages in AD-like mouse models support the hypothesis that adaptive immunity is instrumental in disease pathogenesis127,129.
The gut microbiome, amyloidosis and peripheral cell infiltration
In support of human studies suggesting that the gut microbiota has a modulatory role in AD clinical outcomes, studies in APP/PS1 mice have revealed a remarkable shift in the gut microbiota compared with wild-type animals. Compared with conventionally raised non-germ-free APP/PS1 mice, germ-free APP/PS1 mice exhibited a substantially lower level of cerebral Aβ pathology, which increased on recolonization with microbiota from non-germ-free APP/PS1 mice. Colonization with microbiota from wild-type mice induced a smaller increase in cerebral Aβ levels. These findings support the idea that pathology-related changes in the microbiota have an instrumental role in the development of Aβ pathology130.
Studies in 5×FAD mice also suggest a mechanistic link between gut microbiota dysbiosis and neuroinflammation in AD. Gut microbiota dysbiosis resulted in increased levels of phenylalanine and isoleucine in the faeces and blood, which subsequently promoted the differentiation and proliferation of peripheral TH1 cells131. Infiltration of these peripheral TH1 cells into the brain contributed to increased pro-inflammatory M1-like microglial activation and neuroinflammatory responses, underscoring the potential for imbalances in the gut microbiota to influence peripheral–central immune crosstalk and AD progression131. Of note, gut microbial metabolites of tryptophan have been shown to act in concert with endogenous type I interferons in the CNS to modulate astrocyte activity and CNS autoimmune neuroinflammation132. Whether similar direct modulation of glial cell activity via gut microbiota-derived metabolites occurs in AD remains to be determined.
Other studies have highlighted possible causal links between microbiome alterations, immune cell function and amyloid plaque deposition, with sex-specific differences in results. In a study of an APP/PS1 transgenic model, disruption of the gut microbiome with an antibiotic cocktail was associated with reduced Aβ plaques and astrogliosis only in male mice133. Similar effects on Aβ pathology and changes in microglial morphology were also seen in another independent APP/PS1 mouse model, but these changes were also restricted to male mice134.
Roadmap
Clinical roadmap
Longitudinal, multimodal appraisal
AD is characterized by years, if not decades, of asymptomatic stages. Despite overwhelming evidence for a temporal lag between the initial pathogenic events and subsequent clinical manifestations, our current understanding of these disease trajectories is compromised by ascertainment biases and methodological limitations. The field is rapidly shifting towards appraisal of presymptomatic states, but a large portion of multimodal biomarker data and clinical information is obtained at the time of symptom onset. In addition, most immunological data are cross-sectional and compare healthy individuals in the community with subspecialty clinic-based, convenience samples of symptomatic adults, which might not provide an accurate reflection of the complex presentations of AD across the disease spectrum. Disease pathogenesis and the mechanisms of destruction and protection are likely to undergo dynamic, non-linear changes over time during the presymptomatic and clinical stages of disease. Therefore, a key step towards advancing the field will be the ability to perform comprehensive assessment of longitudinal immunological and AD biomarker information in richly phenotyped, demographically diverse cohorts.
Inherent to this dictum is the need for simultaneous appraisal of immunological changes — innate and adaptive — in peripheral and central compartments over time, and for mapping of these changes onto predictive models of AD pathogenesis and progression. This kind of assessment will require the use of large-scale cohorts of asymptomatic ageing adults in midlife through to late life and of carriers of AD risk alleles (for example, APOE ε4) and causative gene mutations (for example, mutations in PS1 or PS2)135,136,137 before disease onset. This approach will also provide an understanding of whether peripheral immune changes occur in tandem with or precede CNS immune alterations in the development of AD. This knowledge is critical to determining which aspects of peripheral and central immune dysregulation have true causal roles in the development of AD and which aspects are pathological consequences of the disease that affect progression.
To maximize the information gained from multimodal longitudinal studies, capitalizing on machine learning tools and/or other advanced computational approaches to leverage immunological data and develop predictive models of AD could also be beneficial, provided that these models are thoughtfully incorporated into applied conceptual frameworks138. Such information is also likely to aid identification of appropriate windows for treatments targeting immune mechanisms that are fuelling symptom development and disease progression at a specific time point.
