
Abstract
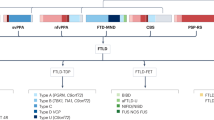
Frontotemporal lobar degeneration (FTLD) represents a group of neurodegenerative brain diseases with highly heterogeneous clinical, neuropathological and genetic characteristics. This high degree of heterogeneity results from the presence of several different underlying molecular disease processes; consequently, it is unlikely that all patients with FTLD will benefit from a single therapy. Therapeutic strategies for FTLD are currently being explored, and tools are urgently needed that enable the selection of patients who are the most likely to benefit from a particular therapy. Definition of the phenotypic characteristics in patients with different FTLD subtypes that share the same underlying disease processes would assist in the stratification of patients into homogeneous groups. The most common subtype of FTLD is characterized by TAR DNA-binding protein 43 (TDP43) pathology (FTLD-TDP). In this group, pathogenic mutations have been identified in four genes: C9orf72, GRN, TBK1 and VCP. Here, we provide a comprehensive overview of the phenotypic characteristics of patients with FTLD-TDP, highlighting shared features and differences among groups of patients who have a pathogenic mutation in one of these four genes.
Key points
-
Comprehension of genotype–phenotype correlations will aid patient selection and stratification for targeted therapeutic strategies.
-
Most individuals with a C9orf72 repeat expansion present with the behavioural variant of frontotemporal dementia (FTD) and frequently have psychotic symptoms, motor neuron disease (MND) and a symmetric pattern of brain impairment that is most predominant in frontotemporal regions.
-
Patients with FTD who carry a GRN mutation are characterized by apathetic behaviour, frequently with language output impairment, parietal lobe dysfunction and parkinsonism, in association with widespread, asymmetric impairment of frontotemporoparietal brain regions.
-
TBK1 mutation in patients with FTD is frequently associated with MND symptomatology and problems with behaviour and language, but the predominant phenotypic features have yet to be distinguished; brain impairment is mostly asymmetric in these individuals.
-
Individuals with FTD who have a VCP mutation can present with or without myopathy or Paget disease of the bone and have characteristic features of apathy, anomia, psychotic signs and a nonspecific pattern of brain impairment.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Hogan, D. B. et al. The prevalence and incidence of frontotemporal dementia: a systematic review. Can. J. Neurol. Sci. 43, S96–S109 (2016).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
Gorno-Tempini, M. L. et al. Classification of primary progressive aphasia and its variants. Neurology 76, 1–10 (2011).
Josephs, K. A. et al. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol. Appl. Neurobiol. 30, 369–373 (2004).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Leigh, P. N. et al. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 93, 197–203 (1988).
Rosso, S. M. et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 126, 2016–2022 (2003).
Goldman, J. S. et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 65, 1817–1819 (2005).
Seelaar, H. et al. Distinct genetic forms of frontotemporal dementia. Neurology 71, 1220–1226 (2008).
Rohrer, J. D. et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 73, 1451–1456 (2009).
Poorkaj, P. et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825 (1998).
Hutton, M. et al. Association of missense and 5´-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 (1998).
Spillantini, M. G. et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl Acad. Sci. USA 95, 7737–7741 (1998).
Cruts, M. et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 (2006).
Baker, M. et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 (2006).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
Gijselinck, I. et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65 (2012).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015).
Pottier, C. et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92 (2015).
Gijselinck, I. et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 85, 2116–2125 (2015).
Watts, G. D. J. et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 (2004).
Skibinski, G. et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nature Genet. 37, 806–808 (2005).
Johnson, J. O. et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 (2010).
MacKenzie, I. R. A. et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879 (2013).
Gendron, T. F. et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844 (2013).
Mizielinska, S. et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857 (2013).
Boeve, B. F. et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 135, 765–783 (2012).
Mahoney, C. J. et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135, 736–750 (2012).
Sha, J. S. et al. Frontotemporal dementia due to C9ORF72 mutations clinical and imaging features. Neurology 79, 1002–1011 (2012).
Simón-Sánchez, J. et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135, 723–735 (2012).
Snowden, J. S. et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135, 693–708 (2012).
Galimberti, D. et al. Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late-onset psychotic clinical presentation. Biol. Psychiatry 74, 384–391 (2013).
Devenney, E. et al. Frontotemporal dementia associated with the C9ORF72 mutation. JAMA Neurol. 71, 1–9 (2014).
Kaivorinne, A.-L. et al. Clinical characteristics of C9ORF72 -linked frontotemporal lobar degeneration. Dement. Geriatr. Cogn. Dis. Extra 3, 251–262 (2013).
