
Abstract
Individuals who are diagnosed with amyotrophic lateral sclerosis (ALS) today face the same historically intransigent problem that has existed since the initial description of the disease in the 1860s — a lack of effective therapies. In part, the development of new treatments has been hampered by an imperfect understanding of the biological processes that trigger ALS and promote disease progression. Advances in our understanding of these biological processes, including the causative genetic mutations, and of the influence of environmental factors have deepened our appreciation of disease pathophysiology. The consequent identification of pathogenic targets means that the introduction of effective therapies is becoming a realistic prospect. Progress in precision medicine, including genetically targeted therapies, will undoubtedly change the natural history of ALS. The evolution of clinical trial designs combined with improved methods for patient stratification will facilitate the translation of novel therapies into the clinic. In addition, the refinement of emerging biomarkers of therapeutic benefits is critical to the streamlining of care for individuals. In this Review, we synthesize these developments in ALS and discuss the further developments and refinements needed to accelerate the introduction of effective therapeutic approaches.
Key points
-
The development of effective treatments for amyotrophic lateral sclerosis (ALS) has been limited by a lack of comprehensive understanding of the biological processes that trigger the disease and promote progression.
-
Causative genetic mutations have been identified, many of which are linked to RNA function and metabolism.
-
Disease heterogeneity suggests that a precision medicine paradigm incorporating extensive phenotypic and genotypic information will be required to realize effective therapy and improve the outcomes for individual patients with ALS.
-
The repurposing of drugs with established safety profiles from their use in other human diseases is a new approach to therapeutic discovery in ALS.
-
Enhanced clinical trial designs, including multi-arm, multi-stage platform trials, that incorporate biomarkers of treatment responses will accelerate drug discovery and increase trial participation.
-
Improved patient stratification and patient-reported outcome measures, including home assessments, will improve the reliability and sensitivity of trial endpoints.
Similar content being viewed by others

Introduction
Neurodegeneration and dementia pose a major public health challenge worldwide owing to their devastating impact on quality of life and the tremendous burden they place on health-care systems1,2. The number of adults with a diagnosis of neurodegenerative disease across the globe is set to increase dramatically in the coming decades — the ageing population harbours a potential epidemic of functional disability, pain and loss of independence3,4,5. Among the neurodegenerative disorders, amyotrophic lateral sclerosis (ALS) is the most rapidly fatal6 — the typical time from symptom onset to death is 2–3 years7,8.
Research in the past 25 years has improved our understanding of the pathophysiology of ALS, but the translation of this knowledge into effective treatments has been disappointing — even today, most patients with ALS do not have the opportunity to participate in clinical trials9. Previously unrecognized bottlenecks in the translational process have been revealed, including a lack of large-scale research infrastructure and of the coordination necessary to initiate trials. In addition, inherent disease characteristics, such as disease heterogeneity, make a one-size-fits-all solution unworkable and long durations of clinical trials are necessary owing to a lack of proven biomarkers and clinical trial outcome measures10. Progress in this era of potential therapeutic advance, in which an unparalleled number of disease-modifying therapies are ready to be trialled or implemented into clinical practice, requires the establishment of stronger collaborative networks across clinical and translational neuroscience domains.
In this Review, we first appraise the current status of the ALS field from a basic neuroscience perspective with respect to recent molecular discoveries and then discuss progress in the development of therapeutics, including biomarkers, drug repurposing strategies and high-throughput drug screening. We critically analyse molecular technology and the emergence of genetic and cellular therapies for patients currently living with ALS. Finally, we consider new trial designs that are facilitated by advances in patient-reported outcome measures.
Deconstructing ALS
Clinical identification of ALS starts with the confirmation of upper and lower motor neuron abnormalities that involve the brain, spinal regions and the peripheral neuromuscular system11. Typical clinical presentations include bulbar-onset ALS with speech and swallowing problems and limb-onset ALS that initially manifests with arm or leg weakness, followed by progressive paralysis7,8,12. Other, less common presentations include isolated type 2 respiratory failure, weight loss, cramps, fasciculations in the absence of muscle weakness, emotional lability and cognitive abnormalities13,14. The altered function of upper motor neurons leads to spasticity and brisk deep reflexes8 and symptoms of lower motor neuron abnormalities include fasciculations, muscle wasting and weakness15,16,17. Given that there are no specific radiological or serological markers of ALS, the diagnosis is established on the basis of clinical features as ratified by consensus criteria11.
Approximately 15% of ALS cases are familial, with a Mendelian pattern of inheritance and a high penetrance18,19. Mutations of C9orf72, SOD1, TARDBP, FUS, ANG and OPTN can produce typical ALS phenotypes (Table 1). Many of these mutations affect proteins that are involved in gene expression and regulation via the regulation of transcription, microRNA processing, RNA maturation and splicing. The mutations linked to ALS are commonly grouped according to whether they alter proteostasis, RNA function or the cytoskeleton20,21,22.
By contrast, sporadic ALS presents in individuals without a family history of the disease. Family aggregation studies have identified overlap between sporadic ALS and other neurodegenerative or developmental disorders, suggesting that variants in some susceptibility genes increase the overall risk of brain and mind disorders within families23,24, similar to findings in familial ALS25 that are consistent with genetic pleiotropy26. Attempts to determine the genetic basis of sporadic ALS have produced limited success, supporting the concept of ALS as a common disease associated with multiple rare variants.
The pathophysiological basis of ALS is typically thought to involve glutamate-induced excitotoxicity, structural abnormalities of mitochondria, autophagy, neuroinflammation and disruption of axonal transport mechanisms1,2 (Fig. 1). Non-neuronal cells, including astrocytes and microglia, also play a role in neurodegeneration in ALS via the secretion of neurotoxic mediators and the modulation of glutamate receptor expression27,28. TAR DNA-binding protein 43 (TDP43) is thought to be pathogenic in ALS. This protein is a major component of the ubiquitylated cytoplasmic protein aggregates that are present in most cases of sporadic and familial ALS. The observation that mutations in highly conserved regions of the TARDBP gene are not evident in controls and segregate with the disease29 provides further evidence for the pathogenicity of TDP43.

Amyotrophic lateral sclerosis (ALS) preferentially involves the descending corticospinal motor neurons that synapse with spinal motor neurons and project to skeletal muscles via the neuromuscular junction. The processes of neurodegeneration in amyotrophic lateral sclerosis involve a complex array of molecular and genetic pathways. Glutamate-induced excitotoxicity can result from the overactivation of ionotropic glutamate receptors that allow excessive influx of Na+ and Ca2+ ions (step 1) and ultimately neurodegeneration through the activation of Ca2+-dependent enzymatic pathways. Glutamate excitotoxicity also generates free radicals, which further contribute to the process of neurodegeneration via oxidative stress. Na+–K+ pump dysfunction (step 2) disrupts the resting membrane potential and leads to secondary effects of altered intracellular Na+ levels. Ion channel dysfunction (step 3) also leads to altered intracellular Na+ levels. Altered Na+ levels result in the reversal of the Na+–Ca2+ exchanger (step 4), thereby increasing the intracellular Ca2+ levels that can cause neuronal toxicity. Defects in RNA processing, RNA metabolism and protein synthesis lead to defects in nucleocytoplasmic trafficking associated with neuronal degeneration (step 5). Mutant SOD1 enzymes increase oxidative stress, induce mitochondrial dysfunction, form intracellular aggregates, and adversely affect neurofilament and axonal transport processes (step 6). Mutations in TARDBP, FUS and C9orf72 can result in the formation of intracellular aggregates of their protein products (step 7), leading to increased oxidative stress, mitochondrial dysfunction, defects in axonal transport and, consequently, in neuronal death. Defects in protein folding and degradation also lead to protein aggregates (step 8). The activation of microglia promotes the secretion of pro-inflammatory cytokines and neurotoxic substances, such as glutamate, which promote neuroinflammation and neuronal death (step 9). Reduced expression and activity of the astrocytic glutamate transporter excitatory amino acid transporter 2 (EAAT2) (step 10) is associated with motor neuron degeneration owing to glutamate toxicity. Accumulation of mutant SOD1 protein in Schwann cells (step 11) are thought to mediate synaptic denervation, which precedes the onset of anterior horn cell degeneration.