Assessment of the human exposome
A full understanding of the immunological contribution to AD also requires a better understanding of the influence of the individual genetic background and the life exposome — the non-genetic lifestyle behaviours and health-related environmental influences that modulate the risk of developing AD. The study of cerebrovascular disease is a prime example of how a comprehensive understanding of health-related behaviours can improve our understanding of disease pathogenesis, prevention and treatment. The same level of rigour is required in AD. Specifically, we need extensive appraisal of how vascular risk factors (for example, midlife obesity, smoking, alcohol consumption and physical inactivity), aseptic insults (for example, surgical procedures), chronic illnesses, mood function (for example, stress), early and midlife environmental exposures, and diet interact with immune-related genes to influence inflammageing processes and the development of (or protection from) disease. The aforementioned risk factors are all known to be independently associated with inflammatory processes, so could create a peripheral milieu that is conducive to or permissive of AD pathology139.
Extending this line of reasoning further, pathological ageing and AD should be viewed as a systemic disease140,141. Dysregulated immune homeostasis and responses occur in the milieu of a highly complex biological system that can be thought of as a confluence of lifespan exposures to protective factors and pro-inflammatory insults. To better understand the dynamic changes in innate and adaptive immune responses associated with AD, we need to delineate how systemic exposures across a human life affects clinical expression of disease. Midlife, in particular, has emerged in the past decade as a critical period of exposure to classic cardiovascular risk factors and inflammatory insults that can lead to immune dysregulation and therefore influence AD trajectories142. A better understanding of how physiological changes and environmental exposure in midlife disproportionately affect ageing trajectories (relative to late-life exposures) will be critical for the development of a comprehensive biological model of peripheral–central crosstalk in AD.
An initial step forward in this area would be to identify the degree to which co-pathologies contribute to immune function and regulation in ageing; examples of co-pathologies include vascular pathology and insulin resistance, both of which occur frequently in AD. Another step would be to explore the roles of single and repeated peripheral events, ranging from acute infections to sepsis and associated organ failure, in the pathological ageing processes and elucidate how these events prospectively relate to immune factors and AD-related pathology55. This knowledge should provide critical insight into whether peripheral insults exacerbate an already primed system or whether they further influence early AD pathological cascades. This aspect is particularly salient given the ongoing COVID-19 pandemic, which has initiated research into the short-term and long-term neurological outcomes of SARS-CoV-2 exposure in adults with and without AD dementia.
Longer-term goals for the field would be to develop testable models of the ways in which lifetime exposures and inflammatory insults — including COVID-19 — affect cognitive ageing trajectories, thereby bridging epidemiology, bioinformatics, clinical neuroscience and immunology perspectives to inform precision medicine approaches. These precision medicine approaches could ultimately aid the development of stratification frameworks for recruitment of patients into clinical trials and the development of personalized immunotherapies.
Expanding the methodological toolkit
A well-founded criticism of clinical research into immune function in AD is the disproportionate focus on circulating protein levels. This focus provides a static window into the immune milieu of an ageing adult, which is important but does not reveal functional changes in the innate and adaptive immune system. To overcome this gap in the clinical research landscape, more in-depth analysis of the function of immune effector cells isolated from the CSF and peripheral blood is critical. Specific functional aspects to study include their inducibility and responsiveness to immune challenges, their transcriptome at the single-cell level, and comparison of immune function across compartments (CSF, blood and gut), and these alterations should be mapped onto clinical trajectories and outcomes. Use of multimodal approaches that make simultaneous use of these techniques (for example, high-dimensional flow cytometry, mass cytometry, RNAseq and single-cell analysis methods) will also be critical for forming a full understanding of peripheral–central immune dysregulation in AD. Similarly, further deciphering of the antigen specificity and functional differentiation profiles of adaptive immune responses associated with the development of AD is critical for a better understanding of their roles in disease pathogenesis, to enable implementation of innovative immunotherapy approaches and to identify new immune-related biomarkers of prognostic interest.
Finally, advances in PET imaging of microglial activation, reactive astrocytes and BBB permeability will be critical for developing better human models of in vivo glial biology and immune crosstalk in ageing adults. The most commonly used PET tracer for imaging of microglia is TSPO, although this ligand has several disadvantages, including a lack of specificity for microglia143 and a lack of sensitivity to different functional states144. Ligands that are specific to the immune cell type (for example, microglia versus astrocytes) and are sensitive to the range of activational states (beyond M1-like and M2-like microglia or A1 and A2 astrocyte phenotypes), and can capture dynamic changes in BBB alterations145 will strengthen our ability to appraise longitudinal communication between the peripheral and central immune systems in ageing adults, particularly when used in tandem with traditional CSF and blood biomarkers of inflammation.
