
Abstract
Post-traumatic headache (PTH) is a highly disabling secondary headache disorder and one of the most common sequelae of mild traumatic brain injury, also known as concussion. Considerable overlap exists between PTH and common primary headache disorders. The most common PTH phenotypes are migraine-like headache and tension-type-like headache. A better understanding of the pathophysiological similarities and differences between primary headache disorders and PTH could uncover unique treatment targets for PTH. Although possible underlying mechanisms of PTH have been elucidated, a substantial void remains in our understanding, and further research is needed. In this Review, we describe the evidence from animal and human studies that indicates involvement of several potential mechanisms in the development and persistence of PTH. These mechanisms include impaired descending modulation, neurometabolic changes, neuroinflammation and activation of the trigeminal sensory system. Furthermore, we outline future research directions to establish biomarkers involved in progression from acute to persistent PTH, and we identify potential drug targets to prevent and treat persistent PTH.
Key points
Post-traumatic headache (PTH) is one of the most common sequelae of traumatic brain injury; the most common headache phenotypes in PTH are migraine-like headache and tension-type-like headache.
PTH is associated with somatic symptoms, including nausea, vomiting, photophobia and phonophobia, and cognitive and psychological symptoms.
Possible disease mechanisms of PTH include impaired descending modulation, neurometabolic changes and activation of the trigeminal sensory system.
The emphasis of future studies of PTH should be on establishing biomarkers of progression from acute PTH to persistent PTH.
Identification of potential treatment targets, such as calcitonin gene-related peptide, should enable randomized controlled trials to be conducted in patients with PTH.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Seifert, T. D. & Evans, R. W. Posttraumatic headache: a review. Curr. Pain Headache Rep. 14, 292–298 (2010).
Nampiaparampil, D. E. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 300, 711–719 (2008).
Mullally, W. J. Concussion. Am. J. Med. 130, 885–892 (2017).
Dewan, M. C. et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 27, 1–18 (2018).
Minen, M. T., Boubour, A., Walia, H. & Barr, W. Post-concussive syndrome: a focus on post-traumatic headache and related cognitive, psychiatric, and sleep issues. Curr. Neurol. Neurosci. Rep. 16, 100 (2016). A review that details the clinical characteristics and associated comorbidities of PTH.
GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 388, 1545–1602 (2016).
Lucas, S. Posttraumatic headache: clinical characterization and management. Curr. Pain Headache Rep. 19, 48 (2015).
Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 38, 1–211 (2018). The newest headache classification.
Baandrup, L. & Jensen, R. Chronic post-traumatic headache–a clinical analysis in relation to the International Headache Classification 2nd edition. Cephalalgia. 25, 132–138 (2005).
Theeler, B., Lucas, S., Riechers, R. G. 2nd & Ruff, R. L. Post-traumatic headaches in civilians and military personnel: a comparative, clinical review. Headache. 53, 881–900 (2013).
Headache Classification Committee of the International Headache Society. Classification and diagnostic criteria for headache disorders, cranial neuralgias, and facial pain. Cephalalgia. 8, 1–96 (1988).
Management of Concussion/mTBI Working Group. VA/DoD clinical practice guideline for management of concussion/mild traumatic brain injury. J. Rehabil Res. Dev. 46, CP1–CP68 (2009).
Aaseth, K. et al. Prevalence of secondary chronic headaches in a population-based sample of 30–44-year-old persons. The Akershus study of chronic headache. Cephalalgia. 28, 705–713 (2008).
Rasmussen, B. K. & Olesen, J. Symptomatic and nonsymptomatic headaches in a general population. Neurology. 42, 1225–1231 (1992).
Zeeberg, P., Olesen, J. & Jensen, R. Efficacy of multidisciplinary treatment in a tertiary referral headache centre. Cephalalgia. 25, 1159–1167 (2005).
Lucas, S., Hoffman, J. M., Bell, K. R. & Dikmen, S. A prospective study of prevalence and characterization of headache following mild traumatic brain injury. Cephalalgia. 34, 93–102 (2014).
Hoffman, J. M. et al. Natural history of headache after traumatic brain injury. J. Neurotrauma. 28, 1719–1725 (2011).
Yilmaz, T. et al. Risk factors and outcomes associated with post-traumatic headache after mild traumatic brain injury. Emerg. Med. J. 34, 800–805 (2017).
