
Abstract
Trained immunity is a functional state of the innate immune response and is characterized by long-term epigenetic reprogramming of innate immune cells. This concept originated in the field of infectious diseases — training of innate immune cells, such as monocytes, macrophages and/or natural killer cells, by infection or vaccination enhances immune responses against microbial pathogens after restimulation. Although initially reported in circulating monocytes and tissue macrophages (termed peripheral trained immunity), subsequent findings indicate that immune progenitor cells in the bone marrow can also be trained (that is, central trained immunity), which explains the long-term innate immunity-mediated protective effects of vaccination against heterologous infections. Although trained immunity is beneficial against infections, its inappropriate induction by endogenous stimuli can also lead to aberrant inflammation. For example, in systemic lupus erythematosus and systemic sclerosis, trained immunity might contribute to inflammatory activity, which promotes disease progression. In organ transplantation, trained immunity has been associated with acute rejection and suppression of trained immunity prolonged allograft survival. This novel concept provides a better understanding of the involvement of the innate immune response in different pathological conditions, and provides a new framework for the development of therapies and treatment strategies that target epigenetic and metabolic pathways of the innate immune system.
Key points
-
Trained immunity is a functional state of the innate immune system that is characterized by long-term epigenetic and metabolic reprogramming of cells associated with potent immune responses.
-
Experimental and clinical studies have demonstrated that exogenous pathogen-associated molecular patterns and endogenous danger-associated molecular patterns induce trained immunity.
-
Trained immunity is a functional adaptation of the innate immune system against secondary infections, but can lead to aberrant inflammatory activity in conditions such as autoimmunity.
-
Sterile inflammation owing to ischaemia–reperfusion injury and organ transplantation induces trained immunity and precipitates allograft rejection.
-
Therapeutic inhibition of trained immunity (for example, in autoimmunity or transplantation) or its induction (for example, in infections or cancer) are promising strategies for treating immunity-related diseases.
Similar content being viewed by others
Introduction
Innate immune defence against infection is largely based on the detection of molecular patterns that are present in infectious agents, such as bacteria, fungi or viruses, and are recognized as danger signals. Recognition of these pathogen signals leads to the activation of different types of immune cells, including T and B lymphocytes, natural killer (NK) cells, monocytes, macrophages, neutrophils and dendritic cells (DCs)1. This multicellular response to infection is coordinated into two distinct phases: the innate and the adaptive immune response. The innate immune response represents the first phase of the immune response and is mediated by physical, chemical and cellular defences in distinct types of myeloid cells (such as monocytes, macrophages and DCs) or lymphoid cells (NK cells and innate lymphoid cells). The second phase of the immune response is mediated by the adaptive response, which is mediated by T and B lymphocytes2.
The innate immune response is evolutionarily conserved and has traditionally been associated with a rapid and non-specific inflammatory response. Innate immune cells recognize pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) expressed on their surface and cytoplasm3. This ancient mechanism of immunological defence can also be triggered by self-derived damage-associated molecular patterns (DAMPs) released in the context of sterile inflammation4. Activation of PRRs in innate immune cells induced by PAMPs and DAMPs rapidly induces the secretion of pro-inflammatory cytokines, such as IL-6, IL-1β and tumour necrosis factor (TNF)5. These pro-inflammatory cytokines subsequently stimulate the generation and presentation of antigenic epitopes that activate the adaptive immune response6. Notably, the adaptive immune response is only present in jawed vertebrates and, although slower than the innate response, generates an antigen-specific response after an initial encounter with a pathogen. This high antigenic specificity is enabled by the recombination of variable (V), diversity (D) and joining (J) genes segments during the T and B cell receptor rearrangement phase of T and B cell development, which results in the generation of a T cell and B cell receptor repertoire with an immense breadth of specificities. Following an encounter with a pathogen, the lymphocytes that can recognize antigens present on that pathogen expand and some of those cells differentiate into long-lived memory T and B cells that can be readily reactivated upon subsequent encounters with the same pathogen7.
The absence of immunological memory historically distinguished innate from adaptive immunity but this concept is now challenged. Although memory T and B lymphocytes undoubtedly provide long-term protection against reinfection, higher vertebrates represent only 5% of all animal species on Earth, including mammals, reptiles, birds, fish and amphibians8. The absence of T and B lymphocytes in the remaining 95% of organisms does not necessarily mean the absence of immune memory. Non-vertebrates have evolved to respond to pathogenic infectious agents and in 2003 one study showed that copepods, which are minute crustaceans that rely solely on an innate immune system, had immunological memory and could prevent reinfection with a parasitic tapeworm9. This innate immune memory phenomenon was subsequently observed in other invertebrate and vertebrate species, including humans, indicating that it is ubiquitous and evolutionarily conserved10.
The term ‘trained immunity’ was first introduced in 2011 and describes the immunological memory response that innate immune cells can mount in response to past insults11. Although cells return to an inactivated state after an initial stimulus, epigenetic changes enable a faster and stronger response upon antigen re-encounter12. PAMPs and DAMPs are classic inducers of such training — they can generate trained immunity in the circulation and tissues (that is, peripheral trained immunity), and in HPSCs in the bone marrow (that is, central trained immunity)13. Importantly, although trained immunity has evolved under continuous evolutionary pressure to provide enhanced protection against pathogens, maladaptive innate immune responses might contribute to chronic inflammatory diseases and autoimmunity14.
In this Review, we examine the concept of trained immunity and describe technical approaches that are currently available to study its underlying molecular mechanisms. Furthermore, we explore the implications of trained immunity in the field of nephrology and discuss potential therapeutic approaches for the manipulation of trained immunity in the context of kidney diseases.
Basic concepts of trained immunity
Trained immunity describes the immunological process by which innate immune cells acquire immunological memory. After exposure to certain stimuli, innate immune cells can adjust their response to subsequent insults, resulting in an enhanced response to previously encountered infectious agents11. This challenging concept was fully demonstrated in 2012, when non-specific protective effects of the Bacillus Calmette–Guérin (BCG) vaccine were reported15. In severe combined immunodeficient mice, which lack T and B lymphocytes and therefore cannot mount an adaptive immune response, BCG vaccination against Mycobacterium tuberculosis conferred cross-protection and reduced mortality caused by a lethal Candida albicans infection. In addition to pathogenic stimuli, self-derived molecules, such as DAMPs (for example, vimentin) and cytokines (for example, granulocyte–macrophage colony-stimulating factor), can also induce trained immunity16. Mechanistically, PAMPs and DAMPs induce trained immunity by eliciting long-term epigenetic reprogramming at the promoters of inflammatory genes and causing metabolic rewiring in memory macrophages15.