Experimental roadmap
Solidifying the role of the peripheral compartment
GWAS have provided clues about innate immune dysfunction in AD by identifying associated genes in brain-resident immune cells, such as microglia, but whether peripheral innate immune cells have important roles in the pathogenesis of AD is unclear. Moreover, the extent to which the dysfunction or altered homeostasis of such innate immune effector cells translates into or synergizes with dysregulation in either humoral or cellular adaptive immunity remains unknown. The importance of the peripheral immune compartment in AD is a key unanswered question, particularly given that chronic, systemic inflammation and peripheral comorbidities, such as metabolic syndrome, type 2 diabetes mellitus and cardiovascular conditions, are known risk factors for AD23,146. Therefore, lineage-tracing, depletion and add-back approaches should be used to further demonstrate the necessity and sufficiency of specific peripheral immune cell populations and their functional subsets in the pathogenesis and progression of relevant AD pathologies.
For any new or repurposed immunomodulatory drug that is translated into clinical settings for the treatment of AD, it will be important to demonstrate that it affords reproducible therapeutic benefits in an animal model of AD-like pathology. This demonstration should include evidence that the drug engages a target or mechanism that is directly involved in peripheral–central neuroimmune processes involved in AD, such as peripheral inflammatory factors, dysregulated homeostasis of peripheral immune effectors, or modulation of immune cell trafficking to the brain.
Systemic inflammation and peripheral infections have been implicated in cognitive decline and acceleration of AD trajectories, but animal studies to directly test the involvement of peripheral immune mechanisms are insufficient and further investigation is warranted. Compelling animal studies also suggest a role for gut microbiota dysbiosis (and microbiome alterations more broadly) in immune-mediated AD progression, but further studies are needed to better understand the timing of the effects, sex differences in outcomes, and mechanisms of peripheral–central crosstalk. Once mechanisms for these associations are found, the key will be to strategically target the process or immune cell subsets that can restore homeostatic immune function and ultimately foster neuronal survival without compromising other beneficial immune processes.
The role of adaptive immunity and the lymphatic–glymphatic system
Studies have shown that adaptive immunity has an important role in AD, but several questions remain with respect to the antigen specificity of T cells that invade the CNS barriers and/or infiltrate the parenchyma. Assessment of whether such T cells are specific for disease-related pathological proteins and/or other self or non-self antigens is critical. These studies should also include further evaluation of the role of the meningeal lymphatic–glymphatic system. This pathway is necessary for drainage of the CSF, brain interstitial fluid and, consequently, clearance of molecules from the brain into the cervical lymph nodes147. Although studies have suggested that age-related dysfunction of the meningeal lymphatic–glymphatic system can exacerbate brain and meningeal Aβ pathology, its impact on the development of disease-related T cell responses remains unknown.
The role of astrocytes and microglia
The involvement of astrocytes in neurodegeneration has been vastly under-studied. As for macrophages, the reactive phenotypes of astrocytes in culture have been described, but further studies are needed to identify their complete range of functional subsets and their roles in neurodegeneration148. Although astrocytes have frequently been studied in tandem with microglia (often from the perspective of neuroinflammatory microglia inducing neurotoxic reactivity in astrocytes), there is clear evidence of bidirectional communication between the cells that suggests greater complexity of astrocyte function in the secretion of inflammatory mediators149. Whether astrocytes are bystanders in degenerative cascades or provide signals to microglia that foster detrimental activity remains unclear. Therefore, the role of astrocytes, both independently and in concert with microglia, in AD pathogenesis and progression needs to be investigated further in animal models of AD-like pathology. The impact of systemic inflammation and peripheral immune effectors on microglia–astrocyte interplay is another key aspect to be addressed.
Tools are lacking for in vivo animal studies of the kinetics of microglia and astrocyte activation in relation both to each other and to progression of AD pathology. Development of reporter mice for high-resolution in vivo imaging of these and other immune cell types would enable substantial advances in the field. These and other tools are needed to study dysregulated central–peripheral crosstalk in vivo to better translate mechanistic animal studies into the clinic.
Translational roadmap
Reconcile differences between species
Animal studies inform us about potential mechanisms that underlie a disease process and enable identification of targets for therapeutic interventions, but immunological differences between rodents and humans limit the extent to which we can extrapolate findings from animal studies to humans. For example, differences in metabolic rates between mice, rats and humans means that dose-finding studies need to be conducted in humans rather than animals to establish safety and toxicity. In light of this knowledge, it is critical to identify innovative ways to translate mechanistic animal studies of central–peripheral immune crosstalk into the clinical research space to move the field forward.
Studies of the turnover of apolipoprotein E (ApoE) isoforms provide a good example of how animal studies can inform and synergize with clinical studies. The isoform and the amount of ApoE in the brain modulate AD pathology by altering the extent of Aβ peptide deposition, so quantification of ApoE isoform production and clearance rates in the brain versus the periphery could improve our understanding of the role of ApoE in neurological diseases. Humanized ApoE mouse models have been seminal in establishing that turnover rates of ApoE isoforms in the central compartment differ substantially from turnover rates in the peripheral compartment, consistent with differences in the pathways that are responsible for ApoE metabolism in the two compartments150.