Jensen, O. K. & Thulstrup, A. M. Gender differences of post-traumatic headache and other post-commotio symptoms. A follow-up study after a period of 9–12 months. Ugeskr Laeger. 163, 5029–5033 (2001).
Kontos, A. P. et al. Posttraumatic migraine as a predictor of recovery and cognitive impairment after sport-related concussion. Am. J. Sports Med. 41, 1497–1504 (2013).
Langlois, J. A. 1, Rutland-Brown, W. & Wald, M. M. The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 31, 375–378 (2006).
Kjeldgaard, D., Forchhammer, H., Teasdale, T. & Jensen, R. H. Chronic post-traumatic headache after mild head injury: a descriptive study. Cephalalgia. 34, 191–200 (2014).
Feigin, V. L. et al. Incidence of traumatic brain injury in New Zealand: a population-based study. Lancet Neurol. 12, 53–64 (2013).
Lew, H. L. et al. Characteristics and treatment of headache after traumatic brain injury: a focused review. Am. J. Phys. Med. Rehabil. 85, 619–627 (2006).
Stacey, A. et al. Natural history of headache five years after traumatic brain injury. J. Neurotrauma. 34, 1558–1564 (2017). A prospective, longitudinal study that details the clinical characteristics and risk factors for self-reported headache attributed to moderate to severe TBI.
Lucas, S., Hoffman, J. M., Bell, K. R., Walker, W. & Dikmen, S. Characterization of headache after traumatic brain injury. Cephalalgia. 32, 600–606 (2012).
Chibnall, J. T. & Duckro, P. N. Post-traumatic stress disorder in chronic post-traumatic headache patients. Headache. 34, 357–361 (1994).
Lieba-Samal, D. et al. Characteristics of acute posttraumatic headache following mild head injury. Cephalalgia. 31, 1618–1626 (2011).
Schwedt, T. J., Chong, C. D., Peplinski, J., Ross, K. & Berisha, V. Persistent post-traumatic headache vs. migraine: an MRI study demonstrating differences in brain structure. J. Headache Pain. 18, 87 (2017). This MRI study provides evidence for cortical differences between patients with PTH and patients with migraine.
Sufrinko, A. M. et al. Using acute performance on a comprehensive neurocognitive, vestibular, and ocular motor assessment battery to predict recovery duration after sport-related concussions. Am. J. Sports Med. 45, 1187–1194 (2017).
Barros, J. et al. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 70, 235–240 (2013).
Kors, E. E. et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 49, 753–760 (2001).
Tottene, A. et al. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 280, 17678–17686 (2005).
Seifert, T. et al. Comprehensive headache experience in collegiate student-athletes: an initial report from the NCAA Headache Task Force. Headache. 57, 877–886 (2017).
You, H. J. et al. Endogenous descending modulation: spatiotemporal effect of dynamic imbalance between descending facilitation and inhibition of nociception. J. Physiol. 588, 4177–4188 (2010).
Vanegas, H. & Schaible, H. G. Descending control of persistent pain: inhibitory or facilitatory? Brain Res. Rev. 46, 295–309 (2004).
Kwon, M., Altin, M., Duenas, H. & Alev, L. The role of descending inhibitory pathways on chronic pain modulation and clinical implications. Pain Pract. 14, 656–667 (2014).
Ossipov, M. H., Morimura, K. & Porreca, F. Descending pain modulation and chronification of pain. Curr. Opin. Support Palliat Care. 8, 143–151 (2014).
Dodick, D. W. Migraine. Lancet. 391, 1315–1330 (2018).
Kurca, E., Sivák, S. & Kucera, P. Impaired cognitive functions in mild traumatic brain injury patients with normal and pathologic magnetic resonance imaging. Neuroradiology. 48, 661–669 (2006).
Chong, C. D. & Schwedt, T. J. Research imaging of brain structure and function after concussion. Headache. 58, 827–835 (2018).
Rau, J. C., Dumkrieger, G. M., Chong, C. D. & Schwedt, T. J. Imaging post-traumatic headache. Curr. Pain Headache Rep. 22, 64 (2018).
Hulkower, M. B., Poliak, D. B., Rosenbaum, S. B., Zimmerman, M. E. & Lipton, M. L. A decade of DTI in traumatic brain injury: 10 years and 100 articles later. Radiology. 267, 231–239 (2013).