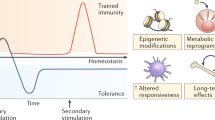
Importantly, monocyte activation, priming and training refer to different processes12 (Fig. 1). Resting monocyte and macrophages can be activated through Toll-like receptor (TLR) signalling, which induces metabolic rewiring and epigenetic changes that are associated with the production of inflammatory cytokines; these changes are transient and dynamic over the course of an inflammatory event17. Of note, macrophages might be re-stimulated before the gene transcription induced by the initial stimulus subsides, which will enhance their inflammatory response; these pre-stimulated cells are considered to be primed. For example, priming studies in vivo demonstrated that mice co-infected with Legionella pneumophila 3 days after influenza virus infection results had significantly higher broncho-alveolar lavage fluid concentrations of IL-1β than single-infected mice18. However, all mice infected with influenza virus and then co-infected with L. pneumophila died within 1 week of priming; signs of morbidity included decreased body weight and hypothermia, despite ongoing inflammation. In contrast to innate immune priming, the induction of trained immunity contains a component of immune memory. In models of trained immunity, the initial gene transcription induced by the primary stimulus ceases before the secondary stimulation and the cells are thus considered to have returned to homeostasis. If restimulation of these innate immune cells at a later time point results in enhanced responsiveness compared with the initial stimulation, the cells are considered to have undergone trained immunity. Training studies in vivo demonstrated that 1 month after influenza virus infection (that is, following elimination of the virus), infection with S. pneumoniae resulted in increased production of IL-6 by monocyte-derived alveolar macrophages compared with animals infected with S. pneumoniae but without a prior influenza challenge19. This enhanced cytokine production was associated with epigenetic changes and conferred a survival advantage, given that IL-6 deficiency abrogated protection. Therefore, although primed monocytes display increased gene expression upon secondary stimulation, trained monocytes maintain long-term epigenetic changes at specific gene promoters after the immune activation status returns to baseline. These epigenetic changes are associated with immune protection after restimulation.

a | Activation of naive macrophages by a primary stimulus is accompanied by histone modifications that lead to an enrichment of open-chromatin in regions containing inflammatory genes and their regulatory elements, which facilitates the transcription of cytokines. Once the activating stimulus is removed, activated macrophages return to a naive epigenetic state characterized by closed chromatin without histone marks. b | However, if restimulation occurs before the gene transcription events induced by the initial stimulus subside, the inflammatory response of these primed cells is enhanced and this effect is also associated with histone modifications. c | By contrast, if the primary stimulus results in persistent epigenetic changes, even after the cell has returned to baseline, and restimulation of that innate immune cell results in an enhanced cytokine response, the cell is considered to have undergone trained immunity. d | Innate immune tolerance is another potential outcome following a primary stimulus. These macrophages might display functional defects associated with non-permanent epigenetic changes for weeks after resolution of the primary challenge, which result in loss of function following restimulation.
Crucially, the opposite of priming and trained immunity can also occur, resulting in blunted innate immune function (Fig. 1). This process, which is termed innate immune tolerance, might be associated with loss of function after restimulation. Supporting this concept, monocytes and macrophages display functional defects for weeks after the resolution of sepsis, independently of metabolic exhaustion or unresponsiveness to endotoxin20. Innate immune tolerance studies in vivo showed that histone H3 Lys27 acetylation (H3K27ac) peak heights correlated significantly with functional paralysis in alveolar macrophages weeks after the resolution of primary E. coli-mediated pneumonia, indicating that this type of pneumonia elicits tolerogenic training of alveolar macrophages21. Immune tolerance data from animal models is consistent with the severe metabolic defects observed in patients with sepsis, which are characterized by multiple deficiencies in glycolytic and oxidative phosphorylation pathways that lead to immunometabolic paralysis20. Interestingly, induction of trained immunity can reverse innate immune tolerance. One study showed that LPS-exposed monocytes in vitro treated with the C. albicans cell wall component β-glucan retain the ability to produce pro-inflammatory cytokines after LPS re-exposure compared with non-β-glucan-treated cells22. In addition, this study demonstrated that ex vivo β-glucan also increases the release of TNF and IL-6 in monocytes from healthy volunteers injected with LPS before LPS re-exposure in vitro, which suggests that it might be possible to modulate immune responses through the regulation of epigenetic histone reprogramming at specific genes and of metabolic rewiring.
Epigenetic reprogramming
Epigenetic control of histone modifications leading to long-term opening of chromatin is the basis of trained immunity17. For example, following a primary stimulus, certain epigenetic modifications enable faster expression of relevant effector genes in response to a second stimulus. Specifically, non-permanent histone modifications associated with gene activation, including H3K4 monomethylation (H3K4me) and trimethylation (H3K4me3), and H3K27ac have been observed in trained macrophages23. These epigenetic marks, which can be detected through several approaches (Fig. 2 and Box 1), result in the opening of chromatin at promoters of genes encoding pro-inflammatory cytokines such as IL-6, IL-1β and TNF and are associated with protection against re-infection24.

Different methodologies can be used to assess trained immunity in innate immune cells. Flow cytometry can be used to quantify the production of pro-inflammatory cytokines167, such as tumour necrosis factor (TNF) and IL-6, in fixed and permeabilized innate immune cells after restimulation with lipopolysaccharide (LPS). Several studies have also used ELISA to measure the pro-inflammatory cytokine response (IL-6, TNF and IL-1β) of trained monocytes in vitro and in vivo168,169. In vitro, trained innate immune cells have higher basal and maximal mitochondrial activity, which can be assessed using colorimetric or fluorometric assays170 to provide insights into trained immunity. For example, cultured trained innate immune cells produce protons via the lactate pathway and the acidification of the culture medium can be assessed by measuring the extracellular acidification rate (ECAR) as an indicator of glycolysis, whereas the oxygen consumption rate (OCR) can be used as a measure of oxidative phosphorylation171,172. Seahorse XF analysers can be used to obtain an in-depth analysis of a variety of mitochondrial functions in single cells in vitro with relatively high throughput through the use of compounds that perturb the cellular bioenergetic profile173. Specifically, the sequential addition of oligomycin (ATP synthase inhibitor), FCCP (mitochondrial uncoupler), and a combination of the complex I inhibitor (rotenone) and complex III inhibitor (antimycin A) provides information on three key parameters of mitochondrial activation — ATP turnover, proton leak and maximal respiration174,175. Finally, induction of trained immunity can also be investigated by evaluating chromatin remodelling, using tools such as chromatin immunoprecipitation (ChIP), assay for transposase-accessible chromatin using sequencing (ATAC-seq), cleavage under targets and release using nuclease (CUT&RUN) and cleavage under targets and tagmentation (CUT&Tag).