Alternative models, such as 3D in vitro models (for example, organoids and 3D matrix cultures) could also have potential to provide mechanistic insights into AD-associated immune dysregulation151. Use of such 3D models is advancing rapidly, but further developments are needed if these experimental models are to improve our understanding of peripheral–central immune crosstalk. These models cannot comprehensively answer questions about central–peripheral interactions in isolation owing to the absence of tissue components that are highly relevant to AD disease processes and related neuroimmune interactions. For example, 3D in vitro models lack the complex vascular systems and components of meningeal immunity, such as meningeal lymphatics, that are present in vivo and are critical for understanding bidirectional interactions between the peripheral and central immune systems. Consequently, until further advances are made in the complexity of these models, their use is mostly limited to studies that complement those in animal models.
Strategic immune-based therapeutics
The potential for therapeutic intervention in AD by targeting immune-mediated processes has been recognized as a result of GWAS that have implicated the immune system in AD152,153 and epidemiological studies that have linked the use of non-steroidal anti-inflammatory drugs (NSAIDs) with a reduced incidence of AD56,154. Despite these compelling findings, results from randomized clinical trials have been disappointing155. As we plan for new clinical trials of immune-based therapies for AD and other neurological conditions, we must take into account the lessons learned.
The simplest explanation for the lack of success with immunotherapy or anti-inflammatory therapy in past trials in AD is the inclusion of patients with advanced dementia and fully established disease and pathology. Treatment failure at this stage of severity could just mean that the spread of pathology cannot be halted and/or too many neurons have been lost to see a therapeutic benefit56. Therefore, earlier intervention is likely to be necessary, and the latest genomic information supports this idea. A genome-wide meta-analysis published in 2019 identified new risk loci and pathways associated with AD, and >60% of the hits were immune-specific, suggesting that immunological processes are part of the causal pathway of AD156. On the basis of this information, it seems imperative that we start to view neurodegenerative diseases in an entirely new light: not as diseases that start with cell-autonomous neuronal dysfunction, but diseases in which immune dysfunction is the primary cause of impaired clearance of toxic protein aggregates and debris that leads to the demise of neurons.
If this view of neurodegenerative diseases is true, then the use of approaches that boost or modulate innate and/or adaptive immune functions to promote clearance of toxic protein aggregates at early stages would be warranted. Such therapeutic strategies would probably need to be modified according to the pathological staging of the disease. Knowledge of which immunomodulatory therapeutics can prevent progression of disease and which slow disease would shed light on the long-standing debate about which aspects of immune dysregulation are a cause of disease and which are pathological consequences and/or exacerbate disease. Given that initial deposition of abnormal proteins begins decades before clinical symptoms of AD, clinical trials have yet to address whether therapeutic intervention in midlife would be more beneficial than intervention once patients are symptomatic. Moreover, numerous questions remain as to whether targeted therapeutics that selectively modulate key immune-mediated processes but do not suppress the entire immune response would yield greater success.
Conclusions
The intersection of immune regulation and neurodegeneration has introduced complexity to our understanding of AD pathogenesis and presents an exciting avenue for the identification of new biomarkers and therapeutic targets and the development of novel interventions. Compelling evidence suggests that AD should be viewed as a systemic disease that involves dynamic responses in the peripheral and central immune compartments. In this context, examining the central immune system without accounting for the role of the periphery and the peripheral–central interface is likely to be insufficient for a complete understanding of the pathophysiology of AD and the immune processes involved. We have underscored the need to examine, appraise and ultimately modulate the crosstalk between peripheral and central immunity and suggested a roadmap for taking the field forward to improve treatment and, ultimately, to the prevention of AD.
Change history
08 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41582-021-00579-5
References
Paouri, E. & Georgopoulos, S. Systemic and CNS inflammation crosstalk: implications for Alzheimer’s disease. Curr. Alzheimer Res. 16, 559–574 (2019).
Morris, G., Berk, M., Maes, M. & Puri, B. K. Could Alzheimer’s disease originate in the periphery and if so how so? Mol. Neurobiol. 56, 406–434 (2019).
Stephenson, J., Nutma, E., van der Valk, P. & Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219 (2018).
Webers, A., Heneka, M. T. & Gleeson, P. A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 98, 28–41 (2020).
Kinney, J. W. et al. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590 (2018).
Le Page, A. et al. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 107, 59–66 (2018).
Jenny, N. S. et al. Long-term assessment of inflammation and healthy aging in late life: the Cardiovascular Health Study All Stars. J. Gerontol. A Biol. Sci. Med. Sci. 67, 970–976 (2012).