Rutgers, D. R. et al. White matter abnormalities in mild traumatic brain injury: a diffusion tensor imaging study. AJNR Am. J. Neuroradiol. 29, 514–519 (2008).
Mayer, A. R. et al. A prospective diffusion tensor imaging study in mild traumatic brain injury. Neurology. 7, 643–650 (2010).
Miller, D. R., Hayes, J. P., Lafleche, G., Salat, D. H. & Verfaellie, M. White matter abnormalities are associated with chronic postconcussion symptoms in blast-related mild traumatic brain injury. Hum. Brain Mapp. 37, 220–229 (2016).
Wada, T., Asano, Y. & Shinoda, J. Decreased fractional anisotropy evaluated using tract-based spatial statistics and correlated with cognitive dysfunction in patients with mild traumatic brain injury in the chronic stage. AJNR Am. J. Neuroradiol. 33, 2117–2122 (2012).
Morey, R. A. et al. Effects of chronic mild traumatic brain injury on white matter integrity in Iraq and Afghanistan war veterans. Hum. Brain Mapp. 34, 2986–2999 (2013).
Mac Donald, C. L. et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 364, 2091–2100 (2011).
Alhilali, L. M., Delic, J. & Fakhran, S. Differences in callosal and forniceal diffusion between patients with and without postconcussive migraine. AJNR Am. J. Neuroradiol. 38, 691–695 (2017). The first MRI study to investigate changes in white matter integrity between patients with PTH and a migraine-like phenotype and patients with mTBI and either a non-migraine-like phenotype or no headache.
Li, X. L. et al. A diffusion tensor magnetic resonance imaging study of corpus callosum from adult patients with migraine complicated with depressive/anxious disorder. Headache. 51, 237–245 (2011).
Lamm, C., Decety, J. & Singer, T. Meta-analytic evidence for common and distinct neural networks associated with directly experienced pain and empathy for pain. NeuroImage 54, 2492–2502 (2011).
Moulton, E. A., Pendse, G., Becerra, L. R. & Borsook, D. BOLD responses in somatosensory cortices better reflect heat sensation than pain. J. Neurosci 32, 6024–6031 (2012).
Schwedt, T. J. & Chong, C. D. Correlations between brain cortical thickness and cutaneous pain thresholds are atypical in adults with migraine. PLoS One 9, e99791 (2014).
Becker, S., Gandhi, W. & Schweinhardt, P. Cerebral interactions of pain and reward and their relevance for chronic pain. Neurosci Lett. 520, 182–187 (2012).
Kong, J. et al. Using fMRI to dissociate sensory encoding from cognitive evaluation of heat pain intensity. Hum. Brain Mapp. 27, 715–721 (2006).
Chong, C. D., Berisha, V., Chiang, C. C., Ross, K. & Schwedt, T. J. Less cortical thickness in patients with persistent post-traumatic headache compared with healthy controls: an MRI study. Headache. 58, 53–61 (2018). This MRI study investigated differences in cortical thickness between patients with PTH and healthy controls.
Obermann, M. et al. Gray matter changes related to chronic posttraumatic headache. Neurology. 73, 978–983 (2009). The first MRI study of PTH; the study revealed cortical changes in pain processing structures in patients with PTH.
Bashir, A., Lipton, R. B., Ashina, S. & Ashina, M. Migraine and structural changes in the brain: a systematic review and meta-analysis. Neurology. 81, 1260–1268 (2013).
Amen, D. G. et al. Impact of playing American professional football on long-term brain function. J. Neuropsychiatry Clin. Neurosci. 23, 98–106 (2011).
Abdel-Dayem et al. SPECT brain perfusion abnormalities in mild or moderate traumatic brain injury. Clin. Nucl. Med. 23, 309–317 (1998).
Goldenberg, G., Oder, W., Spatt, J. & Podreka, I. Cerebral correlates of disturbed executive function and memory in survivors of severe closed head injury: a SPECT study. J. Neurol. Neurosurg. Psychiatry. 55, 362–368 (1992).
Maugans, T. A., Farley, C., Altaye, M., Leach, J. & Cecil, K. M. Pediatric sports-related concussion produces cerebral blood flow alterations. Pediatrics. 129, 28–37 (2012).
Wang, Y. et al. Decreased cerebral blood flow in chronic pediatric mild TBI: an MRI perfusion study. Dev. Neuropsychol. 40, 40–44 (2015).