Development of therapeutics
Precision approaches and biomarkers
In light of the clinical heterogeneity of ALS, the involvement of the upper and lower motor neuron systems, and the underlying molecular complexity that determines disease expression, the concept of therapy that is tailored to an individual’s genetic signature has started to emerge (Fig. 2). An example of how this approach could be beneficial has come from post hoc analysis of three trials of lithium for ALS30, which showed that patients experienced different effects of the therapy according to their UNC13A genotype. For patients with ALS who were homozygous for a specific variant in UNC13A, lithium had a positive disease-modifying effect despite the overall negative results of trials. Among individuals with the UNC13A variant, the probability of survival at 12 months was 28% higher among those who received lithium than among those who received placebo.

A one-size-fits-all approach (left) in amyotrophic lateral sclerosis (ALS) leads to the use of a single treatment in heterogeneous populations. In this scenario, some patients benefit from therapy but others do not. Stratified medicine (centre) improves on the one-size-fits-all approach by enabling patients to be separated into more homogeneous groups based on demographics, clinical phenotype and molecular subtypes of ALS. However, advances in knowledge and technology are enabling a transition to a precision medicine paradigm (right) in ALS. Precision medicine incorporates individual phenotypic and genotypic data to guide individualized therapy. In this scenario, all patients can benefit from treatment.
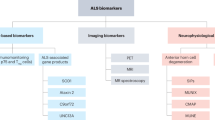
The understanding of individual differences in response to a trial therapy, particularly in a longitudinal manner to assess progression, requires reliable biomarkers. Several candidate biomarkers for ALS have emerged, including biofluid, neurophysiological and neuroimaging measures30,31,32,33,34 that indicate various aspects of disease, from network-level dysfunction to structural changes and basic cellular processes. Most of these markers indicate the loss of either the upper or lower motor neuron systems but rarely of both. However, their clinical utility and ability to enable patient stratification are variable.
A consistent diagnostic biomarker for ALS is cortical hyperexcitability measured with transcranial magnetic stimulation35. Whether the measurement of cortical excitability can contribute to prognostic stratification is less clear — the pharmacodynamic sensitivity of this measure has only been investigated with riluzole, the only established disease-modifying drug for ALS36,37. Structural changes in ALS can also be observed with MRI, which has revealed widespread involvement of cerebral white matter tracts and grey matter. However, the sensitivity of these measures to longitudinal change is inconsistent even at the group level and functional changes are even more difficult to quantify38. Emerging techniques that combine neuroimaging with functional neurophysiological approaches, such as quantitative electroencephalography, evoked responses and source analyses, could capture a true systems-level signature for ALS39.
The most promising biofluid marker of ALS is neurofilament in the cerebrospinal fluid (CSF) and blood, levels of which correlate with the rate of disease progression and are most strongly associated with the involvement of the upper motor neuron system40,41. CSF and serum levels of the neurofilament light chain (NfL) correlate tightly with one another and serum assays have increased in sensitivity to offer a simple, non-invasive measure41. Measurement of serum NfL has been applied in trials of the latest therapies for multiple sclerosis42. In the context of monitoring treatment responses in trials of therapies for ALS, failure of serum NfL levels to decrease during treatment could enable decisions regarding the efficacy of potential therapeutic agents to be made earlier. In a multi-arm, multi-stage (MAMS) platform trial structure (see Enhanced trial design, below), patients could subsequently switch to a new trial agent.
The development of biomarkers for use in trials in ALS is further complicated by the possibility that ALS results from a cascade of multiple pathways. For this reason, multidrug trials will be needed to tackle different aspects of the disease. In such multidrug trials, specific biomarkers are needed for each of the targeted mechanisms to enable the contributions of individual therapies to be assessed as advocated by the Airlie House guidelines10 (See Patient-reported outcomes, below).
Biomarkers are also needed to enable the testing of preventive strategies for at-risk individuals, but biomarkers associated with the symptomatic phase of the disease might not be useful in this context. Currently, the identification of individuals who are at risk of developing ALS can only be done by screening individuals from families that carry highly penetrant mutations. However, some data suggest that predictive biomarkers do exist. A study of individuals who carried an intronic GGGGCC expansion in C9orf72 showed that expansion-derived dipeptides were present in cerebrospinal fluid and peripheral blood mononuclear cells many decades before symptom onset43. Similarly, a large primary care cohort study identified novel metabolic markers, including alterations in carbohydrate, lipid and apolipoprotein profiles, that were associated with an increased risk of ALS in later life44.
Drug repurposing
Alongside efforts to discover new disease-modifying therapies for ALS, drug repurposing is also being investigated to rapidly identify effective therapies in ALS. The repurposing of existing drugs enables the rapid assessment of drug efficacy in phase II trials owing to the established safety profiles of these drugs. In addition, advances in cell reprogramming are revolutionizing drug repurposing strategies in ALS. Human somatic cells, including skin fibroblasts, can be reprogrammed into induced pluripotent stem cells45, which can then be differentiated into neural progenitor cells and ultimately into neuronal and glial cell lines46. This phenotypic approach provides a rich source of neurons and glial cells that are derived from patients with ALS and that retain the key pathophysiological properties of the disease. These properties enable the rapid, ex vivo screening of drugs relevant to an individual’s phenotype. The use of existing drugs that modulate established pathophysiological mechanisms in ALS has already identified several therapeutic candidates47 (Table 2). The most promising of these candidates from a clinical trial perspective are discussed below.
Edaravone
One candidate for repurposing in ALS is edaravone, which was initially discovered as a free-radical scavenger and potent antioxidant in the treatment of acute ischaemic stroke in Japan48. A phase III clinical trial of edaravone in Japanese patients with ALS showed that the drug slowed disease progression in this cohort49. Based on this finding, edaravone was approved for the treatment of ALS by the FDA in 2017. However, a subsequent multicentre study of edaravone in patients with ALS in Italy did not have a positive outcome50 and therefore regulatory approval of edaravone for ALS in Europe is likely to require additional evidence for its efficacy, including evidence of long-term benefits for survival and of efficacy in cohorts of patients with ALS outside Japan49,51.
Ezogabine
Neuronal hyperexcitability is an important pathophysiological mechanism in ALS52 and the antiepileptic agent ezogabine is a potent K+ channel activator that induces membrane hyperpolarization53 and therefore reduces neuronal excitability. Consequently, ezogabine has potential for repurposing in ALS and has been investigated in this context. In an induced pluripotent stem cell model of ALS, ezogabine reduced neuronal hyperexcitability and increased motor neuron survival54. On this basis, a clinical trial has been initiated to assess the efficacy of ezogabine on central and peripheral nerve excitability in ALS — results are expected in late 2020 (ref.55).