Metti, A. L. et al. Change in inflammatory markers and cognitive status in the oldest-old women from the Study of Osteoporotic Fractures. J. Am. Geriatrics Soc. 62, 662–666 (2014).
Sundelof, J. et al. Systemic inflammation and the risk of Alzheimer’s disease and dementia: a prospective population-based study. J. Alzheimers Dis. 18, 79–87 (2009).
Lanzrein, A. S. et al. Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: interleukin-1β, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-a, the soluble tumor necrosis factor receptors I and II, and α1-antichymotrypsin. Alzheimer Dis. Associated Disord. 12, 215–227 (1998).
Ravaglia, G. et al. Blood inflammatory markers and risk of dementia: the Conselice Study of Brain Aging. Neurobiol. Aging 28, 1810–1820 (2007).
Eriksson, U. K. et al. Associations of gene sequence variation and serum levels of C-reactive protein and interleukin-6 with Alzheimer’s disease and dementia. J. Alzheimers Dis. 23, 361–369 (2011).
Swardfager, W. et al. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68, 930–941 (2010).
Lai, K. S. P. et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88, 876–882 (2017).
Darweesh, S. K. L. et al. Inflammatory markers and the risk of dementia and Alzheimer’s disease: a meta-analysis. Alzheimers Dement. 14, 1450–1459 (2018).
Koyama, A. et al. The role of peripheral inflammatory markers in dementia and Alzheimer’s disease: a meta-analysis. J. Gerontol. A Biol. Sci. Med. Sci. 68, 433–440 (2013).
Schmidt, R. et al. Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann. Neurol. 52, 168–174 (2002).
Engelhart, M. J. et al. Inflammatory proteins in plasma and the risk of dementia: the Rotterdam Study. Arch. Neurol. 61, 668–672 (2004).
Tan, Z. S. et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology 68, 1902–1908 (2007).
Weaver, J. D. et al. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 59, 371–378 (2002).
Yaffe, K. et al. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology 61, 76–80 (2003).
Singh-Manoux, A. et al. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology 83, 486–493 (2014).
Yaffe, K. et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 292, 2237–2242 (2004).
Tao, Q. et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw. Open 1, e183597 (2018).
Walker, K. A. et al. Systemic inflammation during midlife and cognitive change over 20 years: the ARIC Study. Neurology 92, e1256–e1267 (2019).
Gottesman, R. F. et al. Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) cohort. JAMA Neurol. 74, 1246–1254 (2017).
Bettcher, B. M. et al. Increases in a pro-inflammatory chemokine, MCP-1, are related to decreases in memory over time. Front. Aging Neurosci. 11, 25 (2019).
Marsland, A. L. et al. Interleukin-6 covaries inversely with cognitive performance among middle-aged community volunteers. Psychosom. Med. 68, 895–903 (2006).
Wersching, H. et al. Serum C-reactive protein is linked to cerebral microstructural integrity and cognitive function. Neurology 74, 1022–1029 (2010).
Jefferson, A. L. et al. Inflammatory biomarkers are associated with total brain volume: the Framingham Heart Study. Neurology 68, 1032–1038 (2007).
Bettcher, B. M. et al. C-reactive protein is related to memory and medial temporal brain volume in older adults. Brain Behav. Immun. 26, 103–108 (2012).
Walker, K. A. et al. Association of peripheral inflammatory markers with connectivity in large-scale functional brain networks of non-demented older adults. Brain Behav. Immun. 87, 388–396 (2020).
Walker, K. A. et al. The association of mid- and late-life systemic inflammation with brain amyloid deposition: the ARIC-PET Study. J. Alzheimers Dis. 66, 1041–1052 (2018).
Shen, X. N. et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 90, 590–598 (2019).
Diniz, B. S. et al. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 22, 1305–1311 (2010).
Saleem, M., Herrmann, N., Swardfager, W., Eisen, R. & Lanctot, K. L. Inflammatory markers in mild cognitive impairment: a meta-analysis. J. Alzheimers Dis. 47, 669–679 (2015).
Leung, R. et al. Inflammatory proteins in plasma are associated with severity of Alzheimer’s disease. PLoS ONE 8, e64971 (2013).
Galimberti, D. et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 27, 1763–1768 (2006).
Morgan, A. R. et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 15, 776–787 (2019).
Motta, M., Imbesi, R., Di Rosa, M., Stivala, F. & Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: correlation with the disease progression. Immunol. Lett. 114, 46–51 (2007).
Brosseron, F., Krauthausen, M., Kummer, M. & Heneka, M. T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol. Neurobiol. 50, 534–544 (2014).