Metabolic rewiring
Under steady-state conditions, immune cells have low biosynthetic activity and their energy requirements are predominantly met through oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO). Upon activation, the energy demand of innate immune cells increases and aerobic glycolysis, glutaminolysis, cholesterol metabolism and fatty acid synthesis can be used to meet those additional needs. Metabolic intermediates, such as acetyl-CoA, fumarate, succinate, nicotinamide adenine dinucleotide (NAD+) and mevalonate, which are produced as a result of this activation-induced metabolic rewiring, regulate the epigenetic landscape25. Epigenetic rewiring is associated with a metabolic shift in trained macrophages, which includes an increase in aerobic glycolysis compared with untrained macrophages that is dependent on the activation of the mammalian target of rapamycin (mTOR) via the dectin-1–AKT–hypoxia-inducible factor 1α (HIF-1α) pathway26. mTOR detects cellular nutrient, oxygen and energy levels via various upstream inputs and then activates transcriptional regulators (for example, HIF1α, transcriptional repressor protein YY1 and peroxisome proliferator-activated receptor-γ coactivator 1-α (PCG1α)) that stimulate glycolysis and mitochondrial oxidative metabolism27. Inhibition of the mTOR pathway or glycolysis with rapamycin, 2-deoxyglucose (2-DG) or metformin during training interferes with pro-inflammatory cytokine production in macrophages and results in loss of protection against infection. The upregulation of glycolysis in trained monocytes is associated with both mitochondrial respiration and accumulation of lactate13. Of note, in BCG-induced trained macrophages, the basal and maximal extracellular acidification rate, which is an indicator of glycolysis, and the oxygen consumption rate, which reflects oxidative phosphorylation, increased before LPS re-stimulation compared with untrained macrophages. These findings indicate upregulation of both aerobic glycolysis and oxidative phosphorylation in trained macrophages28 (Fig. 2).
Long-lasting effects of trained immunity
Evidence for the long-lasting protective effects of the BCG vaccine that are mediated by innate immune cells support the existence of long-term trained immunity, which has been investigated in longitudinal population-based cohort studies29,30. The long-lasting effect of trained immunity was also observed in a randomized controlled trial in which older patients hospitalized for a new infection were randomly assigned to receive the BCG vaccine or placebo31. These patients were followed for 1 year after discharge to determine the rate of reinfections. Interestingly, the rate of infections was 79% lower in the BCG group, primarily owing to fewer viral pulmonary infections. The role of trained immunity in this protection is supported by epigenetic analyses that revealed an enrichment of H3K4me3 at the promoters for IL6 and TNF compared with the placebo group, which was associated with pro-inflammatory cytokine secretion in macrophages. These analyses also demonstrated that BCG had a cross-protective effect against C. albicans and Staphylococcus aureus infections for at least 3 months after vaccination. A 6-month follow-up phase III placebo-controlled randomized clinical trial of BCG vaccination against SARS-CoV-2 demonstrated a 68% relative reduction in the risk of developing COVID-19 in older individuals 6 months after vaccination compared with those in the placebo group32. Another study demonstrated that BCG induces an increase in the peripheral blood mononuclear cell production of TNF and IL-1β after LPS stimulation in vitro 1 year after vaccination compared with pre-vaccination levels, which supports the sustained training effects of BCG vaccination33.
The longevity of trained immunity can be explained by its effects in the bone marrow. One study showed that intravenous BCG injection induced the expansion of HSPCs and promoted their differentiation towards myelopoiesis13. Moreover, macrophages derived from the bone marrow of BCG-vaccinated mice (collected 1 or 5 months post-infection) were more resistant to M. tuberculosis in vitro than those derived from unvaccinated mice; this difference was associated with distinct gene expression signatures. Importantly, vaccination induced epigenetic modifications in myeloid precursor cells and transfer of bone marrow from vaccinated to naive mice enhanced the protection provided by myeloid cells against pulmonary infection by M. tuberculosis13. These data suggest that epigenetic modifications induced on bone marrow myeloid precursors are retained throughout development and are maintained once the cells migrate out of the bone marrow and infiltrate inflamed tissue in response to certain infections. Supporting this hypothesis, another study demonstrated that BCG leads to imprinting of persistent transcriptomic signatures on human HSPCs that skew them towards myeloid development. These changes in transcription were associated with specific epigenetic modifications and both alterations could still be detected in peripheral monocytes 3 months after vaccination34. In addition to BCG, other stimuli can induce trained immunity, including β-glucan, which induces trained immunity via its receptor, C-type lectin domain family 7 member A (also known as dectin-1). β-glucan-mediated induction of trained immunity occurs via modulation of HSPCs in the bone marrow. Specifically, β-glucan enhanced myelopoiesis by increasing the numbers and frequency of haematopoietic progenitors, such as Lin−Sca1+c-Kit+ (LSK) cells, and inducing metabolic and transcriptomic alterations associated with trained immunity35. For example, β-glucan binding to dectin-1 activated the mTOR pathway and calcium-dependent nuclear factor of activated T cells (NF-AT) signalling, resulting in epigenetic reprogramming of immune gene promoters25,36. These alterations were protective in mice challenged with a lethal secondary cytotoxic stress (that is, lethal C. albicans infection). β-glucan-induced trained immunity can also reprogram granulopoiesis and neutrophil development through transcriptomic and epigenetic modulation to promote an antitumour phenotype37. This protective effect against tumour development was dependent on reactive oxygen species (ROS) and type I interferon signalling, and could still be observed in mice lacking B and T cells. Of note, although initially described in monocytes and macrophages, trained immunity can also be induced in neutrophils38, NK cells39, innate lymphoid cells40 and non-immune cells41, including endothelial cells42, epithelial cells43 and haematopoietic precursors13. For example, BCG vaccination of healthy humans also induced long-lasting enhancement of neutrophil function, which was associated with genome-wide epigenetic modifications in H3K4me3 in neutrophils38. Here, we focus mainly on the potential role of trained monocytes and macrophages (Box 2).
Trained immunity and immunopathology
Many kidney diseases are associated with immune pathological conditions — several immunological disorders (for example, anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis or systemic lupus erythematosus (SLE)) can cause damage to the kidney and lead to chronic kidney disease (CKD). Moreover, patients with CKD often have impaired immunity owing to the effects of uraemic toxicity, as well as treatment-associated immune alterations, especially in patients with kidney failure who require kidney replacement therapy (KRT) such as dialysis or kidney transplantation. These immune alterations associated with kidney disease and KRT include chronic inflammation and premature immune ageing, and patients with advanced CKD are more susceptible to infections, have a diminished response to vaccination and display profound innate immune system alteration compared with the general population44. Below, we discuss the molecular regulation of trained immunity in the context of pathological conditions that involve the kidney.
Autoimmune diseases
Several systemic autoimmune diseases, such as SLE, are complicated by kidney involvement. Although SLE can affect any organ of the body, the kidneys are involved in ~50% of patients45. The pathogenesis of SLE is not fully understood and the mechanisms underlying the observed loss of self-tolerance in this disease are unclear. SLE is characterized by an imbalance between the induction of apoptosis and the removal of apoptotic cells. Apoptotic material can originate from any cell type, including neutrophils, of which approximately one billion undergo apoptosis every day46. Moreover, neutrophils in patients with SLE have an increased propensity to form neutrophil extracellular traps46. Apoptotic cellular debris and neutrophil extracellular traps contain nucleic antigens that can trigger an inflammatory response by activating cytosolic nucleic acid sensors and TLRs in innate immune cells. Their activation induces type I interferon production, which can stimulate the adaptive immune system, and therefore has the potential to activate self-reactive T and B cells45,46. Engagement of self-reactive lymphocytes drives the production of autoantibodies against nuclear components, such as double-stranded DNA, RNA, histones and small nuclear ribonucleoproteins. Binding of nuclear antigens to the subendothelial space of the glomerular capillaries might lead to the formation and deposition of immune complexes in the kidneys, leading to the activation of complement and recruitment of leukocytes that characterize lupus nephritis45.