Immunomodulatory therapies
Neuroinflammatory pathways have been implicated in the pathophysiology of ALS and immunomodulatory therapies are under investigation for repurposing in ALS. Two of these agents — dimethyl fumarate and IL-2 — target regulatory T (Treg) cell function. Treg cells have neuroprotective effects via the suppression of toxic neuroinflammation in the CNS56 and autologous infusion of expanded Treg cells in a small cohort of patients with ALS slowed disease progression57. Dimethyl fumarate, which is approved for the treatment of relapsing–remitting multiple sclerosis, increases Treg cell numbers and function58 and could therefore be beneficial in ALS. A phase II clinical trial is currently under way to assess the efficacy of dimethyl fumarate in this context59. IL-2 is a cytokine that is important for Treg cell development and homeostasis60 and recombinant analogues of IL-2 are used in the treatment of cancers. Currently, two trials in Europe are in progress to assess the effects of IL-2 in ALS61,62. Results from one of these studies — the IMODALS study — showed that low-dose IL-2 was well tolerated and immunologically effective in ALS63.
Microglia are another potential neuroinflammatory target in ALS. Masitinib, which was originally developed as a therapy for cancer, is a tyrosine kinase inhibitor that inhibits microglial activation. In a phase II–III trial in patients with ALS, masitinib as an add-on therapy to riluzole slowed disease progression50, underscoring the importance of neuroinflammation as a treatment target in ALS64.
AMX0035
AMX0035 is a combination of sodium phenylbutyrate and tauroursodeoxycholic acid. These agents have been used individually in the treatment of various disorders — sodium phenylbutyrate for urea cycle disorders65 and tauroursodeoxycholic acid for familial amyloid polyneuropathy, cholelithiasis and cholestatic liver disease66. In combination, they act synergistically to prevent neuronal death and oxidative, bioenergetic and metabolic stress67 and could therefore be beneficial in ALS. A trial to evaluate the efficacy of AMX0035 in 137 people with ALS has been completed68. Treatment with this combination therapy resulted in a significant 25% slowing of disease progression over 6 months68. Secondary outcomes of vital capacity and quantitative strength trended in a positive direction. When the randomized controlled study was combined with a period of open-label access, survival was also improved in patients who originally received placebo: overall, random assignment to receive AMX0035 conferred a 6.5-month survival advantage compared with random assignment to receive placebo69. Separately, the antioxidant effects of tauroursodeoxycholic acid alone in ALS are being assessed in a European consortium trial (TUDCA-ALS)70.
Rasagiline
Rasagiline is a monoamine oxidase B inhibitor used for the treatment of Parkinson disease, but a study in the SOD1 mouse model of ALS indicated a benefit of rasagiline in this context71. However, two subsequent trials of rasagiline in patients with ALS did not demonstrate its efficacy71,72,73 (although post hoc analysis identified a possible effect in patients with rapid disease progression) suggesting that, overall, repurposing of this drug will not be a useful strategy in ALS. This lack of translation from the SOD1 mouse model to humans with ALS could be a result of differences in pathophysiology. For instance, in SOD1 mice, the loss of motor neurons and associated weakness progresses over a short period of time74, whereas symptoms in humans do not emerge for decades, apparently triggered by a multi-step process that has not yet been replicated in animal models75,76,77,78,79,80.
Arimoclomol
Arimoclomol is a co-inducer of heat shock proteins that is under investigation for therapeutic use in insulin resistance. Preclinical data suggest that arimoclomol could be beneficial in ALS, as it reduced misaggregated proteins in ALS models and in models of lysosomal storage diseases81. The safety and tolerability of arimoclomol has been established in patients with ALS82 and larger trials are now being conducted to assess its efficacy; whether translational difficulties similar to those encountered with rasagiline arise remains to be seen.
High-throughput drug screening
High-throughput drug screening (HTDS) methodologies represent an alternative to drug repurposing strategies for the identification of effective therapeutic agents in ALS. The main advantage of HTDS is the ability to identify novel compounds that exert unexpected effects on the pathogenic mechanisms of ALS. Advances in computational techniques and artificial intelligence have led to the development of powerful platforms that enable the screening of millions of compounds and hundreds of thousands of cell assays83.
In ALS, in silico approaches have predominantly been used to screen millions of drugs for predicted effects such as receptor binding or protein stabilization. Drug-focused and disease-focused databases and online tools that associate drugs with disease mechanisms are being developed and are likely to result in additional therapeutic breakthroughs for ALS83. However, the increase in potential therapies that these platforms are expected to identify could lead to bottlenecks if large numbers of drugs subsequently need to be tested in patients.
Engineering tools that simulate developmental systems, such as cell-based in silico HTDS approaches, are increasingly being used in ALS drug discovery, largely driven by advances in computational and cell reprogramming technologies. Many cell-based HTDS studies have been undertaken to identify compounds that exert anti-glutamatergic activity84, downregulate mutant SOD1 transcription85, reduce TDP43 protein levels and aggregation86, reduce oxidative stress87, and improve motor neuron survival88. Several promising drug candidates have been identified in these studies but none have yet been translated into effective therapies for ALS.
Cell-free in silico HTDS strategies have also been used in ALS drug discovery to identify drugs that modulate protein interactions and misfolding. This cell-free approach has identified drugs that inhibit TDP43 protein misfolding and aggregation but whether these drugs are beneficial in ALS remains to be determined89. Similarly, the disruption of molecular pathways, including interactions between mutant SOD1 protein and proteins that trigger motor neuron death via the resident endoplasmic reticulum protein Derlin 1, could represent a further therapeutic option90. However, a critical limitation of cell-free HTDS approaches is that biological interactions are assessed outside of the cellular environment and in the absence of other pathogenic processes that could have a role in the onset and progression of ALS; therefore, the translational relevance remains unclear.
Genetic and cellular therapy
The success of targeted therapies for spinal muscular atrophy — antisense oligonucleotides (ASOs) or small molecules and τηε use of viral vectors for gene replacement — has demonstrated the power of genetic therapy91. Genetic mutations are present in ~15% of patients with ALS, but the multiplicity of mutation types that cause ALS, including missense mutations, repeat expansions and loss-of-function mutations, means that a variety of gene therapy approaches would be needed to target all the causal mutations. In addition, although many causal mutations are associated with the toxicity of the mutant protein and therefore reducing the expression of the toxic protein seems a logical approach, many different mutations have been identified in single genes, making it difficult to design ASOs to selectively target the mutant allele. Furthermore, the safe inactivation of gene expression with ASOs is likely to be impossible in some cases; for example, the inactivation of TARDBP would be lethal owing to the physiological function of TDP43 in splicing and the contribution of the protein to other pathways that are critical for cellular survival92.
Antisense oligonucleotides
Evidence from mouse studies suggests that knockdown of the SOD1 protein is tolerated without severe phenotypic consequences. Whether this tolerance would be maintained over decades, particularly in the context of human ageing, is unknown, but these findings have encouraged the development of ASO therapy directed against wild-type and mutant SOD1 (ref.93). Initial reports suggest that such an approach is well tolerated and could be effective. Results of the phase I–II trial of the ASO tofersen, which mediates the degradation of SOD1 mRNA, were published in 2020 and confirmed that the treatment reduced CSF concentrations of SOD1 protein in patients with ALS94. Separate studies in two patients with ALS with SOD1 mutations have raised the possibility that intrathecal delivery of an adeno-associated virus that encodes a microRNA that targets SOD1 could be a potential therapy in these patients95. To maximize the benefits of these ASO therapies, various routes of administration have been considered, from systemic intravenous administration to intraparenchymal brain delivery, although intrathecal delivery has been used most frequently to date. Commonly observed lumbar puncture-related adverse events could be mitigated in the future through the development of specialized nanotechnology drug delivery systems96.