Riphagen, J. M. et al. Linking APOE-epsilon4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol. Aging 85, 96–103 (2020).
Popp, J. et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav. Immun. 62, 203–211 (2017).
Bettcher, B. M. et al. Cerebrospinal fluid and plasma levels of inflammation differentially relate to CNS markers of Alzheimer’s disease pathology and neuronal damage. J. Alzheimers Dis. 62, 385–397 (2018).
Jack, C. R. Jr. et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018).
Yang, H. S. et al. Plasma IL-12/IFN-γ axis predicts cognitive trajectories in cognitively unimpaired older adults. Alzheimers Dement https://doi.org/10.1002/alz.12399 (2021).
Thome, A. D. et al. Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener. 13, 61 (2018).
Saresella, M. et al. IL-33 and its decoy sST2 in patients with Alzheimer’s disease and mild cognitive impairment. J. Neuroinflammation 17, 174 (2020).
Dong, Y. et al. Neutrophil hyperactivation correlates with Alzheimer’s disease progression. Ann. Neurol. 83, 387–405 (2018).
Linton, P. J. & Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 5, 133–139 (2004).
Pawelec, G. et al. T cells and aging, January 2002 update. Front. Biosci. 7, d1056–d1183 (2002).
Danielson, M. et al. Neuroinflammatory markers associate with cognitive decline after major surgery: findings of an explorative study. Ann. Neurol. 87, 370–382 (2020).
Holmes, C., Cunningham, C., Zotova, E., Culliford, D. & Perry, V. H. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 77, 212–218 (2011).
Cunningham, C. Systemic inflammation and delirium: important co-factors in the progression of dementia. Biochem. Soc. Trans. 39, 945–953 (2011).
Iwashyna, T. J., Ely, E. W., Smith, D. M. & Langa, K. M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304, 1787–1794 (2010).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Holmes, C. et al. Systemic infection, interleukin 1β, and cognitive decline in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 788–789 (2003).
Holmes, C. et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774 (2009).
Sipila, P. N. et al. Hospital-treated infectious diseases and the risk of dementia: a large, multicohort, observational study with a replication cohort. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(21)00144-4 (2021).
Rakic, S. et al. Systemic infection modifies the neuroinflammatory response in late stage Alzheimer’s disease. Acta Neuropathol. Commun. 6, 88 (2018).
Westin, K. et al. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 7, e30525 (2012).
Pillai, J. A. et al. Inflammatory pathway analytes predicting rapid cognitive decline in MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 7, 1225–1239 (2020).
Ewers, M. et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl Med. 11, eaav6221 (2019).
Taipa, R. et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 76, 125–132 (2019).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292 (2011).
Meyer, P. F. et al. Bi-directional association of cerebrospinal fluid immune markers with stage of Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 63, 577–590 (2018).
Bradburn, S., Murgatroyd, C. & Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. Ageing Res. Rev. 50, 1–8 (2019).
Hamelin, L. et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer’s disease. Brain 141, 1855–1870 (2018).
Whelan, C. D. et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 7, 169 (2019).
Larbi, A. et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J. Alzheimers Dis. 17, 91–103 (2009).
Pellicano, M. et al. Immune profiling of Alzheimer patients. J. Neuroimmunol. 242, 52–59 (2012).
McManus, R. M., Mills, K. H. & Lynch, M. A. T cells–protective or pathogenic in Alzheimer’s disease? J. Neuroimmune Pharmacol. 10, 547–560 (2015).
Saresella, M. et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav. Immun. 25, 539–547 (2011).
Ciccocioppo, F. et al. The characterization of regulatory T-cell profiles in Alzheimer’s disease and multiple sclerosis. Sci. Rep. 9, 8788 (2019).
Saresella, M. et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 21, 927–938 (2010).
Le Page, A. et al. Differential phenotypes of myeloid-derived suppressor and T regulatory cells and cytokine levels in amnestic mild cognitive impairment subjects compared to mild Alzheimer diseased patients. Front. Immunol. 8, 783 (2017).
Lueg, G. et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol. Aging 36, 81–89 (2015).
Gate, D. et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020).
Zotova, E. et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 136, 2677–2696 (2013).
Merlini, M., Kirabali, T., Kulic, L., Nitsch, R. M. & Ferretti, M. T. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener. Dis. 18, 49–56 (2018).
Zhuang, Z. Q. et al. Gut microbiota is altered in patients with Alzheimer’s disease. J. Alzheimers Dis. 63, 1337–1346 (2018).
Vogt, N. M. et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7, 13537 (2017).
Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017).
Wilmanski, T. et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 3, 274–286 (2021).