Historically, SLE research has focused largely on the adaptive immune system. However, monocytes and macrophages are increasingly recognized to have a key role in this disease47. For example, phagocytosis of apoptotic cells is reduced in macrophages from patients with SLE compared with healthy individuals, and their ability to clear immune complexes is also impaired48,49; these macrophages also show signs of enhanced activation50. Of note, circulating cytokine levels, including IL-6, TNF and IL-1β, are higher in patients with SLE compared with healthy individuals and blood levels of these cytokines correlate positively with disease activity and autoantibody levels51,52. In lupus nephritis, macrophage infiltration is prognostic for disease progression53.
Several studies have begun to elucidate the molecular mechanisms that underlie the increased activation of macrophages in SLE, including immunometabolic and epigenetic reprogramming. In a mouse model of lupus nephritis (MRL-lpr), kidney myeloid cells were isolated for single-cell RNA sequencing, which revealed a marked enrichment of glycolysis-related gene expression in kidney-derived macrophages54. The study demonstrated, through transcriptional and metabolic assays, that Fc-gamma receptor (FcyR) cross-linking induces a switch to aerobic glycolysis in macrophages that is regulated through mTOR–HIF1α signalling. This work also demonstrated that metabolic reprogramming is required to induce IL-1β production, and that inhibition of this glycolytic switch with 2-Deoxy-d-glucose (2-DG) suppresses IL-1β production in primary human kidney macrophages54. In addition, in MRL-lpr mice infused with IgG immune complexes, treatment with 2-DG decreased expression of IL-6, TNF and IL-1β in kidney tissue and decreased kidney neutrophil infiltration, demonstrating that these immunometabolic circuits, which are known to be involved in trained immunity, participate in SLE54.
Other studies focused on the epigenome of monocytes and macrophages, and found substantial alterations in histone acetylation and methylation in the context of SLE. H4 acetylation, which increases chromatin accessibility for gene transcription, was increased in the TNF locus in circulating monocytes of patients with SLE compared with healthy individuals55. Whole-genome chromatin immunoprecipitation showed that H4 acetylation is higher overall in patients with SLE than in controls, and pathway analyses revealed that H4 acetylation was enriched at the promoter regions of genes that drive inflammatory pathways56,57. Interestingly, >60% of genes with increased H4 acetylation were potentially regulated by the interferon regulatory factor 1 (IRF1) transcription factor58. Subsequent studies demonstrated that IRF1 interacts directly with histone acetyltransferases, which promotes H4 acetylation56,59; overexpression of IRF1 results in H4 hyperacetylation. Of note, innate immune memory could be induced in macrophages through exposure to IFNγ, which induced changes in the histone chromatin marks of interferon-stimulated genes60.
Although the study of trained immunity in autoimmune diseases is currently largely unexplored, the involvement of training pathways suggests a potential role for trained immunity in SLE (Fig. 3). Further functional, metabolic and epigenetic studies on myeloid cells in different anatomical sites might provide additional mechanistic insights into the potential role of trained immunity in SLE. For example, one study investigated the transcriptional regulation of bone marrow progenitors in a murine SLE model (NZBW/F1) and in patients with SLE, and demonstrated alterations in haematopoiesis, with a skewing towards myelopoiesis in both mice and humans61. Interestingly, these changes were associated with a ‘training’ LSK signature, consistent with data on the effects of β-glucan training on LSK progenitors and granulopoiesis35,37. Induction of trained immunity in the bone marrow of patients with SLE might therefore promote the development of pro-inflammatory granulocytes that home to peripheral tissues, including the kidneys62,63. Future studies are needed to confirm the presence of histone modifications in the bone marrow progenitors and circulating monocytes of patients to confirm this hypothesis. Furthermore, longitudinal studies are needed to assess peripheral and central trained immunity at different stages of the disease, and to investigate how trained immunity might relate to disease outcomes. Interestingly, therapeutic interventions that target metabolic and epigenetic processes can ameliorate SLE and inhibition of trained immunity might underlie some of this protection54,64. For example, a 2020 study reported that hydroxychloroquine, which is commonly used to prevent SLE flares, inhibited trained immunity by suppressing H3K27ac and H3K4me3 of inflammation-related genes65. Trained immunity was also reported to modulate inflammation-induced fibrosis in systemic sclerosis, suggesting that it might be involved in a broad range of systemic autoimmune diseases66.

Nucleic antigens from apoptotic cells and neutrophil extracellular traps (NETs) induce inflammation and the production of type I interferon (IFN-I), IL-1β, IL-6 and tumour necrosis factor (TNF). IFN-I and IL-1β cause metabolic and epigenetic reprogramming of myeloid and granulopoietic progenitors in the bone marrow. This reprogramming creates trained monocytes and neutrophils with an enhanced inflammatory phenotype and increased capacity for cytokine production after stimulation by nucleic antigens, autoantibodies and inflammatory cytokines. This overproduction of cytokines also stimulates the adaptive immune system, contributing to the activation of autoreactive T and B cells, and the formation of anti-nuclear antibodies. Collectively, these processes generate an inflammatory cycle that leads to tissue destruction. CMP, common myeloid progenitor; HSC, haematopoietic stem cell.
CKD and dialysis
The uraemic state in patients with CKD, including those receiving dialysis, can have a profound effect on the immune system67. This CKD-associated immune dysregulation, which also compromises responses to vaccination68, was highlighted by the COVID-19 pandemic69 — patients with CKD and organ transplant recipients were at one of the highest risks of SARS-CoV-2-associated morbidity and mortality70,71. In addition, patients with advanced CKD are more susceptible to other immunity-mediated diseases, including virus-associated cancers, periodontitis and atherosclerosis, compared with the general population72,73,74,75.
The exact mechanisms underlying immune dysregulation in patients with kidney disease are not completely defined, but oxidative stress induced by the retention of uraemic toxins and decreased clearance of inflammatory cytokines seem to have an important role76. Oxidative stress results in the production of advanced glycation end-products (AGEs), which are recognized as DAMPs through the receptor for AGE (RAGE) and can trigger innate immune activation. The oxidation of low-density lipoprotein (oxLDL) is another well-described consequence of oxidative stress, and patients with CKD have significantly higher serum levels of oxLDL than healthy individuals77,78. Accumulation of oxLDL in the intima of arterial walls leads to aberrant inflammation owing to inflammasome activation in innate immune cells. These processes eventually lead to plaque rupture, which manifests as acute myocardial infarction or stroke79. In addition, oxLDL promotes the production of pro-inflammatory myeloid cells in the bone marrow, contributing to a systemic pro-inflammatory state80. Patients with advanced CKD can also have elevated serum uric acid levels, which not only induce acute inflammation in the context of gout, but are also associated with chronic systemic inflammation81.