Stephens, J. A., Liu, P., Lu, H. & Suskauer, S. J. Cerebral blood flow after mild traumatic brain injury: associations between symptoms and post-injury perfusion. J. Neurotrauma. 35, 241–248 (2018).
Barlow, K. M. et al. Cerebral perfusion changes in post-concussion syndrome: a prospective controlled cohort study. J. Neurotrauma. 34, 996–1004 (2017).
Shumskaya, E., Andriessen, T. M., Norris, D. G. & Vos, P. E. Abnormal whole-brain functional networks in homogeneous acute mild traumatic brain injury. Neurology. 79, 175–182 (2012).
Mayer, A. R., Mannell, M. V., Ling, J., Gasparovic, C. & Yeo, R. A. Functional connectivity in mild traumatic brain injury. Hum. Brain Mapp. 32, 1825–1835 (2011).
Sharp, D. J. Default mode network functional and structural connectivity after traumatic brain injury. Brain. 134, 2233–2247 (2011).
McAllister, T. W. et al. Effect of head impacts on diffusivity measures in a cohort of collegiate contact sport athletes. Neurology. 82, 63–69 (2014).
Zhou, Y. et al. Default-mode network disruption in mild traumatic brain injury. Radiology. 265, 882–892 (2012).
Johnson, B. et al. Alteration of brain default network in subacute phase of injury in concussed individuals: resting-state fMRI study. Neuroimage. 59, 511–518 (2012).
Messe et al. Specific and evolving resting-state network alterations in post-concussion syndrome following mild traumatic brain injury. PLoS One. 8, e65470 (2013).
Katayama, Y., Becker, D. P., Tamura, T. & Hovda, D. A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900 (1990).
Choe, M. C. The pathophysiology of concussion. Curr. Pain Headache Rep. 20, 42 (2016).
Yoshino, A., Hovda, D. A., Kawamata, T., Katayama, Y. & Becker, D. P. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 561, 106–119 (1991).
Barkhoudarian, G., Hovda, D. A. & Giza, C. C. The molecular pathophysiology of concussive brain injury – an update. Phys. Med. Rehabil Clin. N. Am. 27, 373–393 (2016).
Hill, C. S., Coleman, M. P. & Menon, D. K. Traumatic axonal injury: mechanisms and translational opportunities. Trends Neurosci. 39, 311–324 (2016).
Pettus, E. H. & Povlishock, J. T. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 722, 1–11 (1996).
Povlishock, J. T. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 2, 1–12 (1992).
Burtscher, I. M. & Holtås, S. Proton MR spectroscopy in clinical routine. J. Magn. Reson. Imaging. 13, 560–567 (2001).
Kirov, I. I., Whitlow, C. T. & Zamora, C. Susceptibility-weighted imaging and magnetic resonance spectroscopy in concussion. Neuroimaging Clin. N. Am. 28, 91–105 (2018).
Gasparovic, C. et al. Neurometabolite concentrations in gray and white matter in mild traumatic brain injury: an 1H-magnetic resonance spectroscopy study. J. Neurotrauma. 26, 1635–1643 (2009).
Manning, K. Y. et al. Multiparametric MRI changes persist beyond recovery in concussed adolescent hockey players. Neurology. 89, 2157–2166 (2017).
Al-Karagholi, M. M., Hansen, J. M., Guo, S., Olesen, J. & Ashina M. Opening of ATP sensitive channels causes migraine attacks: a new target for the treatment of migraine. Brain https://doi.org/10.1093/brain/awz199 (2019).
Borkum, J. M. Migraine triggers and oxidative stress: a narrative review and synthesis. Headache. 56, 12–35 (2016).
Aytaç, B. et al. Decreased antioxidant status in migraine patients with brain white matter hyperintensities. Neurol. Sci. 35, 1925–1929 (2014).
Dalkara, T., Nozari, A. & Moskowitz, M. A. Migraine aura pathophysiology: the role of blood vessels and microembolisation. Lancet Neurol. 9, 309–317 (2010).
Sanchez-Del-Rio, M., Reuter, U. & Moskowitz, M. A. New insights into migraine pathophysiology. Curr. Opin. Neurol. 19, 294–298 (2006).
Bolay, H. et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 8, 136–142 (2002).
Schock, S. C. et al. Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res. 1168, 129–138 (2007).
Strong, A. J. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 33, 2738–2743 (2002).