Walker, K. A., Ficek, B. N. & Westbrook, R. Understanding the role of systemic inflammation in Alzheimer’s disease. ACS Chem. Neurosci. 10, 3340–3342 (2019).
Dantzer, R., Konsman, J. P., Bluthe, R. M. & Kelley, K. W. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? Auton. Neurosci. 85, 60–65 (2000).
Engelhardt, B., Vajkoczy, P. & Weller, R. O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131 (2017).
Wendeln, A. C. et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018).
Tejera, D. et al. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 38, e101064 (2019).
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P. & Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502 (2006).
El Khoury, J. et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438 (2007).
Town, T. et al. Blocking TGF-β–Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687 (2008).
Naert, G. & Rivest, S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 6208–6220 (2011).
Varvel, N. H. et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 212, 1803–1809 (2015).
Prokop, S. et al. |Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 212, 1811–1818 (2015).
Kelly, R. J. et al. Glial activation in AβPP/PS1 mice is associated with infiltration of IFNγ-producing cells. J. Alzheimers Dis. 37, 63–75 (2013).
Zenaro, E. et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886 (2015).
Minogue, A. M. et al. Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol. Aging 35, 1442–1452 (2014).
Cruz Hernandez, J. C. et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 22, 413–420 (2019).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Okroglic, S., Widmann, C. N., Urbach, H., Scheltens, P. & Heneka, M. T. Clinical symptoms and risk factors in cerebral microangiopathy patients. PLoS ONE 8, e53455 (2013).
Semmler, A. et al. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp. Neurol. 204, 733–740 (2007).
Sarlus, H. & Heneka, M. T. Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249 (2017).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013).
Tancredi, V. et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 146, 176–178 (1992).
Murray, C. A., Clements, M. P. & Lynch, M. A. Interleukin-1 induces lipid peroxidation and membrane changes in rat hippocampus: an age-related study. Gerontology 45, 136–142 (1999).
McAlpine, F. E. et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 34, 163–177 (2009).
MacPherson, K. P. et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95 (2017).
Hewett, S. J., Csernansky, C. A. & Choi, D. W. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron 13, 487–494 (1994).
Franklin, B. S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737 (2014).
Venegas, C. et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361 (2017).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Ising, C. et al. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019).
Tejera, D. & Heneka, M. T. In vivo phagocytosis analysis of amyloid beta. Methods Mol. Biol. 2034, 287–292 (2019).
Marsh, S. E. et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl Acad. Sci. USA 113, E1316–E1325 (2016).
Spani, C. et al. Reduced β-amyloid pathology in an APP transgenic mouse model of Alzheimer’s disease lacking functional B and T cells. Acta Neuropathol. Commun. 3, 71 (2015).
Togo, T. et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 124, 83–92 (2002).
Itagaki, S., McGeer, P. L. & Akiyama, H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci. Lett. 91, 259–264 (1988).
Rogers, J., Luber-Narod, J., Styren, S. D. & Civin, W. H. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 9, 339–349 (1988).
Rosset, M. B., Lui, G., Dansokho, C., Chaigneau, T. & Dorothee, G. Vaccine-induced Aβ-specific CD8+ T cells do not trigger autoimmune neuroinflammation in a murine model of Alzheimer’s disease. J. Neuroinflammation 12, 95 (2015).
Monsonego, A. et al. Aβ-induced meningoencephalitis is IFN-γ-dependent and is associated with T cell-dependent clearance of Aβ in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 103, 5048–5053 (2006).
Browne, T. C. et al. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 190, 2241–2251 (2013).
Cao, C. et al. Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiol. Dis. 34, 63–70 (2009).
Ethell, D. W. et al. Aβ-specific T-cells reverse cognitive decline and synaptic loss in Alzheimer’s mice. Neurobiol. Dis. 23, 351–361 (2006).
Toly-Ndour, C. et al. MHC-independent genetic factors control the magnitude of CD4+ T cell responses to amyloid-β peptide in mice through regulatory T cell-mediated inhibition. J. Immunol. 187, 4492–4500 (2011).
Dansokho, C. et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139, 1237–1251 (2016).
Baruch, K. et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6, 7967 (2015).
Laurent, C. et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140, 184–200 (2017).
Harach, T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7, 41802 (2017).
Wang, X. et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 29, 787–803 (2019).
Rothhammer, V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Minter, M. R. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 6, 30028 (2016).
Dodiya, H. B. et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp. Med. 216, 1542–1560 (2019).
Farfara, D. et al. γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 69, 170–180 (2011).
Ledo, J. H. et al. Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Mol. Psychiatry https://doi.org/10.1038/s41380-020-0856-8 (2020).