Benussi, L. et al. C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: a genotype-phenotype correlation study. J. Alzheimer’s Dis. 38, 799–808 (2014).
Cooper-Knock, J. et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764 (2012).
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012).
Dobson-Stone, C. et al. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 79, 995–1001 (2012).
Van Langenhove, T. et al. Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol. 70, 365–373 (2013).
Millecamps, S. et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 49, 258–263 (2012).
Hsiung, G. Y. R. et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722 (2012).
Van Mossevelde, S., van der Zee, J., Cruts, M. & Van Broeckhoven, C. Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 44, 117–124 (2017).
Majounie, E. et al. Repeat expansion in C9ORF72 in Alzheimer’s Disease. N. Engl. J. Med. 366, 283–284 (2012).
Harms, M. et al. C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 70, 736–741 (2013).
Cacace, R. et al. C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 34, 1712.e1–1712.e7 (2013).
Kohli, M. A. et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer’s disease in Caucasians. Neurobiol. Aging 34, 1519.e5–1519.e12 (2013).
Lesage, S. et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136, 385–391 (2013).
Beck, J. et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353 (2013).
Chiò, A. et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135, 784–793 (2012).
van der Zee, J. et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 34, 363–373 (2013).
Xi, Z. et al. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch. Neurol. 69, 1583–1590 (2012).
Snowden, J. S. et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph. Lateral Scler. Frontotemporal Degener. 8421, 1–9 (2015).
Mahoney, C. J. et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers. Res. Ther. 4, 41 (2012).
Murray, M. E. et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 122, 673–690 (2011).
Irish, M. et al. Neural substrates of episodic memory dysfunction in behavioural variant frontotemporal dementia with and without C9ORF72 expansions. Neuroimage Clin. 2, 836–843 (2013).
Kertesz, A. et al. Psychosis and hallucinations in FTD with C9orf72 mutation: a detailed clinical cohort. Cogn. Behav. Neurol. 26, 146–154 (2013).
Galimberti, D. et al. C9ORF72 hexanucleotide repeat expansion as a rare cause of bipolar disorder. Bipolar Disord. 16, 448–449 (2014).
Huey, E. D. et al. C9orf72 repeat expansions not detected in a group of patients with schizophrenia. Neurobiol. Aging 34, 1309.e9–1309.e10 (2013).
Nuytemans, K. et al. Absence of C9ORF72 expanded or intermediate repeats in autopsy-confirmed Parkinson’s disease. Mov. Disord. 29, 827–830 (2014).
Theuns, J. et al. Global investigation and meta-analysis of the C9orf72 (G4C2)n repeat in Parkinson disease. Neurology 83, 1906–1913 (2014).
Black, H. A. et al. Genetic epidemiology of motor neuron disease-associated variants in the Scottish population. Neurobiol. Aging 51, 178.e11–178.e20 (2017).
Stewart, H. et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 123, 409–417 (2012).
Van Rheenen, W. et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 79, 878–882 (2012).
Van Mossevelde, S. et al. Clinical evidence of disease anticipation in families segregating a C9orf72 repeat expansion. JAMA Neurol. 74, 445–452 (2017).
Whitwell, J. L. et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135, 794–806 (2012).
Floeter, M. K. et al. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. 12, 1035–1043 (2016).
Bigio, E. H. et al. Frontotemporal lobar degenertation with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9orf72: clinicopathologic correlation. Neuropathology 33, 122–133 (2013).
Mori, K. et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893 (2013).
Mann, D. M. et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1, 68 (2013).
Bieniek, K. F. et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 125, 289–302 (2013).
Ghidoni, R., Benussi, L., Glionna, M., Franzoni, M. & Binetti, G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239 (2008).
Sleegers, K. et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann. Neurol. 65, 603–609 (2009).
Finch, N. et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591 (2009).
Lopez de Munain, A. et al. Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol. Psychiatry 63, 946–952 (2008).
van der Zee, J. et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum. Mutat. 28, 416 (2007).
Petkau, T. L. & Leavitt, B. R. Progranulin in neurodegenerative disease. Trends Neurosci. 37, 388–398 (2014).
Beck, J. et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131, 706–720 (2008).
Gass, J. et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001 (2006).
Le Ber, I. et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum. Mutat. 28, 846–855 (2007).
Yu, C.-E. et al. The spectrum of mutations in progranulin. Arch. Neurol. 67, 161–170 (2010).
Bronner, I. F. et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur. J. Hum. Genet. 15, 369–374 (2007).