In ALS caused by C9orf72 mutations, ASOs against apparently toxic RNA species that arise from the hexanucleotide repeat expansion are currently in early phase clinical trials. Preclinical research suggests that RNA repeats can be successfully targeted in this way without downregulating the C9orf72 protein, which could contribute to toxicity independently of the RNA97.
The efficiency of ASOs is currently limited by a failure to traverse the blood–brain barrier, a generic problem that will need to be solved for all forms of precision-based molecular therapies in ALS. However, ASOs are also limited by complexities in intracellular processing that limit their direct engagement with mRNA species. Advances in the development of chemical modifications that increase the stability of ASOs and their ability to penetrate cells will result in a new generation of ASOs that could be delivered via the systemic circulation98.
Gene editing
Besides the use of ASOs, genome editing could form the basis of another gene-based therapy for ALS. Rapid advances in genome editing have greatly advanced the cellular modelling of ALS but also raise the possibility that precise targeting of genetic mutations with systems such as CRISPR–Cas9 could be possible in vivo; such gene editing could effectively prevent ALS from developing. A key requirement for such an approach is the ability to safely target constructs to the nervous system, thereby avoiding off-target effects. However, the use of gene editing in ALS poses similar challenges as the use of ASOs as many of the mutations in genes commonly linked to ALS are unique to the individual; therefore, a bespoke approach would be required that also avoids the inactivation of the normal gene copy. Gene editing is a rapidly advancing field and the technical challenges in developing genome editing as a therapy for diseases such as ALS are gradually being addressed — studies are moving from cell and rodent models into larger animals, including dogs99.
Cellular therapy
Expectations in most therapeutic trials in ALS are limited to slowing or arresting disease progression, but patients understandably place a high value on the search for treatments that could restore function. Whether targeting the cause of ALS will influence disease progression in symptomatic individuals remains unclear; if this is not the case, then gene therapy that targets causal genes would be pointless in these people. So-called regenerative medicine approaches, in which various forms of stem cells or neural progenitor cells are used, are more plausibly based around the notion that transplanted cells could provide trophic support to slow the disease process and raise hopes for restorative therapy for people already living with ALS. However, given the immense complexity of the functional networks required to produce voluntary movement and the fact that these networks are produced via a complex developmental programme that is inactivated in adulthood, the restoration of function with cellular therapy is currently a theoretical rather than practical option. Several early phase research programmes are under way to explore the potential of transplantation of neural progenitors into the spinal cord, but results of small-scale tolerability and safety studies have not yet provided any clear evidence of disease-modifying effects100.
Improving trial design
Challenges with traditional trials
In ALS, as in other neurodegenerative diseases, the mainstay of clinical research has been the randomized controlled trial in which the effects of a therapeutic candidate are compared with those of a placebo101. Typically, a single agent is tested in a homogeneous population, with a null hypothesis that treatment with this agent is no better than standard care. Such trials are costly and inefficient given that only a small proportion of treatments make it from initial testing to the commercial market — the FDA reports that just 8% of treatments tested in a phase I trial make it to market102. This sequential approach has been used in more than 40 clinical trials of potential disease-modifying therapies for ALS. The approach limits the speed with which treatments for ALS can be evaluated owing to the finite capacity of trial networks in terms of personnel and infrastructure, the limited number of patients with ALS who are eligible, and limited financial resources, especially for academic-led clinical trials. In addition, inherent problems exist in this approach, such as patient heterogeneity and long trial durations, even when the ALS Functional Rating Scale is used as the primary outcome measure.
The inefficiencies in the current approach to ALS clinical trials become more evident when considering the time commitment and expense required for staff recruitment, deployment and training, contract negotiations, regulatory approvals, monitoring arrangements, and infrastructure; these costs must be reconstituted anew for each trial102. After trial completion, staff move elsewhere, skills are lost and approvals lapse. A further inefficiency, and perhaps the most problematic from a patient perspective, is the fact that each study has a separate placebo arm. Such an approach is costly and is a major disincentive for patients to participate in a clinical trial, particularly as their disease is terminal. Indeed, patients with ALS are known to be particularly motivated to participate in trials and report being less dissuaded by the possibility of adverse effects and privacy and confidentiality issues than are people with other diseases103. However, as their disease progresses, patients with ALS face increasing challenges, including the logistics of attending clinic appointments104.
Advanced trial design
Interest is increasing in methods that maintain the rigour of randomized controlled trials while maximizing their efficiency, reducing costs and providing answers regarding efficacy. One approach that is gaining traction is the use of a master protocol for the simultaneous evaluation of multiple compounds105,106 (Fig. 3). With a master protocol, a common system is used for patient selection, logistics, templates and data management. This approach can incorporate precision genotyping and molecular markers and can be implemented in different trial designs, including basket trials (in which the therapeutic target is a specific genetic mutation) and MAMS platform trials107. Such designs have been successfully used in cancer, HIV and some neurological diseases such as multiple sclerosis107,108,109.

In this multi-arm, multi-stage (MAMS) platform trial, eligible patients are randomly assigned to one of four sub-studies and subsequently randomly assigned to receive active treatment or placebo. A master protocol determines patient selection criteria, logistics, outcome measures, biomarkers and data management in all four sub-studies. Genotype and molecular markers can also be collected systematically. The platform consists of five arms (treatments 1–4 and a pooled placebo arm). Pre-planned interim analyses are built into the design at points A and B. At point A, treatment 2 is found to have a favourable efficacy signal, so the arm seamlessly moves into phase III and more patients are recruited into that arm (thicker arrow). At the same point, futility criteria are met with treatment 3 and this arm is dropped (cross). New arms can be added over time such as treatment 4 here.
In a MAMS trial with a sequential design, the sample size is not fixed in advance and data are sequentially analysed, with pre-determined futility or superiority analyses built in, which enable treatment arms to be discontinued owing to a lack of efficacy or to graduate to the next phase of study (Fig. 3). Consequently, adaptive, seamless phase II–III designs can be used to reduce the delays that are usually encountered after an early promising signal in a phase II trial. Typically, criteria for the analysis of accumulating data are pre-planned and, if a favourable signal is detected in one sub-study, recruitment into that arm is increased to provide phase III efficacy data for regulatory approval. As data accumulate, response-adaptive randomization can be used to allocate more patients to a treatment arm that is showing more promise than others and to reduce random assignment to arms with inferior outcomes.
These approaches work well in diseases for which readouts are robust, for example, tumour load in cancer or white matter lesion burden on MRI in multiple sclerosis. Such a robust biomarker does not yet exist for ALS and therefore adaptive decisions will be limited by existing metrics such as ALS Functional Rating Scale — Revised (ALSFRS-R) scores and survival. However, a further advantage of the MAMS platform is that patients can be randomly assigned to a particular sub-study for which genetic or other molecular data predicts a favourable response and can be automatically excluded from sub-studies for which they are not suitable. This possibility promotes personalized stratification in a biologically heterogeneous disease.
The advantages of a master protocol platform design include efficiencies in terms of cost (fixed costs shared across a number of trials), time and infrastructure that enable more patients to participate in clinical trials. However, these designs are not a panacea and they bring their own challenges, including highly complex Bayesian statistical analyses and the fact that several small sub-studies being conducted in parallel can increase the rate of false positive findings106,107. Additional challenges of platform trials include their large-scale, long-term nature, the associated costs of managing and executing the trial, the need to build organizations or frameworks that can operate these trials perpetually, and the need for an independent data monitoring committee106. Regulatory authorities are open to discussing such approaches: the FDA has published its guidance for adaptive platform master protocol designs and the EMA has established a taskforce to explore the issue110.