Fung, S. et al. Early-onset familial Alzheimer disease variant PSEN2 N141I heterozygosity is associated with altered microglia phenotype. J. Alzheimers Dis. 77, 675–688 (2020).
Glymour, M. M. & Bibbins-Domingo, K. The future of observational epidemiology: improving data and design to align with population health. Am. J. Epidemiol. 188, 836–839 (2019).
Heneka, M. T., Golenbock, D. T. & Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236 (2015).
Morris, J. K., Honea, R. A., Vidoni, E. D., Swerdlow, R. H. & Burns, J. M. Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 1842, 1340–1349 (2014).
Wang, J., Gu, B. J., Masters, C. L. & Wang, Y. J. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 13, 612–623 (2017).
Ritchie, K., Ritchie, C. W., Yaffe, K., Skoog, I. & Scarmeas, N. Is late-onset Alzheimer’s disease really a disease of midlife? Alzheimers Dement. 1, 122–130 (2015).
Gui, Y., Marks, J. D., Das, S., Hyman, B. T. & Serrano-Pozo, A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. 30, 151–164 (2020).
Boche, D., Gerhard, A., Rodriguez-Vieitez, E. & Faculty, M. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 46, 2831–2847 (2019).
Turkheimer, F. E. et al. Increased serum peripheral C-reactive protein is associated with reduced brain barriers permeability of TSPO radioligands in healthy volunteers and depressed patients: implications for inflammation and depression. Brain Behav. Immun. 91, 487–497 (2021).
Gottesman, R. F. et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317, 1443–1450 (2017).
Da Mesquita, S., Fu, Z. & Kipnis, J. The meningeal lymphatic system: a new player in neurophysiology. Neuron 100, 375–388 (2018).
Liddelow, S. A. & Barres, B. A. Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967 (2017).
McAlpine, C. S. et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595, 701–706 (2021).
Wildsmith, K. R. et al. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS ONE 7, e38013 (2012).
Cenini, G. et al. Dissecting Alzheimer’s disease pathogenesis in human 2D and 3D models. Mol. Cell Neurosci. 110, 103568 (2021).
Shi, Y. & Holtzman, D. M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772 (2018).
Scheiblich, H., Trombly, M., Ramirez, A. & Heneka, M. T. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 41, 300–312 (2020).
McGeer, P. L., Rogers, J. & McGeer, E. G. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimers Dis. 9, 271–276 (2006).
Arvanitakis, Z. et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 70, 2219–2225 (2008).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Acknowledgements
This manuscript was facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART), through the Immunity and Neurodegeneration Professional Interest Area (PIA). The views and opinions expressed by the authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer’s Association. B.M.B. is a funded investigator of the NIH/NIA (R01 AG058772) and the Department of Defense CDMRP. M.G.T. is a funded investigator of the NIH/NIA (7RF1AG057247), the Parkinson’s Foundation and the Michael J. Fox Foundation for Parkinson’s Research. G.D. is a funded investigator of the Fondation Plan Alzheimer, Fondation pour la Recherche Medicale, LECMA/Fondation Vaincre Alzheimer and Alzheimer Research UK. M.T.H. is a funded investigator of the German Research Council (DFG) Cluster of Excellence and has received support from this work through an NIH grant (R01 AG059752-02).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
M.G.T. is a member of the Medical Scientific Advisory Group to the Alzheimer’s Association and a consultant to INmune Bio. G.D. is listed as an inventor on patent WO2014206899A1 related to a peripheral immunomodulatory approach for treating Alzheimer disease and related disorders, and on patent WO2018172540A1 related to peripheral immune biomarkers for predicting the progression of Alzheimer disease. The other authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Neurology thanks O. Garaschuk and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bettcher, B.M., Tansey, M.G., Dorothée, G. et al. Peripheral and central immune system crosstalk in Alzheimer disease — a research prospectus. Nat Rev Neurol 17, 689–701 (2021). https://doi.org/10.1038/s41582-021-00549-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-021-00549-x
This article is cited by
-
Peripheral blood amyloid-β involved in the pathogenesis of Alzheimer’s disease via impacting on peripheral innate immune cells
Journal of Neuroinflammation (2024)
-
Modeling the neuroimmune system in Alzheimer’s and Parkinson’s diseases
Journal of Neuroinflammation (2024)
-
Exercise mimetics: a novel strategy to combat neuroinflammation and Alzheimer’s disease
Journal of Neuroinflammation (2024)
-
Genetic overlap between Alzheimer’s disease and immune-mediated diseases: an atlas of shared genetic determinants and biological convergence
Molecular Psychiatry (2024)
-
Current understanding of the Alzheimer’s disease-associated microbiome and therapeutic strategies
Experimental & Molecular Medicine (2024)