CKD is associated with increased production of CD14++CD16+ pro-inflammatory monocytes in the bone marrow82. This activated profile is accompanied by elevated circulating levels of pro-inflammatory cytokines, such as IL-6, IL-1β and TNF83. Interestingly, this enhanced inflammatory activity observed in CKD is retained after patients receive a kidney transplant and uraemia is resolved84,85. Although not investigated as such, this sustained propensity for increased production of pro-inflammatory cytokines could be mediated by trained immunity. Notably, uraemia is associated with epigenetic changes, such as changes in DNA methylation, in leukocytes86. Moreover, maladaptive training of myelopoiesis underlies the development of inflammatory comorbidities87, which might have a role in the strong susceptibility of patients with advanced kidney disease to other immune-mediated pathological conditions, including periodontitis and atherosclerosis.
Although the concept of trained immunity has not been fully investigated in the context of CKD and dialysis, several studies suggest that it might have a direct role in the immune alterations observed in affected patients. For example, monocytes stimulated with oxLDL have an enhanced capacity for production of IL-6 and TNF in response to TLR 2 and 4 agonists, compared with untreated cells. This effect is accompanied by upregulation of H3K4me3 on the promoters of the genes encoding these proteins and was completely abolished when monocytes were pre-incubated with the methyltransferase inhibitor methylthioadenosine88. Uric acid-stimulated monocytes also produced higher levels of IL-1 and IL-6 than untreated cells upon stimulation with TLR 2 and 4 agonists; the enhanced capacity for cytokine production could be reversed by pharmacological inhibition of histone methyl transferases, suggesting a trained immunity process89. Consequently, therapies that inhibit trained immunity through histone methyltransferase inhibition, have been proposed to prevent chronic innate immune activation in hyperuricaemia90, which is common in patients with CKD91. Future research will need to explore the role of trained immunity in patients with advanced CKD further, including its contribution to inflammatory comorbidities and therapeutic targeting potential92.
Organ transplantation
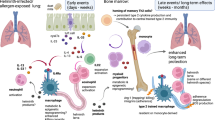
Several factors that contribute to allograft rejection, including ischaemia–reperfusion injury (IRI) and the presence of infection, involve immune mechanisms that have been implicated in trained immunity (Fig. 4).

a | Events that activate innate immunity in organ transplantation include ischaemia–reperfusion injury (IRI) and the release of damage-associated molecular patters (DAMPs), such as high-mobility group box 1 (HMGB1) or advanced glycation end products (AGEs), which are produced under conditions of oxidative stress and the presence of viral, bacterial and fungal infections. In the context of infection, microbial pathogen-associated molecular patterns (PAMPs) can reprogram macrophages into pro-inflammatory cells that respond to and can kill a broad range of infectious pathogens, and offer protection to susceptible transplant recipients. However, these trained macrophages produce high levels of pro-inflammatory cytokines that can also amplify inflammation and lead to acute rejection. Innate immune cells can also develop a memory response to specific alloantigens, including CD47, which is recognized by signal-regulatory protein-α (SIRPα), and MHC class I (MHC-I) detected via Ig-like receptor A (PIR-A). b | Early after transplantation, DAMPs are released from the donor kidney into the circulation (1). Trained immunity-inducing DAMPs reach the bone marrow and reprogram innate immune cell precursors (2). Reprogrammed innate immune cells migrate to the graft where they encounter additional training signals (3). Once in the transplanted organ, reprogrammed innate immune cells interact with adaptive immune cells and contribute to organ rejection (4). β2M, β2-microglobulin; PRR, pattern-recognition receptor; TLR4, Toll-like receptor 4; TSP1, thrombospondin.
Ischaemia–reperfusion injury
In transplantation, the donor kidney is subjected to ischaemia during organ retrieval, which leads to pathophysiological events such as hypoxia and tissue injury owing to limited blood supply. Following transplantation, the return of the blood supply to the transplanted organ induces reperfusion injury, which is mediated by the production of ROS, alterations in intracellular calcium, endothelial cell dysfunction, complement activation and cell death93. Multiple stimuli generated during this process of IRI can activate the innate immune system and trigger potent inflammatory responses94. IRI-derived DAMPs released into the extracellular space, such as mitochondrial ATP, which is released from damaged cells, induce metabolic changes in innate immune cells95,96. Extracellular ATP activates the macrophage cell surface purinergic receptor P2X7 and induces the production of pro-inflammatory cytokines, such as TNF and IL-1β, which are associated with allograft rejection97,98. Interestingly, P2X7 blockade abrogated the T helper 1 (TH1) and TH17 T cell immune response, thereby reducing the number of effector T cells, and promoted long-term transplant survival in mice99. Thrombospondin (TSP1), which is a glycoprotein that regulates nitric oxide and has an important role in cellular metabolism100, is also rapidly upregulated during IRI and mediates acute kidney injury101. TSP1 binds to TLR4 and triggers multiple pathways of inflammation, including the production of IL-6, TNF and IL-1β in monocytes and macrophages102,103,104. Of note, oxidative stress-triggered IL-6 production in mouse myeloid cells enhanced their capacity to activate allogeneic T cells in a mixed lymphocyte reaction105. Moreover, targeting IL-6 reduced inflammation following myocardial ischaemia–reperfusion106,107, and early blockade of IL-6 in the cardiac post-transplant setting reduces infiltration of adaptive allogeneic leukocytes and extends graft survival in mice108. Overall, these data indicate that IRI, which is associated with poor graft survival109, upregulates innate immune pathways that are known to be associated with trained immune memory110. Preconditioning and postconditioning of donor organs through pharmaceuticals and novel strategies such as RNA interference favours innate immune tolerance94. In addition to kidney transplantation, IRI also occurs in thrombotic diseases, sepsis and trauma, all of which can affect the kidney111. Consequently, the potential role of trained immunity in these IRI outcomes has broad therapeutic implications.
Innate allorecognition
Although kidney transplantation is a life-saving procedure for patients with kidney failure, it requires the lifelong use of immunosuppressive drugs. These therapies currently used in the clinic to prevent allograft rejection primarily target the adaptive immune system because B and T lymphocytes are directly responsible for antibody- and cell-mediated immunity against the graft112. However, increasing evidence indicates that innate immune cells also have an important role113. Of note, the long-term viability of kidney transplants remains suboptimal despite a notable improvement in high-risk groups114 and targeting the innate immune system could act synergistically with immunosuppressive drugs that target adaptive immunity to improve patient outcomes.
The surgical procedure required for kidney transplantation inevitably causes vascular tissue damage, which promotes the recruitment and extravasation of inflammatory monocytes into the allograft115. Once in the transplanted kidney, circulating monocytes can differentiate into different subsets of macrophages following recognition of DAMPs and non-self alloantigens, and their phenotype is also influenced by the systemic presence of immunosuppressive drugs. One study reported that the innate alloresponse worsened if recipient mice had been previously primed with donor cells116. After initial priming of lymphocyte deficient (RAG−/−) mice with allogeneic splenocytes or skin grafts, monocytes from the recipient mice identify and eliminate allogeneic spleen cells injected subcutaneously 4 weeks later116, demonstrating that monocytes and macrophages develop a memory response against alloantigens. Further evaluation of the mechanisms that mediate non-self-recognition by monocytes demonstrated that polymorphic differences in signal-regulatory protein-α (SIRPα) between donor and recipient mice, which were recognized by CD47 expressed on host monocytes, drives activation of the innate immune response against the allograft117. Moreover, a subsequent study found that paired Ig-like receptor A (PIR-A) expressed in recipient innate immune cells recognizes donor allogeneic MHC class I antigens, which leads to the development of innate immune memory to foreign tissues118.