Elliott, M. B., Oshinsky, M. L., Amenta, P. S., Awe, O. O. & Jallo, J. I. Nociceptive neuropeptide increases and periorbital allodynia in a model of traumatic brain injury. Headache. 52, 966–984 (2012).
Feliciano, D. P. et al. Nociceptive sensitization and BDNF up-regulation in a rat model of traumatic brain injury. Neurosci Lett. 583, 55–59 (2014).
Rogatsky, G. G., Sonn, J., Kamenir, Y., Zarchin, N. & Mayevsky, A. Relationship between intracranial pressure and cortical spreading depression following fluid percussion brain injury in rats. J. Neurotrauma. 20, 1315–1325 (2003).
Kane, M. J. et al. A mouse model of human repetitive mild traumatic brain injury. J. Neurosci. Methods. 203, 41–49 (2012).
Goddeyne, C., Nichols, J., Wu, C. & Anderson, T. Repetitive mild traumatic brain injury induces ventriculomegaly and cortical thinning in juvenile rats. J. Neurophysiol. 113, 3268–3280 (2015).
Packard, R. C. The relationship of neck injury and post-traumatic headache. Curr. Pain Headache Rep. 6, 301–307 (2002).
Mayer, C. L., Huber, B. R. & Peskind, E. Traumatic brain injury, neuroinflammation, and post-traumatic headaches. Headache. 53, 1523–1530 (2013).
Charles, A. Migraine: a brain state. Curr. Opin. Neurol. 26, 235–239 (2013).
Loane, D. J. & Byrnes, K. R. Role of microglia in neurotrauma. Neurotherapeutics. 7, 366–377 (2010).
Frugier, T., Morganti-Kossmann, M. C., O’Reilly, D. & McLean, C. A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma. 27, 497–507 (2010).
Hains, B. C. & Waxman, S. G. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 26, 4308–4317 (2006).
Waxman, S. G. & Hains, B. C. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci. 29, 207–215 (2006).
Gursoy-Ozdemir, Y. et al. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Invest. 113, 1447–1455 (2004).
Vilalta, A. et al. Brain contusions induce a strong local overexpression of MMP-9. Results of a pilot study. Acta Neurochir Suppl. 102, 415–419 (2008).
Imamura, K., Takeshima, T., Fusayasu, E. & Nakashima, K. Increased plasma matrix metalloproteinase-9 levels in migraineurs. Headache. 48, 135–139 (2008).
Martins-Oliveira, A. et al. Specific matrix metalloproteinase 9 (MMP-9) haplotype affect the circulating MMP-9 levels in women with migraine. J. Neuroimmunol. 252, 89–94 (2012).
Ashina, M. et al. Matrix metalloproteinases during and outside of migraine attacks without aura. Cephalalgia. 30, 303–310 (2010).
Zhang, X. et al. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 69, 855–865 (2011).
Karatas, H. et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 339, 1092–1095 (2013).
Schain, A. J. et al. Activation of pial and dural macrophages and dendritic cells by cortical spreading depression. Ann. Neurol. 83, 508–521 (2018).
Levy, D. et al. Responses of dural mast cells in concussive and blast models of mild traumatic brain injury in mice: potential implications for post-traumatic headache. Cephalalgia. 36, 915–923 (2016).
Benromano, T. et al. Mild closed head injury promotes a selective trigeminal hypernociception: implications for the acute emergence of post-traumatic headache. Eur. J. Pain. 19, 621–628 (2015).
Defrin, R., Gruener, H., Schreiber, S. & Pick, C. G. Quantitative somatosensory testing of subjects with chronic post-traumatic headache: implications on its mechanisms. Eur. J. Pain. 14, 924–931 (2010).
Burstein, R., Yarnitsky, D., Goor-Aryeh, I., Ransil, B. J. & Bajwa, Z. H. An association between migraine and cutaneous allodynia. Ann. Neurol. 47, 614–624 (2000).
Gracely, R. H., Lynch, S. A. & Bennett, G. J. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 51, 175–194 (1992).
Burstein, R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 89, 107–110 (2001).
Roth, T. L. et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 505, 223–228 (2014).
Chen, G., Shi, J., Hu, Z. & Hang, C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008, 716458 (2008).
Ellis, E. F., Dodson, L. Y. & Police, R. J. Restoration of cerebrovascular responsiveness to hyperventilation by the oxygen radical scavenger n-acetylcysteine following experimental traumatic brain injury. J. Neurosurg. 75, 774–779 (1991).