Huey, E. D. et al. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann. Med. 60, 374–380 (2006).
Pickering-Brown, S. M. et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131, 721–731 (2008).
Le Ber, I. et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131, 732–746 (2008).
Bruni, A. C. et al. Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69, 140–147 (2007).
Rademakers, R. et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C→T (Arg493X) mutation: an international initiative. Lancet Neurol. 6, 857–868 (2007).
Brouwers, N. et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446 (2007).
Moreno, F. et al. Frontotemporoparietal dementia: clinical phenotype associated with the c.709-1G> A PGRN mutation. Neurology 73, 1367–1374 (2009).
Kelley, B. J. et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol. Aging 30, 739–751 (2009).
Snowden, J. S. et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129, 3091–3102 (2006).
Benussi, L. et al. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 33, 379–385 (2009).
van der Zee, J. et al. TBK1 mutation spectrum in an extended European patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum. Mutat. 38, 297–309 (2017).
Benussi, L. et al. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging 29, 427–435 (2008).
Benussi, L. et al. Estimating the age of the most common italian grn mutation: walking back to Canossa times. J. Alzheimer’s Dis. 33, 69–76 (2013).
Josephs, K. A. et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J. Neuropathol. Exp. Neurol. 66, 142–151 (2007).
Sassi, C. et al. A novel splice-acceptor site mutation in GRN (c.709-2A>T) causes frontotemporal dementia spectrum in a large family from southern Italy. J. Alzheimer’s Dis. 53, 475–485 (2016).
Van Mossevelde, S. et al. Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain 139, 452–467 (2016).
Kelley, B. J. et al. Alzheimer disease-like phenotype associated with the c.154delA mutation in progranulin. Arch. Neurol. 67, 171–177 (2010).
Chen-Plotkin, A. A. S. et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch. Neurol. 68, 488–497 (2011).
Mesulam, M. et al. Progranulin mutations in primary progressive aphasia. Arch. Neurol. 64, 43 (2007).
Spina, S. et al. Corticobasal syndrome associated with the A9D progranulin mutation. J. Neuropathol. Exp. Neurol. 66, 892–900 (2007).
Pires, C. et al. Phenotypic variability of familial and sporadic progranulin p. Gln257Profs*27 mutation. J. Alzheimer’s Dis. 37, 335–342 (2013).
Puoti, G. et al. A mutation in the 5´-UTR of GRN gene associated with frontotemporal lobar degeneration: phenotypic variability and possible pathogenetic mechanisms. J. Alzheimer’s Dis. 42, 939–947 (2014).
Boeve, B. F. et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G→A mutation in progranulin: a clinicopathologic study. Brain 129, 3103–3114 (2006).
Whitwell, J. L. et al. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch. Neurol. 64, 371–376 (2007).
Rohrer, J. D., Crutch, S. J., Warrington, E. K. & Warren, J. D. Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia 48, 288–297 (2010).
Caso, F. et al. The progranulin (GRN) Cys157LysfsX97 mutation is associated with nonfluent variant of primary progressive aphasia clinical phenotype. J. Alzheimer’s Dis. 28, 759–763 (2012).
Milan, G. et al. GRN deletion in familial frontotemporal dementia showing association with clinical variability in 3 familial cases. Neurobiol. Aging 53, 193.e9–193.e16 (2017).
Rohrer, J. D., Rossor, M. N. & Warren, J. D. Syndromes of nonfluent primary progressive aphasia: a clinical and neurolinguistic analysis. Neurology 75, 603–610 (2010).
Flanagan, E. P. et al. Dominant frontotemporal dementia mutations in 140 cases of primary progressive aphasia and speech apraxia. Dement Geriatr. Cogn. Disord. 39, 281–286 (2015).
Kim, G. et al. Asymmetric pathology in primary progressive aphasia with progranulin mutations and TDP inclusions. Neurology 86, 627–636 (2016).
Josephs, K. A. et al. Progranulin-associated PiB-negative logopenic primary progressive aphasia. J. Neurol. 261, 604–614 (2014).
Spina, S. et al. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology 68, 820–827 (2007).
Kuuluvainen, L. et al. A novel loss-of-function GRN mutation p.(Tyr229∗): clinical and neuropathological features. J. Alzheimer’s Dis. 55, 1167–1174 (2017).
Schymick, J. C. et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 78, 754–756 (2007).
Sleegers, K. et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259 (2008).
Gijselinck, I., Van Broeckhoven, C. & Cruts, M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum. Mutat. 29, 1373–1386 (2008).