We also found evidence that innate immune memory is involved in rejection in a vascularized heart transplant model in mice119. When trained immunity was inhibited with myeloid cell-specific mTOR-inhibiting nanobiologics, chromatin immunoprecipitation (ChIP) of allograft monocytes showed reduced H3K4me3 of inflammation-related genes, including TNF and IL-6, which was associated with reduced secretion of these cytokines. In this setting, vimentin and high-mobility group box 1 (HMGB1) were the inflammatory mediators that induced trained macrophages in the transplanted heart. Vimentin is an endogenous intermediate filament protein that activates the dectin-1 receptor, which is highly expressed on endothelial cells, whereas extracellular HMGB1 is a DAMP that binds to TLR4 and induces a robust TNF-mediated immune response in vivo120,121. Both of these inflammatory mediators are upregulated in organ transplantation122,123 and they both bind cell surface receptors known to induce trained immunity (that is, dectin-1 and TLR4) — we confirmed that sequential treatment with vimentin and HMGB1 induces trained immunity in the context of organ transplantation119. Importantly, we found that inhibition of trained immunity using a myeloid cell-specific mTOR-inhibiting nanobiologic promoted a tolerogenic milieu by enhancing the number of regulatory T cells in the allograft, and induced indefinite graft survival without the need for continuous immunosuppression. In another study of cardiac allografts in mice123, both allogeneic and syngeneic transplantation induced upregulation of HMGB1. However, whereas this upregulation decreased in syngeneic grafts over the first week post-transplant, high HMGB1 levels were sustained in allogeneic grafts, which was accompanied by interstitial infiltration and acute allograft rejection. Overall, these findings suggest that induction of trained innate immunity, through activation of PRRs during ischaemia–reperfusion injury, contributes to allograft rejection and indicates that the endogenous DAMPs that induce innate immune memory affect graft survival.
Infection
Organ transplant recipients are more susceptible to infections than the general population owing to their lifelong immunosuppressive treatment124. In these patients, macrophages can contribute to acute and chronic allograft immunopathology via antigen processing and presentation, co-stimulation, pro-inflammatory cytokine production and tissue injury125,126. However, these innate immune cells also have important protective roles in the context of infection, including the intracellular killing of pathogens. Importantly, infection can lead to kidney transplant complications and graft loss owing to the production of inflammatory cytokines by innate immune cells. For example, bacterial infection with Staphylococcus aureus at the time of skin transplantation prevents the induction of organ transplant acceptance in mice because of excessive IL-6 production127. Therefore, although macrophages can induce potent IL-6 cytokine responses that confer protection against S. aureus infection128, this response represents a potential risk in the context of kidney transplantation because an excessive inflammatory immune response can lead to graft loss. Notably, immunosuppressive therapy with either cyclosporine or sirolimus could not prevent graft loss driven by the response to S. aureus infection in mice127, which suggests that common immunosuppressive drugs fail to regulate inflammatory cytokine production by macrophages in response to a bacterial infection. Similar results were obtained using the bacteria Listeria monocytogenes, which prevented transplantation tolerance in mice despite therapeutic treatment with co-stimulation blockade129,130. These findings are consistent with data indicating that IL-6 produced by macrophages mediates costimulatory blockade-resistant graft rejection in murine models131. Given that trained macrophages produce high levels of IL-6 following re-stimulation, these studies suggest that macrophages trained by bacterial infections might represent a risk to kidney transplanted patients.
Viral infections have a similar effect on macrophage-mediated inflammatory cytokine production. Although latent cytomegalovirus (CMV) infection is present in the majority of the general immunocompetent population, CMV is an important cause of morbidity and is mainly observed in immunosuppressed hosts, including kidney transplant recipients132. CMV induces expression of nucleotide-binding oligomerization domain-containing protein 2 (NOD2), which had been previously shown to mediate epigenetic reprogramming of innate immune cells and trained immunity15,133. CMV is present in the myeloid lineage of mice with a latent infection134 and induces the production of pro-inflammatory IL-6 in CMV-infected macrophages135. Of note, IL-6 induces reactivation of CMV-infected monocytes that produce CMV virions at higher rates than monocytes treated with other cytokines136. Accordingly, circulating CMV-infected monocytes can extravasate to tissues and significantly increase their production of IL-6 upon restimulation with bacterial products, such as LPS137. Unbiased bulk and single-cell transcriptomics paired with functional assays (fungal killing) of cells from patients infected with CMV demonstrated that human CMV-infected monocytes do not effectively phagocytose opportunistic fungal pathogens, and that this functional impairment occurs in the context of decreased expression of fungal PRRs138. Moreover, CMV-infected monocytes upregulate the expression of phagocytic receptors, pro-inflammatory chemokines, activate the inflammasome and induce the expression of innate immune transcripts associated with allograft rejection138. Notably, latent infection with CMV prevents the induction of prolonged allograft survival following transplantation and therefore represents a major barrier in kidney transplantation139. Considering that latent CMV is prevalent worldwide — estimated seroprevalence of 83% in the general population140 — and is reactivated by inflammation, CMV infection is a relevant clinical pathology associated with decreased long-term graft function132.
Therapeutic modulation of trained immunity
Therapeutically targeting trained immunity represents a promising approach to regulating innate immunity and, consequently, the adaptive immune responses that are triggered and modulated by innate immune cell surface molecules and soluble mediators. Trained immunity can be manipulated to either enhance immune response against infections and malignancies or to inhibit the responses that drive autoimmune diseases and allograft rejection. Trained immunity is regulated at multiple levels, including ligand–receptor interaction, as well as metabolic and epigenetic regulation. These different regulatory levels present several targets for therapeutic intervention (Fig. 5).

Trained immunity depends on histone modifications that enhance the production of pro-inflammatory cytokines and can be induced through different receptors, including the dectin-1 receptor, the IL-1 receptor (IL-1R), and the nucleotide-binding oligomerization domain-containing protein 2 (NOD2). Dectin-1 can be activated by β-glucan (component of fungal cell walls) and blocked by receptor-blocking antibodies or laminarin. IL-1R can be activated by IL-1α and IL-1β, and can be blocked by anakinra, whereas canakinumab can be used to prevent IL-1β binding to IL-1R. The intracellular pattern recognition receptor NOD-2 recognizes peptidoglycans containing muramyl dipeptide (MDP; component of bacterial cell walls) and can be blocked by the small-molecule inhibitors GSK669 and GSK717. Engagement of the aforementioned receptors by their ligands triggers activation of mechanistic target of rapamycin (mTOR), which is a key regulator of cell metabolism, and stimulates glycolysis and mitochondrial oxidative metabolism. The metabolic intermediates derived from these metabolic processes (acetyl-CoA, fumarate, succinate, nicotinamide adenine dinucleotide (NAD+) and mevalonate), are directly involved in mediating the epigenetic changes that underpin trained immunity. Mevalonate-induced training is mediated through increased function of insulin-like growth factor-1 receptor (IGF-1R). Trained immunity can also be suppressed with mTOR inhibitors (for example, rapamycin and metformin). Moreover, mTOR is activated at the surface of lysosomes, which can be prevented with chloroquine (CQ) and hydroxychloroquine (HCQ), which interfere with lysosomal function. Lipid metabolism also has an important role in activating trained immunity. Statins and nuclear liver receptor (LXR) blockers can inhibit trained immunity by blocking mevalonate production.