In addition to the trial platform, the complex pathophysiological mechanisms in ALS and the involvement of multiple pathways mean that the clinical benefits might not be apparent with a single drug. The use of a multi‐drug regimen could enable the detection of a more universal treatment effect by simultaneously addressing multiple disease mechanisms. However, designing trials that test multiple drugs remains complex and determining the mechanisms of action of a synergistic combination of therapeutics would logically require a factorial design111.
Patient stratification
In the context of disease heterogeneity in ALS, the stratification of patients as an inclusion criterion for clinical trials is becoming an important consideration when determining the efficacy of a compound. A lead-in phase during which patients are monitored before being randomly assigned to receive a treatment (typically after patient inclusion but before initiation of therapy) might provide a more accurate indication of disease progression in individuals and thereby enable the assessment of investigational drug efficacy112,113,114. In addition, patient stratification can be based on clinical parameters, genetics, disease stage and disease trajectory.
The purpose of pre-selection criteria for trial entry is to facilitate the use of tailored treatments. In ALS, pre-selection is a technique that is still in its infancy and remains largely confined to selection on this basis of genetic causes. However, some studies suggest that modifier genes, such as those that affect prognosis, can also be used as enrolment criteria20 and specific clinical features, such as respiratory, nutritional or emotional symptoms, have been used for pre-selection115,116. For example, patients with respiratory impairment have been the focus of trials of enhancers of troponin C (which activates muscle contraction) to improve diaphragmatic function117. Pre-selection is also an obvious approach for clinical trials of nutritional interventions and approaches to saliva control, which have so far only been tested in exploratory studies. For example, dextromethorphan–quinidine is beneficial for emotional lability in ALS, which occurs in a subset of patients, often in association with bulbar impairment. Anecdotal patient reports also suggest that dextromethorphan–quinidine might be useful for improving bulbar function and a phase II trial has indicated benefits on all aspects of self-reported bulbar function, including speech, swallowing and salivation118.
Stratification at the time of recruitment also enables patients to be matched according to their likely clinical course. Ideally, the rate of disease progression for an individual would be established at the initial clinic visit and classified as fast, medium or slow119. However, the clinical course remains heterogeneous and unpredictable and, therefore, attempts to homogenize cohorts on the basis of likely disease course at trial entry lead to restrictive inclusion criteria for most trials. The ENCALS Prediction model, published in 2018, is based on data from over 11,000 patients with ALS in population-based registers and has improved the accuracy of personalized prediction of clinical decline and survival8. The model is based on eight factors: age, El Escorial classification, site of onset, vital capacity, genetic status for C9orf72 expansion, diagnostic delay, cognitive status and functional score. In the context of clinical trial enrolment, the model generates a personalized prediction of survival for each patient and enables the inclusion of those in whom an effect of the trial treatment is most likely (for example, minimizing the number of patients with very slow or very rapid disease progression). The use of the model as an inclusion criterion could enable up to 80% of patients to participate in trials while minimizing the impact of disease variance. This model was recently used in ALS in a phase II clinical trial of dolutegravir, abacavir and lamivudine (Triumeq)120.
Another emerging approach to stratification for clinical trials in ALS is disease staging. Staging is important because ALS is a continuously evolving process and treatments might need to be administered at precise time points in the disease course to be beneficial — staging provides information about this timing. The benefits of staging were shown in a post hoc analysis of the original riluzole trial data; specifically, post hoc analysis showed that riluzole has a beneficial effect late in the disease course121,122,123. No information was available in the original trial data about patients in the very early stages of disease and therefore the post hoc analysis could not examine whether there was an effect early in the disease. A subsequent analysis of data from the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database, which includes patients in the early stages of the disease, confirmed the original finding and showed that riluzole also has an effect in the early stages of ALS121.
The two main staging systems currently under consideration in ALS are the King’s Clinical Staging system and the Milano–Torino Staging (MiToS) system124. The King’s system includes five stages based on disease burden, assessed according to the muscle groups that are clinically involved, and incorporates feeding difficulties and respiratory failure123; stage 1 is symptom onset and stage 5 is death. The MiToS system involves six stages (0–5) based on functional ability assessed with the ALSFRS-R; stage 0 is defined as normal function and stage 5 is death. The MiToS stage at diagnosis correlates with survival at 18 months122. The two staging systems are complementary — the King’s system has a higher resolution in early disease to mid-disease, when clinical or disease burden is increasing, and the MiToS system has a higher resolution in late disease, when functional involvement has developed. Cognitive change, which is a feature of ALS, is not accounted for in either the King’s or MiToS systems115,125. However, use of the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) tool has demonstrated that increasing impairment in executive function and behavioural changes correlate with later King’s stages126 and a cognitive protocol is likely to form part of future staging systems.
Patient-reported outcomes
The requirement for participants in clinical trials to make episodic visits to study centres so that progression of their disease can be measured imposes substantial restrictions on the conduct of clinical trials. First, it limits participation to patients who live close enough to a study centre to attend mandatory visits on a monthly or bimonthly schedule. In an analysis of the reasons for poor adherence in ALS clinical trials, travel difficulties and caregiver burden were contributing factors to the withdrawal of 27% of patients who withdrew their consent127. Second, the requirement for visits to study centres means that substantial variability is seen in outcome measures that are commonly used in phase II and III trials128; some of this variability might be due to true fluctuations in patient status and some due to measurement error. Both of these issues could be addressed if data could be collected through more frequent assessment of patients because it would reduce variability by increasing the sampling frequency; however, this would be possible only if undertaken in patients’ homes. Other aspects of trial participation, such as safety assessments and drug dispensing, could also be accommodated at patients’ homes, thereby enabling the participation of patients who have previously been deemed inaccessible129.
The value of self-reported outcomes has previously been studied through the use of the PatientsLikeMe website, where patients directly entered their symptoms and the data were subsequently analysed130. Such patient-reported outcomes are valuable as they are cost-effective, can be implemented from a distance, can provide comprehensive assessments and can capture an element of personal well-being that is not captured with other approaches131. The limitations of self-reported outcomes include a lack of objectivity and unfamiliarity with the use of digital platforms that are necessary for this approach to be successfully implemented.
The assessment of patients at home with the ALSFRS-R has frequently been performed in clinical trials and in clinical practice, usually via a telephone assessment by a trained evaluator132. Furthermore, the reliability of an online tool for assessment with the ALSFRS-R that requires no evaluator assistance has been established — values were close to those from assessments in the clinic133. The self-reporting of ALSFRS-R scores was done via secure online portals and this approach was reliable and quick.
In an ongoing study, the results of which are not yet available129, the evaluation of ALS trial endpoints at home was extended to include novel endpoints as well as commonly used endpoints. In that study, patients were recruited via online tools, diagnosis was confirmed from medical records and consent was obtained from patients through an interactive webinar. Tools for endpoint measurement were sent to patients and demonstration and training in the use of these tools was conducted online. These tools included an activity band, a spirometer, a handgrip metre, a personal electrical impedance myography device and a voice evaluation tool that included an app for most smartphones. ALSFRS-R scores were assessed and recorded via a secure website. Once trained, participants were asked to perform all assessments — except the ALSFRS-R — every day for 3 months and then twice weekly for an additional 6 months. Overall, 114 patients and 30 healthy controls were recruited from 40 states in the USA. At 3 months and at the end of the study, patient views on the experience were assessed with an online questionnaire. The results of this study will be used to determine whether home assessments are a viable strategy for clinical trials in ALS and the extent to which frequent sampling improves the reliability and sensitivity of a range of trial endpoints.