Chiang, H. H. et al. Progranulin mutation causes frontotemporal dementia in the Swedish Karolinska family. Alzheimer’s Dement. 4, 414–420 (2008).
Premi, E. et al. Subcortical and deep cortical atrophy in frontotemporal dementia due to granulin mutations. Dement. Geriatr. Cogn. Dis. Extra 4, 95–102 (2014).
Rohrer, J. D. et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53, 1070–1076 (2010).
Whitwell, J. L. et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 72, 813–820 (2009).
Caroppo, P. et al. Extensive white matter involvement in patients with frontotemporal lobar degeneration. JAMA Neurol. 71, 1562 (2014).
Pietroboni, A. et al. Phenotypic heterogeneity of the progranulin gene Asp22 fs mutation in a large Italian kindred. J. Alzheimer’s Dis. 258, 253–259 (2011).
Paternico, D. et al. White matter hyperintensities characterize monogenic frontotemporal dementia with granulin mutations. Neurobiol. Aging 38, 176–180 (2016).
Whitwell, J. L. et al. Clinical and neuroimaging biomarkers of amyloid-negative logopenic primary progressive aphasia. Brain Lang. 142, 45–53 (2015).
Mackenzie, I. R. A. et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090 (2006).
Hosokawa, M. et al. Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation. Sci. Rep. 7, 1513 (2017).
Gleason, C. E., Ordureau, A., Gourlay, R., Arthur, J. S. C. & Cohen, P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J. Biol. Chem. 286, 35663–35674 (2011).
Weidberg, H. & Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 4, e39 (2011).
Caroppo, P. et al. Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimer’s dement. 1, 481–486 (2015).
Le Ber, I. et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging 36, 3116.e5–3116.e8 (2015).
Oakes, J. A., Davies, M. C. & Collins, M. O. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10, 5 (2017).
Williams, K. L. et al. Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol. Aging 36, 3334.e1–3334.e5 (2015).
Tsai, P.-C. et al. Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 40, 191.e11–191.e16 (2016).
Borghero, G. et al. TBK1 is associated with ALS and ALS-FTD in Sardinian patients. Neurobiol. Aging 43, 180.e1–180.e5 (2015).
Kim, Y.-E. et al. Genetic and functional analysis of TBK1 variants in Korean patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 50, 170.e1–170.e6 (2017).
Koriath, C. A. M. et al. The clinical, neuroanatomical, and neuropathologic phenotype of TBK1-associated frontotemporal dementia: a longitudinal case report. Alzheimer’s Dement. 6, 75–81 (2017).
Schönecker, S. et al. Ein Geschwisterpaar mit frontotemporaler Lobärdegeneration und amyotropher Lateralsklerose und einer neuen Mutation im TBK1-Gen (Thr462Lysfs). Fortschritte Neurol. Psychiatr. 84, 494–498 (2016).
Pozzi, L. et al. TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J. Neurol. Neurosurg. Psychiatry 88, 869–875 (2017).
Wilke, C. et al. Beyond ALS and FTD: the phenotypic spectrum of TBK1 mutations includes PSP-like and cerebellar phenotypes. Neurobiol. Aging 62, 244.e9–244.e13 (2017).
Tohnai, G. et al. Frequency and characteristics of the TBK1 gene variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 64, 158.e15–158.e19 (2017).
Verheijen, J. et al. Common and rare TBK1 variants in early-onset Alzheimer disease in a European cohort. Neurobiol. Aging 62, 245.e1–245.e7 (2017).
Meyer, H. & Weihl, C. C. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883 (2014).
Weihl, C. C., Dalal, S., Pestronk, A. & Hanson, P. I. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15, 189–199 (2006).
Van Der Zee, J. et al. Clinical heterogeneity in 3 unrelated families linked to VCP p. Arg159His. Neurology 73, 626–632 (2009).
Mehta, S. G. et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin. Genet. 83, 422–431 (2013).
Kumar, K. R. et al. Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul. Disord. 20, 330–334 (2010).
Guyant-Maréchal, L. et al. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 67, 644–651 (2006).
Fanganiello, R. D., Kimonis, V. E., Côrte, C. C., Nitrini, R. & Passos-Bueno, M. R. A. Brazilian family with hereditary inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Braz. J. Med. Biol. Res. 44, 374–380 (2011).
Djamshidian, A. et al. A novel mutation in the VCP gene (G157R) in a German family with inclusion-body myopathy with Paget disease of bone and frontotemporal dementia. Muscle Nerve 39, 389–391 (2009).