Blocking ligand–receptor interactions
Different receptors can mediate the induction of trained immunity. One of the best described is dectin-1, which is expressed mainly by DCs, monocytes and macrophages141, and the dectin-1 signalling cascade triggered by binding to β-glucan can be blocked with receptor-blocking antibodies or laminarin142,143. Another well-described pathway that induces trained immunity is mediated by the intracellular PRR NOD-2, which recognizes peptidoglycans containing muramyl dipeptide (MDP)15; MDP is a cell wall component in both Gram-positive and Gram-negative bacteria144,145. NOD-2 receptor activation to induce trained immunity can be achieved with a synthetic small peptide conjugate comprising N-acetyl muramic acid and the short amino acid chain of l-alanine d-isoglutamine dipeptide146,147. By contrast, the small-molecule inhibitors GSK669 and GSK717 can inhibit NOD-2 receptor activation148.
The NOD-2 receptor is also activated by BCG, which is widely delivered through intravesical instillation as immunotherapy for high-risk non-muscle-invasive bladder cancer149. The induction of trained immunity has an important role in the BCG-mediated antitumour effects150. The working hypothesis is that inducing trained immunity can counter the immunosuppressive tumour microenvironment, thereby allowing the immune system to recognize and eliminate tumour cells. Of note, repeated administration of intravesical BCG increases the concentration of pro-inflammatory cytokines151. Accordingly, a myeloid-specific nanobiologic could induce trained immunity through NOD-2 activation and mediated potent suppression of tumour growth in a mouse model of melanoma152. Treatment with BCG is also used to prevent recurrence of superficial bladder tumours153, which improves overall patient survival154. Moreover, BCG vaccination might have a broad effect against infection as it can enhance protection against a variety of viruses, including herpes simplex virus and human papilloma virus155.
IL-1 is another receptor signalling pathway with a fundamental role in the induction and modulation of innate immune responses that is associated with trained immunity. The receptors of this family signal through a Toll/IL‐1 receptor (TIR) domain that leads to nuclear factor-κB (NF‐κB) activation156. The IL-1 pathway can be activated (for example, through IL‐1α, IL‐1β, IL‐18, IL‐33, IL‐36α, IL‐36β, IL‐36γ), or antagonized (for example, via IL‐1Ra, IL‐36Ra, IL‐38) depending on the cytokines and receptors that are triggered. Trained immunity induced by β-glucan depends on IL‐1β production, and blocking antibodies against IL-1R (for example, anakinra) and anti-IL-1β antibodies (for example, canakinumab) prevented the development of peripheral trained macrophages157. Another study showed that anakinra prevented central trained immunity through inhibition of cell-cycle progression and increased glycolysis in bone marrow progenitors35. These compounds are effective in the treatment of a wide range of conditions, including rheumatoid arthritis, familial Mediterranean fever, macrophage activation syndrome, gout and atherosclerosis157,158 and their beneficial effects might be partly mediated through regulation of trained immunity. Of note, IL-1 also has an important role in the systemic inflammation observed in patients receiving haemodialysis159. A randomized, placebo-controlled study showed that, in patients treated with haemodialysis, 4-week treatment with anakinra markedly reduced serum C-reactive protein (CRP) and IL-6 levels160. Moreover, in patients with stage 3–4 CKD, treatment with the IL-1 inhibitor rilonacept for 12 weeks reduced serum CRP concentrations and improved vascular function measured by flow-mediated vasodilation161. These effects might also be partly mediated through effects on immune training, although blocking IL-1 has immunomodulating effects beyond trained immunity157.
Immunometabolic targeting
Cellular metabolic pathways provide another opportunity to intervene and control the epigenetic reprogramming that underlies trained immunity. Trained immunity can be potently suppressed by inhibiting mTOR with rapamycin or metformin162. Moreover, mTOR is activated at the lysosomal surface and lysosomes have a pivotal role in coordinating immunometabolism163. Interestingly, the trained immunity phenotype is characterized by activation of key regulators of lysosome genes22, and chloroquine and hydroxychloroquine, which are weak bases that diffuse passively to the lysosome where they interfere with its function, are potent inhibitors of trained immunity65. Compounds that affect cellular lipid metabolism might also modulate trained immunity. Statins and nuclear liver receptor (LXR) blockers inhibit the cholesterol synthesis pathway and thereby the production of mevalonate, which is a mediator of trained immunity via mTOR164,165. By contrast, the synthesis of fatty acid induced by aldosterone can induce trained immunity and this effect can be inhibited by aldosterone receptor blockers166.
Conclusions
In the past decade, trained immunity, which is characterized by the induction of epigenetic memory in innate immune cells, has emerged as a new concept in immunology, providing new insights into the immune response to infections and the pathophysiology of immunity-mediated diseases. Although originally described as an evolutionary adaptation to protect organisms from reinfection, new evidence shows that this immunological concept extends to other areas of pathology. In the context of kidney diseases, the research field of trained immunity is still in its infancy. Currently, little is known about the role of trained immunity in patients with kidney failure receiving KRT. In autoimmune diseases, evidence of trained immunity has been reported in SLE and systemic sclerosis, although further investigations are required to determine the mechanisms by which trained immunity mediates disease activity, and whether inhibition of trained immunity can prevent disease flares. In other autoimmune disorders such as ANCA vasculitis, which frequently leads to kidney failure, the role of trained immunity is largely unknown, although the involvement of pathways associated with innate immune training suggest a potential contribution to the pathological development of the disease. In organ transplantation, animal studies have demonstrated the role of trained immunity in graft rejection, although human data are still lacking. Important questions remain about the effect of IRI on innate immune training and how trained immunity relates to short-term outcomes, such as delayed graft function, and long-term outcomes, such as formation of donor-specific antibodies and graft survival. Understanding the mechanisms underlying trained immunity in these clinical scenarios has important therapeutic implications. For example, emerging experimental and clinical studies suggest that activation of receptors such as NOD2 might be an effective strategy to induce trained immunity, whereas ligand–receptor blockade or intervention at the metabolic level might help to suppress the pathological effects of trained immune cells. Future research should clarify whether targeting the development of innate immune memory and modulating the trained immune response represents an innovative and effective therapeutic approach in nephrology.
References
Janeway, C. A. Jr. & Medzhitov, R. Introduction: the role of innate immunity in the adaptive immune response. Semin. Immunol. 10, 349–350 (1998).
Murphy, K. & Weaver, C. Janeway’s Immunobiology (Garland Science, 2016).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Bianchi, M. E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 (2007).
Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353 (2015).
Guermonprez, P., Valladeau, J., Zitvogel, L., Thery, C. & Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 20, 621–667 (2002).