Eakin, K. et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 9, e90617 (2014).
Hoffer, M. E., Balaban, C., Slade, M. D., Tsao, J. W. & Hoffer, B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 8, e54163 (2013).
Ashina, H., Schytz, H. W. & Ashina M. CGRP in human models of migraine. Handb. Exp. Pharmacol. https://doi.org/10.1007/164_2018_128 (2018).
Asghar, M. S. et al. Evidence for a vascular factor in migraine. Ann. Neurol. 69, 635–645 (2011).
Daiutolo, B. V., Tyburski, A., Clark, S. W. & Elliott, M. B. Trigeminal pain molecules, allodynia, and photosensitivity are pharmacologically and genetically modulated in a model of traumatic brain injury. J. Neurotrauma. 15, 748–760 (2016).
Khan, S., Olesen, A. & Ashina, M. CGRP, a target for preventive therapy in migraine and cluster headache: systematic review of clinical data. Cephalalgia 39, 374–389 (2017).
Lennerz, J. K. et al. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J. Comp. Neurol. 507, 1277–1299 (2008).
McCulloch, J., Uddman, R., Kingman, T. A. & Edvinsson, L. Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc. Natl Acad. Sci. U.S.A. 83, 5731–5735 (1986).
Goadsby, P. J., Edvinsson, L. & Ekman, R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann. Neurol. 23, 193–196 (1988).
Bree, D. & Levy, D. Development of CGRP-dependent pain and headache related behaviours in a rat model of concussion: implications for mechanisms of post-traumatic headache. Cephalalgia. 38, 246–258 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03347188 (2019).
Hilz, M. J. Valsalva maneuver unveils central baroreflex dysfunction with altered blood pressure control in persons with a history of mild traumatic brain injury. BMC Neurol. 16, 61 (2016).
Wei, X. et al. Meningeal norepinephrine produces headache behaviors in rats via actions both on dural afferents and fibroblasts. Cephalalgia. 35, 1054–1064 (2015).
Lindholt, M. et al. Lack of effect of norepinephrine on cranial haemodynamics and headache in healthy volunteers. Cephalalgia. 29, 384–387 (2009).
Ray, B. S. & Wolff, H. G. Experimental studies on headache; pain-sensitive structures of the head and their significance in headache. Arch. Surg. 41, 813 (1940).
Olesen, J., Burstein, R., Ashina, M. & Tfelt-Hansen, P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 8, 679–690 (2009).
Schueler, M., Messlinger, K., Dux, M., Neuhuber, W. L. & De Col, R. Extracranial projections of meningeal afferents and their impact on meningeal nociception and headache. Pain. 154, 1622–1631 (2013).
Kosaras, B., Jakubowski, M., Kainz, V. & Burstein, R. Sensory innervation of the calvarial bones of the mouse. J. Comp. Neurol. 515, 331–348 (2009).
Ashina, S. et al. Prevalence of neck pain in migraine and tension-type headache: a population study. Cephalalgia. 35, 211–219 (2015).
Ropper, A. H. & Gorson, K. C. Clinical practice. Concussion. N. Engl. J. Med. 356, 166–172 (2007).
Johnston, M. M., Jordan, S. E. & Charles, A. C. Pain referral patterns of the C1 to C3 nerves: implications for headache disorders. Ann. Neurol. 74, 145–148 (2013).
Bartsch, T. & Goadsby, P. J. Stimulation of the greater occipital nerve induces increased central excitability of dural afferent input. Brain. 125, 1496–1509 (2002).
Le Doaré, K. et al. Occipital afferent activation of second order neurons in the trigeminocervical complex in rat. Neurosci. Lett. 403, 73–77 (2006).
Bree, D. & Levy, D. Strides toward better understanding of post-traumatic headache pathophysiology using animal models. Curr. Pain Headache Rep. 22, 67 (2018). An up-to-date review of animal models of PTH.
Andreou A. P., Oshinsky M. L. Animal models of migraine. Ashina M., Geppetti P., editors. Pathophysiology of Headaches From Molecule to Man. Switzerland: Springer International Publishing. pp. 31–66 (2015).
Stovner, L. J., Schrader, H., Mickeviciene, D., Surkiene, D. & Sand, T. Headache after concussion. Eur. J. Neurol. 16, 112–120 (2009).