In another study, published in 2019, a hybrid design was used in which a traditional in-clinic trial was combined with a ‘virtual’ study to test the nutritional supplement lunasin134, which was previously identified as a candidate for ALS therapy135. Fifty participants enrolled at one centre for the 12-month, open-label study. Historical controls were used rather than a control group. The self-reported ALSFRS-R scores closely agreed with confirmatory clinical assessment (Lin’s concordance correlation coefficient, 0.94–0.99). The study produced no evidence that lunasin can lead to “ALS reversal”136, but lessons learned from this hybrid virtual trial could provide ideas for ways to reduce the burden on participants by enabling clinical study visits to be reduced.
Given that each trial is unique, the 2019 Airlie House Clinical Trial guidelines recommend that patients and caregivers are included in trial design groups to minimize the trial burden and ensure that recruitment and retention strategies will be successful10. These guidelines were developed to accelerate progress in ALS, from preclinical research through to trial design and statistical approaches, and to advocate the incorporation of biomarkers in all future trials and the development of home-based outcome measures10. Such measures are included in large genetic and biomarker studies in which the goal is to enrol more than 1,000 patients with ALS and assess phenotypic information in the clinic and at home (ANSWER ALS, TARGET ALS and ALS Therapy Development Institute). Home-based outcome measures are also becoming increasingly relevant for patients with ALS who have substantial disease-related disability, particularly in the context of a shift towards telemedicine owing to the COVID-19 pandemic.
Conclusions and future directions
ALS is a complex neurodegenerative disorder mediated by interactions between genetic, epigenetic and environmental factors, with multiple steps probably being required to trigger disease onset. Consequently, effective treatment strategies might require the use of novel compounds acting synergistically, potentially in different phases of the disease. Advances in clinical trials seem critical to accelerate the translation of such novel therapies into the clinic; novel trial designs are needed to enable the assessment of multiple compounds with a common placebo group and to better determine target engagement and the effect on disease trajectory (Box 1). The implementation of these approaches for the assessment of current and future treatment strategies in ALS could shorten trial durations and reduce costs and burden on patients, thereby providing hope that effective therapies can be rapidly translated into the ALS clinic.
References
Alzheimer’s Disease International. World Alzheimer report 2019: attitudes to dementia (ADI, 2019).
Hurd, M. D., Martorell, P., Delavande, A., Mullen, K. J. & Langa, K. M. Monetary costs of dementia in the United States. N. Engl. J. Med. 368, 1326–1334 (2013).
World Health Organization. Global action plan on the public health response to dementia 2017–2025 (WHO, 2017).
Hebert, L. E., Weuve, J., Scherr, P. A. & Evans, D. A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80, 1778 (2013).
Huynh, W. et al. The impact of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Expert Rev. Neurother. 20, 281–293 (2020).
Kiernan, M. C. et al. Amyotrophic lateral sclerosis. Lancet 377, 942–955 (2011).
Hardiman, O., van den Berg, L. H. & Kiernan, M. C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639 (2011).
Westeneng, H.-J. et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 17, 423–433 (2018).
Bedlack, R. S., Pastula, D., Welsh, E., Pulley, D. & Cudkowicz, M. E. Scrutinizing enrollment in ALS clinical trials: room for improvement? Amyotroph. Lateral Scler. 9, 257–265 (2008).
van den Berg, L. H. et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology 92, e1610–e1623 (2019).
Shefner, J. M. et al. A proposal for new diagnostic criteria for ALS. Clin. Neurophysiol. 113, 1975–1978 (2020).
Dharmadasa, T., Matamala, J. M., Howells, J., Vucic, S. & Kiernan, M. C. Early focality and spread of cortical dysfunction in amyotrophic lateral sclerosis: a regional study across the motor cortices. Clin. Neurophysiol. 131, 958–966 (2020).
Vucic, S., Rothstein, J. D. & Kiernan, M. C. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci. 37, 433–442 (2014).
Swash, M. et al. Occasional essay: upper motor neuron syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 91, 227–234 (2020).
Huynh, W. et al. Assessment of the upper motor neuron in amyotrophic lateral sclerosis. Clin. Neurophysiol. 127, 2643–2660 (2016).
Simon, N. G. et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann. Neurol. 76, 643–657 (2014).
de Carvalho, M., Kiernan, M. C. & Swash, M. Fasciculation in amyotrophic lateral sclerosis: origin and pathophysiological relevance. J. Neurol. Neurosurg. Psychiatry 88, 773–779 (2017).
Al-Chalabi, A. & Hardiman, O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617 (2013).
Turner, M. R. et al. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 88, 1042 (2017).
Blair, I. P. et al. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 81, 639–645 (2010).
Williams, K. L. et al. Pathophysiological insights into ALS with C9ORF72 expansions. J. Neurol. Neurosurg. Psychiatry 84, 931–935 (2013).
Brown, R. H. & Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172 (2017).
Byrne, S. et al. Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: a population-based case–control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann. Neurol. 74, 699–708 (2013).
Huisman, M. H. B. et al. Family history of neurodegenerative and vascular diseases in ALS. Neurology 77, 1363-1369 (2011).
Devenney, E. M. et al. Psychiatric disorders in C9orf72 kindreds: study of 1,414 family members. Neurology 91, e1498–e1507 (2018).
O’Brien, M. et al. Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol. 74, 1425–1430 (2017).
Lin, C.-L. G. et al. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602 (1998).
Trotti, D., Rolfs, A., Danbolt, N. C., Brown, R. H. & Hediger, M. A. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 2, 427–433 (1999).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
van Eijk, R. P. et al. Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology 89, 1915–1922 (2017).
De Schaepdryver, M. et al. Comparison of elevated phosphorylated neurofilament heavy chains in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 89, 367–373 (2018).
Kassubek, J. et al. Imaging the pathoanatomy of amyotrophic lateral sclerosis in vivo: targeting a propagation-based biological marker. J. Neurol. Neurosurg. Psychiatry 89, 374–381 (2018).
Turner, M. R., Kiernan, M. C., Leigh, P. N. & Talbot, K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 8, 94–109 (2009).
van Eijk, R. P. et al. Monitoring disease progression with plasma creatinine in amyotrophic lateral sclerosis clinical trials. J. Neurol. Neurosurg. Psychiatry 89, 156–161 (2018).
Menon, P. et al. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. 14, 478–484 (2015).
Geevasinga, N. et al. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 580–588 (2016).
Vucic, S. et al. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 136, 1361–1370 (2013).
Menke, R. A., Agosta, F., Grosskreutz, J., Filippi, M. & Turner, M. R. Neuroimaging endpoints in amyotrophic lateral sclerosis. Neurotherapeutics 14, 11–23 (2017).
McMackin, R. et al. Measuring network disruption in neurodegenerative diseases: new approaches using signal analysis. J. Neurol. Neurosurg. Psychiatry 90, 1011–1020 (2019).
Lu, C.-H. et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84, 2247–2257 (2015).
Verde, F. et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 157–164 (2019).
Sormani, M. P. et al. Blood neurofilament light as a potential endpoint in Phase 2 studies in MS. Ann. Clin. Transl. Neurol. 6, 1081–1089 (2019).
Gendron, T. F. et al. Poly (GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 9, eaai7866 (2017).
Mariosa, D. et al. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: a more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 81, 718–728 (2017).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Mertens, J., Marchetto, M. C., Bardy, C. & Gage, F. H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17, 424 (2016).
Martinez, A., Del Valle Palomo Ruiz, M., Perez, D. I. & Gil, C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 26, 403–414 (2017).
Yoshida, H. et al. Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 12, 9–20 (2006).
Abe, K. et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512 (2017).
Lunetta, C. et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 267, 3258–3267 (2020).