Kimonis, V. E. et al. Clinical studies in familial VCP myopathy associated with paget disease of bone and frontotemporal dementia. Am. J. Med. Genet. A 146, 745–757 (2008).
De Bot, S. T., Schelhaas, H. J., Kamsteeg, E. J. & Van De Warrenburg, B. P. C. Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain 135, e223 (2012).
Segers, K., Glibert, G., Callebaut, J., Kevers, L. & Alcan, I. Involvement of peripheral and central nervous systems in a valosin-containing protein mutation. J. Clin. Neurol. 10, 166–170 (2014).
Gonzalez, M. A. et al. A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain 137, 2897–2902 (2014).
Spina, S. et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur. J. Neurol. 20, 251–258 (2013).
Stojkovic, T. et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul. Disord. 19, 316–323 (2009).
Rohrer, J. D. et al. A novel exon 2 I27V valosin-containing protein variant is associated with dissimilar clinical syndromes. J. Neurol. 258, 1494–1496 (2011).
Kovach, M. J. et al. Clinical delineation and localization to chromosome 9p13.3–p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol. Genet. Metab. 74, 458–475 (2001).
Viassolo, V. et al. Inclusion body myopathy, Paget’s disease of the bone and frontotemporal dementia: recurrence of the VCP R155H mutation in an Italian family and implications for genetic counselling. Clin. Genet. 74, 54–60 (2008).
Kim, E.-J. et al. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia linked to VCP p. Arg155Cys in a Korean family. Arch. Neurol. 68, 787–796 (2011).
Forman, M. S. et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J. Neuropathol. Exp. Neurol. 65, 571–581 (2006).
Neumann, M. et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 66, 152–157 (2007).
Ghetti, B. et al. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46 (2015).
Gasca-Salas, C. et al. Characterization of movement disorder phenomenology in genetically proven, familial frontotemporal lobar degeneration: a systematic review and meta-analysis. PLoS ONE 11, 1–20 (2016).
Sudre, C. H. et al. White matter hyperintensities are seen only in GRN mutation carriers in the GENFI cohort. Neuroimage Clin. 15, 171–180 (2017).
Rohrer, J. D., Warren, J. D., Fox, N. C. & Rossor, M. N. Presymptomatic studies in genetic frontotemporal dementia. Rev. Neurol. 169, 820–824 (2013).
Borroni, B. et al. Founder effect and estimation of the age of the Progranulin Thr272fs mutation in 14 Italian pedigrees with frontotemporal lobar degeneration. Neurobiol. Aging 32, 555.e1–555.e8 (2011).
Gijselinck, I. et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry. 21, 1112–1124 (2016).
Shi, J. et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 110, 501–512 (2005).
Sampathu, D. M. et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 169, 1343–1352 (2006).
Cairns, N. J. et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 171, 227–240 (2007).
Davidson, Y. et al. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533 (2007).
Mackenzie, I. R. A., Bigio, E. H., Cairns, N. J. & Kril, J. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 117, 15–18 (2009).
Mackenzie, I. R. A. et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113 (2011).
Lee, E. B. et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 134, 65–78 (2017).
Acknowledgements
Research by the authors is funded in part by the Belgian Science Policy Office Interuniversity Attraction Poles programme, the Flemish-government-initiated Flanders Impulse Program on Networks for Dementia Research (VIND) and the Methusalem Excellence Program, the Research Foundation Flanders (FWO) and the University of Antwerp Research Fund, Belgium.
Author information
Authors and Affiliations
Contributions
All authors contributed to the research, discussion, writing, review and editing of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Van Mossevelde, S., Engelborghs, S., van der Zee, J. et al. Genotype–phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol 14, 363–378 (2018). https://doi.org/10.1038/s41582-018-0009-8
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-018-0009-8
This article is cited by
-
Progranulin haploinsufficiency mediates cytoplasmic TDP-43 aggregation with lysosomal abnormalities in human microglia
Journal of Neuroinflammation (2024)
-
Reduced progranulin increases tau and α-synuclein inclusions and alters mouse tauopathy phenotypes via glucocerebrosidase
Nature Communications (2024)
-
Physiological and pathological effects of phase separation in the central nervous system
Journal of Molecular Medicine (2024)
-
Amplifying the Heat Shock Response Ameliorates ALS and FTD Pathology in Mouse and Human Models
Molecular Neurobiology (2023)
-
Case report: coexistence of C9orf72 expansion and progranulin mutation in a case of genetic frontotemporal dementia—clinical features and neuroimaging correlates
Journal of Neurology (2023)