Olesen, J. et al. The economic cost of brain disorders in Europe. Eur. J. Neurol. 19, 155–162 (2012).
Bhattacharjee, Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science. 319, 406–408 (2008).
Acknowledgements
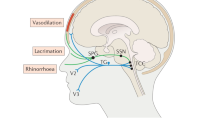
The authors thank PhD student T. P. Do, University of Copenhagen, for drawing a preliminary sketch of figure 2. No compensation was received for this contribution.
Authors contributions
All authors researched data for the article, discussed the content, wrote the text, and reviewed and edited the manuscript before submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
F.M.A. is a lecturer or scientific adviser for Novartis and Teva. M.A. is a consultant, speaker or scientific adviser for Alder, Allergan, Amgen, Eli Lilly, Novartis and Teva. H.W.S. is a lecturer for Novartis. D.W.D. reports the following competing interests: personal fees from Alder, Allergan, Amgen, Association of Translational Medicine, Autonomic Technologies, Aural Analytics, Biohaven, Charleston Laboratories, Daniel Edelman, Axsome, Dr Reddy’s Laboratories/Promius, Electrocore, Eli Lilly, eNeura, Foresite Capital, Impel, Ipsen, Neurolief, Nocira, Novartis, Oppenheimer, PSL Group Services, Satsuma, Sun Pharma (India), Supernus, Teva, Theranica, University of British Columbia, University Health Network, Vedanta, WL Gore, XoC, Zosano and ZP Opco; CME fees or royalty payments from Academy for Continued Healthcare Learning, Cambridge University Press, Chameleon, Global Access Meetings, Global Life Sciences, Global Scientific Communications, Haymarket, Healthlogix, Medicom Worldwide, Medlogix Communications, Mednet, Miller Medical, Oxford University Press, PeerView, Universal Meeting Management, UpToDate (Elsevier), WebMD Health/Medscape and Wolters Kluwer Health; stock options with Aural Analytics, Epien, GBS/Nocira, Healint, King-Devick Technologies, Matterhorn/Ontologics, Second Opinon/Mobile Health and Theranica; consulting without fee for Aural Analytics, Epien, Healint and Second Opinion/Mobile Health; position on the board of directors for Epien, King-Devick Technologies and Matterhorn/Ontologics; patent 17189376.1-1466: vTitle: Botulinum Toxin Dosage Regimen for Chronic Migraine Prophylaxis without fee; research funding from American Migraine Foundation, Henry Jackson Foundation, Patient-Centered Outcomes Research Institute and US Department of Defence; professional society fees or reimbursement for travel from American Academy of Neurology, American Brain Foundation, American Headache Society, American Migraine Foundation, Canadian Headache Society and International Headache Society; and use agreement through employer for Myndshft. The other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Review criteria
Articles discussed in this Review were identified by PubMed searches performed between 1 May 2018 and 1 September 2018 with no restrictions on the date of publication. The search terms used were ‘post-traumatic headache’, ‘PTH’, ‘concussion’, ‘traumatic brain injury’, ‘pathophysiology’ and ‘imaging’. The reference lists of identified papers were searched for further relevant articles, and related citations for identified papers as listed on the PubMed site were evaluated. The final references included were chosen based on the relevance to the scope of this Review.
Rights and permissions
About this article

Cite this article
Ashina, H., Porreca, F., Anderson, T. et al. Post-traumatic headache: epidemiology and pathophysiological insights. Nat Rev Neurol 15, 607–617 (2019). https://doi.org/10.1038/s41582-019-0243-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-019-0243-8
This article is cited by
-
Effects of PDE-3 inhibition in persistent post-traumatic headache: evidence of cAMP-dependent signaling
The Journal of Headache and Pain (2024)
-
White matter hyperintensities and cerebral microbleeds in persistent post-traumatic headache attributed to mild traumatic brain injury: a magnetic resonance imaging study
The Journal of Headache and Pain (2023)
-
Imaging the brain and vascular reactions to headache treatments: a systematic review
The Journal of Headache and Pain (2023)
-
Repeat mild traumatic brain injuries (RmTBI) modify nociception and disrupt orexinergic connectivity within the descending pain pathway
The Journal of Headache and Pain (2023)
-
Idiopathic intracranial hypertension: a step change in understanding the disease mechanisms
Nature Reviews Neurology (2023)