Al-Chalabi, A. et al. July 2017 ENCALS statement on edaravone. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 471–474 (2017).
Geevasinga, N., Menon, P., Özdinler, P. H., Kiernan, M. C. & Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 12, 651 (2016).
Rudzinski, L. A. et al. New antiepileptic drugs: focus on ezogabine, clobazam, and perampanel. J. Investig. Med. 64, 1087–1101 (2016).
Wainger, B. J. et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11 (2014).
Wainger, B. J. et al. Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis. A randomized clinical trial. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.4300 (2020).
Sheean, R. K. et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. 75, 681–689 (2018).
Thonhoff, J. R. et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol. Neuroimmunol. Neuroinflamm. 5, e465 (2018).
Ghadiri, M. et al. Dimethyl fumarate–induced lymphopenia in MS due to differential T-cell subset apoptosis. Neurol. Neuroimmunol. Neuroinflamm. 4, e340 (2017).
Vucic, S. et al. Phase 2 randomized placebo controlled double blind study to assess the efficacy and safety of tecfidera in patients with amyotrophic lateral sclerosis (TEALS Study): Study protocol clinical trial (SPIRIT Compliant). Medicine 99, e18904 (2020).
Burchill, M. A., Yang, J., Vang, K. B. & Farrar, M. A. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol. Lett. 114, 1–8 (2007).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03039673 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02059759 (2016).
Camu, W. et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): a phase 2a randomised, double-blind, placebo-controlled trial. EBioMedicine 59, 102844 (2020).
Mora, J. S. et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 21, 5–14 (2020).
Burrage, L. C. et al. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol. Genet. Metab. 113, 131–135 (2014).
Obici, L. et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 19, 34–36 (2012).
Cudkowicz, M. E. et al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph. Lateral Scler. 10, 99–106 (2009).
Paganoni, S. et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 383, 919–930 (2020).
Paganoni, S. et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in ALS. Muscle Nerve https://doi.org/10.1002/mus.27091 (2020).
TUDCA ALS. New clinical trial for ALS/MND. TUDCA https://www.tudca.eu/ (2020).
Waibel, S., Reuter, A., Malessa, S., Blaugrund, E. & Ludolph, A. C. Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J. Neurol. 251, 1080–1084 (2004).
Statland, J. M. et al. Rasagiline for amyotrophic lateral sclerosis: a randomized, controlled trial. Muscle Nerve 59, 201–207 (2019).
Ludolph, A. C. et al. Safety and efficacy of rasagiline as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomised, double-blind, parallel-group, placebo-controlled, phase 2 trial. Lancet Neurol. 17, 681–688 (2018).
Turner, M. R. et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 (2013).
Ahmed, R. M. et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 87, 1234 (2016).
Eisen, A. et al. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 88, 917 (2017).
Eisen, A., Kiernan, M., Mitsumoto, H. & Swash, M. Amyotrophic lateral sclerosis: a long preclinical period? J. Neurol. Neurosurg. Psychiatry 85, 1232 (2014).
Henderson, R. D., Garton, F. C., Kiernan, M. C., Turner, M. R. & Eisen, A. Human cerebral evolution and the clinical syndrome of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 570–575 (2019).
Kiernan, M. C., Ziemann, U. & Eisen, A. Amyotrophic lateral sclerosis: origins traced to impaired balance between neural excitation and inhibition in the neonatal period. Muscle Nerve 60, 232–235 (2019).
Vucic, S. et al. ALS is a multistep process in South Korean, Japanese, and Australian patients. Neurology 94, e1657 (2020).
Lanka, V., Wieland, S., Barber, J. & Cudkowicz, M. Arimoclomol: a potential therapy under development for ALS. Expert Opin. Investig. Drugs 18, 1907–1918 (2009).
Cudkowicz, M. E. et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38, 837–844 (2008).
Cha, Y. et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 175, 168–180 (2018).
Cudkowicz, M. E. et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 13, 1083–1091 (2014).
Wright, P. D. et al. A high-throughput screen to identify inhibitors of SOD1 transcription. Front. Biosci. 4, 2801-2808 (2012).
Boyd, J. D. et al. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J. Biomol. Screen. 19, 44–56 (2014).
Mead, R. J. et al. S [+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 61, 438–452 (2013).
Benmohamed, R. et al. Identification of compounds protective against G93A-SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 87–96 (2011).
Oberstadt, M. et al. TDP-43 self-interaction is modulated by redox-active compounds Auranofin, Chelerythrine and Riluzole. Sci. Rep. 8, 2248 (2018).
Nishitoh, H. et al. ALS-linked mutant SOD1 induces ER stress-and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464 (2008).
Groen, E. J., Talbot, K. & Gillingwater, T. H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 14, 214 (2018).
Tan, R. H. et al. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain 138, 3110–3122 (2015).
Miller, T. M. et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013).
Miller, T. et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119 (2020).
Mueller, C. et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N. Engl. J. Med. 383, 151–158 (2020).
Kariyawasam, D., Alexander, I. E., Kurian, M. & Farrar, M. A. Great expectations: virus-mediated gene therapy in neurological disorders. J. Neurol. Neurosurg. Psychiatry 91, 849–860 (2020).
Donnelly, C. J. et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013).
Smith, C. E. & Zain, R. Therapeutic oligonucleotides: state of the art. Annu. Rev. Pharmacol. Toxicol. 59, 605–630 (2019).
Amoasii, L. et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91 (2018).
Goutman, S. A. et al. Long-term Phase 1/2 intraspinal stem cell transplantation outcomes in ALS. Ann. Clin. Transl. Neurol. 5, 730–740 (2018).
Nicholson, K. A., Cudkowicz, M. E. & Berry, J. D. Clinical trial designs in amyotrophic lateral sclerosis: does one design fit all? Neurotherapeutics 12, 376–383 (2015).
Swash, M. Clinical trials in the ALS syndrome: it is time for change. J. Neurol. Neurosurg. Psychiatry 90, 1308 (2019).
DasMahapatra, P., Raja, P., Gilbert, J. & Wicks, P. Clinical trials from the patient perspective: survey in an online patient community. BMC Health Serv. Res. 17, 166 (2017).
Collet, M. How much does distance limit the pool of potential clinical trial participants in the United States? F1000Research https://doi.org/10.7490/f1000research.1115158.1 (2017).
Cecchini, M. et al. Challenges with novel clinical trial designs: master protocols. Clin. Cancer Res. 25, 2049–2057 (2019).
Hirakawa, A., Asano, J., Sato, H. & Teramukai, S. Master protocol trials in oncology: review and new trial designs. Contemp. Clin. Trials Commun. 12, 1–8 (2018).
Saville, B. R. & Berry, S. M. Efficiencies of platform clinical trials: a vision of the future. Clin. Trials 13, 358–366 (2016).
Connick, P. et al. Multiple sclerosis-secondary progressive multi-arm randomisation trial (MS-SMART): a multiarm phase IIb randomised, double-blind, placebo-controlled clinical trial comparing the efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis. BMJ Open 8, e021944 (2018).
Stern, A. D. & Mehta, S. Adaptive platform trials: the clinical trial of the future? Harvard Business School https://www.hbs.edu/faculty/Pages/item.aspx?num=53315 (2017).
US Food and Drug Administration. Master protocols: efficient clinical trial design strategies to expedite development of oncology drugs and biologics guidance for industry (FDA, 2018).
Rosenfeld, J. Multi-drug therapy in amyotrophic lateral sclerosis: the case for a multi-drug approach. Muscle Nerve 30, 673–675 (2004).
Park, S. B. et al. Flecainide in amyotrophic lateral sclerosis as a neuroprotective strategy (FANS): a randomized placebo-controlled trial. EBioMedicine 2, 1916–1922 (2015).
de Carvalho, M. & Swash, M. Can selection of rapidly progressing patients shorten clinical trials in amyotrophic lateral sclerosis? Arch. Neurol. 63, 557–560 (2006).
Moore, D. H. II & Miller, R. G. Improving efficiency of ALS clinical trials using lead-in designs. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 5 (Suppl. 1), 57–60 (2004).
Al-Chalabi, A. et al. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–1194 (2016).
Balendra, R. et al. Use of clinical staging in amyotrophic lateral sclerosis for phase 3 clinical trials. J. Neurol. Neurosurg. Psychiatry 86, 45–49 (2015).
Al-Chalabi, A. et al. Oral levosimendan in amyotrophic lateral sclerosis: a phase II multicentre, randomised, double-blind, placebo-controlled trial. J. Neurol. Neurosurg. Psychiatry 90, 1165–1170 (2019).
Smith, R. et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the Nuedexta treatment trial. Neurotherapeutics 14, 762–772 (2017).
Labra, J., Menon, P., Byth, K., Morrison, S. & Vucic, S. Rate of disease progression: a prognostic biomarker in ALS. J. Neurol. Neurosurg. Psychiatry 87, 628–632 (2016).
Gold, J. et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the Lighthouse trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 595–604 (2019).
Atassi, N. et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 83, 1719–1725 (2014).
Tramacere, I. et al. The MITOS system predicts long-term survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 86, 1180–1185 (2015).
Fang, T. et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 17, 416–422 (2018).
Fang, T. et al. Comparison of the King’s and MiToS staging systems for ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 227–232 (2017).
Iazzolino, B. et al. Validation of the revised classification of cognitive and behavioural impairment in ALS. J. Neurol. Neurosurg. Psychiatry 90, 734 (2019).
Crockford, C. et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 91, e1370–e1380 (2018).
Atassi, N. et al. Analysis of start-up, retention, and adherence in ALS clinical trials. Neurology 81, 1350–1355 (2013).
Kaji, R. et al. Ultra-high-dose methylcobalamin in amyotrophic lateral sclerosis: a long-term phase II/III randomised controlled study. J. Neurol. Neurosurg. Psychiatry 90, 451–457 (2019).
Rutkove, S. B. et al. ALS longitudinal studies with frequent data collection at home: study design and baseline data. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 61–67 (2019).
Wicks, P., Vaughan, T. E., Massagli, M. P. & Heywood, J. Accelerated clinical discovery using self-reported patient data collected online and a patient-matching algorithm. Nat. Biotechnol. 29, 411 (2011).
Rutkove, S. B. Clinical measures of disease progression in amyotrophic lateral sclerosis. Neurotherapeutics 12, 384–393 (2015).
Shefner, J. M. et al. A phase 2, double-blind, randomized, dose-ranging trial of Reldesemtiv in patients with ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. https://doi.org/10.1080/21678421.2020.1822410 (2020).
Maier, A. et al. Online assessment of ALS functional rating scale compares well to in-clinic evaluation: a prospective trial. Amyotroph. Lateral Scler. 13, 210–216 (2012).
Bedlack, R. et al. Lunasin does not slow ALS progression: results of an open-label, single-center, hybrid-virtual 12-month trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 285–293 (2019).
ALSUntangled Group. ALSUntangled no. 26: lunasin. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 622–626 (2014).
Bedlack, R. S. et al. How common are ALS plateaus and reversals? Neurology 86, 808–812 (2016).
Paganoni, S. et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 383, 919–930 (2020).
Oskarsson, B. et al. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: a randomized, double-blind crossover trial. Muscle Nerve 58, 42–48 (2018).
Weiss, M. D. et al. A randomized trial of mexiletine in ALS: safety and effects on muscle cramps and progression. Neurology 86, 1474–1481 (2016).
Writing Group, Edaravone (MCI-186) ALS Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512 (2017).
Chen, P. C., Hsieh, Y. C., Huang, C. C. & Hu, C. J. Tamoxifen for amyotrophic lateral sclerosis: a randomized double-blind clinical trial. Medicine 99, e20423 (2020).
Babu, S. et al. Selection design phase II trial of high dosages of tamoxifen and creatine in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 21, 15–23 (2020).
Levine, T. D., Bowser, R., Hank, N. & Saperstein, D. A pilot trial of memantine and riluzole in ALS: correlation to CSF biomarkers. Amyotroph. Lateral Scler. 11, 514–519 (2010).
de Carvalho, M. et al. A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 11, 456–460 (2010).
Macchi, Z. et al. A multi-center screening trial of rasagiline in patients with amyotrophic lateral sclerosis: possible mitochondrial biomarker target engagement. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 345–352 (2015).
Ludolph, A. C. et al. Safety and efficacy of rasagiline as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomised, double-blind, parallel-group, placebo-controlled, phase 2 trial. Lancet Neurol. 17, 681–688 (2018).
Benatar, M. et al. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 90, e565–e574 (2018).
Acknowledgements
This manuscript was prepared by members of ForeFront, a large collaborative research group dedicated to the study of neurodegenerative diseases and funded by the National Health and Medical Research Council of Australia Program Grant (#1132524), a Dementia Research Team Grant (#1095127) and a Partnership Project (1153439). M.C.K. is supported by an NHMRC Practitioner Fellowship (1156093). J.M.S. receives funding from ALS Finding a Cure Foundation. A.A.-C. is supported through the United Kingdom Medical Research Council (MR/R024804/1) under the aegis of JPND (www.jpnd.eu), the Motor Neurone Disease Association, and the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. P.W. is employed by Wicks Digital Health, which has received funding from Ada Health, AstraZeneca, Baillie Gifford, Bold Health, Camoni, Compass Pathways, Coronna, EIT, Happify, HealthUnlocked, Inbeeo, Kheiron Medical, Sano Genetics, Self Care Catalysts, The Learning Corp, The Wellcome Trust, VeraSci and Woebot. M.R.T. is supported by the Motor Neurone Disease Association.
Author information
Authors and Affiliations
Contributions
M.C.K. researched data for the article. M.C.K. and S.V. made substantial contributions to discussion of the content. M.C.K., S.V., K.T., C.J.M., O.H., J.M.S., A.A.-C. and M.R.T. contributed to the writing of the article. All authors reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Neurology thanks M. de Carvalho, C. Lunetta, S. Petri and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ALS Therapy Development Institute: www.als.net
ANSWER ALS: www.answerals.org
PatientsLikeMe: www.patientslikeme.com
TARGET ALS: www.targetals.org
Rights and permissions
About this article

Cite this article
Kiernan, M.C., Vucic, S., Talbot, K. et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol 17, 104–118 (2021). https://doi.org/10.1038/s41582-020-00434-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-020-00434-z
This article is cited by
-
Accelerating drug development for amyotrophic lateral sclerosis: construction and application of a disease course model using historical placebo group data
Orphanet Journal of Rare Diseases (2024)
-
Heterogeneity of cortical pTDP-43 inclusion morphologies in amyotrophic lateral sclerosis
Acta Neuropathologica Communications (2023)
-
Asynchronous online focus groups for research with people living with amyotrophic lateral sclerosis and family caregivers: usefulness, acceptability and lessons learned
BMC Medical Research Methodology (2023)
-
Opinion: more mouse models and more translation needed for ALS
Molecular Neurodegeneration (2023)
-
The role of placebo control in clinical trials for neurodegenerative diseases
Nature Medicine (2023)