Netea, M. G., Schlitzer, A., Placek, K., Joosten, L. A. B. & Schultze, J. L. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe 25, 13–26 (2019).
Titley, M. A., Snaddon, J. L. & Turner, E. C. Scientific research on animal biodiversity is systematically biased towards vertebrates and temperate regions. PLoS One 12, e0189577 (2017).
Kurtz, J. & Franz, K. Innate defence: evidence for memory in invertebrate immunity. Nature 425, 37–38 (2003).
Netea, M. G. et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388 (2020).
Netea, M. G., Quintin, J. & van der Meer, J. W. Trained immunity: a memory for innate host defense. Cell Host Microbe 9, 355–361 (2011).
Divangahi, M. et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat. Immunol. 22, 2–6 (2021).
Kaufmann, E. et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176–190 e119 (2018).
Bekkering, S., Dominguez-Andres, J., Joosten, L. A. B., Riksen, N. P. & Netea, M. G. Trained immunity: reprogramming innate immunity in health and disease. Annu. Rev. Immunol. 39, 667–693 (2021).
Kleinnijenhuis, J. et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl Acad. Sci. USA 109, 17537–17542 (2012).
Ochando, J., Fayad, Z. A., Madsen, J. C., Netea, M. G. & Mulder, W. J. M. Trained immunity in organ transplantation. Am. J. Transpl. 20, 10–18 (2020).
Zhao, S. et al. H3K4 methylation regulates LPS-induced proinflammatory cytokine expression and release in macrophages. Shock 51, 401–406 (2019).
Jamieson, A. M. et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 340, 1230–1234 (2013).
Aegerter, H. et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 21, 145–157 (2020).
Cheng, S. C. et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 17, 406–413 (2016).
Roquilly, A. et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat. Immunol. 21, 636–648 (2020).
Novakovic, B. et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167, 1354–1368 e1314 (2016).
Saeed, S. et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014).
Quintin, J. et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232 (2012).
Fanucchi, S., Dominguez-Andres, J., Joosten, L. A. B., Netea, M. G. & Mhlanga, M. M. The intersection of epigenetics and metabolism in trained immunity. Immunity 54, 32–43 (2021).
Cheng, S. C. et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684 (2014).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Arts, R. J. W. et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 17, 2562–2571 (2016).
Abubakar, I. et al. Systematic review and meta-analysis of the current evidence on the duration of protection by bacillus Calmette-Guerin vaccination against tuberculosis. Health Technol. Assess. 17, 1–372 (2013).
Nguipdop-Djomo, P., Heldal, E., Rodrigues, L. C., Abubakar, I. & Mangtani, P. Duration of BCG protection against tuberculosis and change in effectiveness with time since vaccination in Norway: a retrospective population-based cohort study. Lancet Infect. Dis. 16, 219–226 (2016).
Giamarellos-Bourboulis, E. J. et al. Activate: randomized clinical trial of BCG vaccination against infection in the elderly. Cell 183, 315–323.e9 (2020).
Tsilika, M. et al. ACTIVATE-2: a double-blind randomized trial of BCG vaccination against COVID-19 in individuals at risk. Front. Immunol. 13, 873067 (2022).
Kleinnijenhuis, J. et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J. Innate Immun. 6, 152–158 (2014).
Cirovic, B. et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe 28, 322–334.e5 (2020).
Mitroulis, I. et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161.e12 (2018).
Brown, G. D. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 6, 33–43 (2006).
Kalafati, L. et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell 183, 771–785.e12 (2020).
Moorlag, S. et al. BCG vaccination induces long-term functional reprogramming of human neutrophils. Cell Rep. 33, 108387 (2020).
Kleinnijenhuis, J. et al. BCG-induced trained immunity in NK cells: role for non-specific protection to infection. Clin. Immunol. 155, 213–219 (2014).
Placek, K., Schultze, J. L. & Netea, M. G. Immune memory characteristics of innate lymphoid cells. Curr. Opin. Infect. Dis. 32, 196–203 (2019).
Hamada, A., Torre, C., Drancourt, M. & Ghigo, E. Trained immunity carried by non-immune cells. Front. Microbiol. 9, 3225 (2018).
Shao, Y. et al. Vascular endothelial cells and innate immunity. Arterioscler. Thromb. Vasc. Biol. 40, e138–e152 (2020).
Bigot, J. et al. Respiratory epithelial cells can remember infection: a proof-of-concept study. J. Infect. Dis. 221, 1000–1005 (2020).
Babel, N., Hugo, C. & Westhoff, T. H. Vaccination in patients with kidney failure: lessons from COVID-19. Nat. Rev. Nephrol. https://doi.org/10.1038/s41581-022-00617-5 (2022).
Yu, F., Haas, M., Glassock, R. & Zhao, M. H. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat. Rev. Nephrol. 13, 483–495 (2017).
Tsokos, G. C., Lo, M. S., Costa Reis, P. & Sullivan, K. E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 12, 716–730 (2016).
Ma, W. T., Gao, F., Gu, K. & Chen, D. K. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front. Immunol. 10, 1140 (2019).
Baumann, I. et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 46, 191–201 (2002).
Frank, M. M., Hamburger, M. I., Lawley, T. J., Kimberly, R. P. & Plotz, P. H. Defective reticuloendothelial system Fc-receptor function in systemic lupus erythematosus. N. Engl. J. Med. 300, 518–523 (1979).
Labonte, A. C. et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS One 13, e0208132 (2018).
Aringer, M. et al. Increased bioactive TNF in human systemic lupus erythematosus: associations with cell death. Lupus 11, 102–108 (2002).
Umare, V. et al. Effect of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) on clinical manifestations in Indian SLE patients. Mediators Inflamm. 2014, 385297 (2014).
Hill, G. S. et al. Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int. 59, 304–316 (2001).
Jing, C. et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc. Natl Acad. Sci. USA 117, 15160–15171 (2020).
Sullivan, K. E. et al. The TNFα locus is altered in monocytes from patients with systemic lupus erythematosus. Clin. Immunol. 123, 74–81 (2007).
Leung, Y. T. et al. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics 10, 191–199 (2015).
Zhang, Z., Maurer, K., Perin, J. C., Song, L. & Sullivan, K. E. Cytokine-induced monocyte characteristics in SLE. J. Biomed. Biotechnol. 2010, 507475 (2010).
Zhang, Z., Song, L., Maurer, K., Petri, M. A. & Sullivan, K. E. Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes. Immun. 11, 124–133 (2010).
Zhang, Z. et al. Interferon regulatory factor 1 marks activated genes and can induce target gene expression in systemic lupus erythematosus. Arthritis Rheumatol. 67, 785–796 (2015).
Kamada, R. et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl Acad. Sci. USA 115, E9162–E9171 (2018).
Grigoriou, M. et al. Transcriptome reprogramming and myeloid skewing in haematopoietic stem and progenitor cells in systemic lupus erythematosus. Ann. Rheum. Dis. 79, 242–253 (2020).
Kokkinopoulos, I. et al. Patrolling human SLE haematopoietic progenitors demonstrate enhanced extramedullary colonisation; implications for peripheral tissue injury. Sci. Rep. 11, 15759 (2021).
Nakou, M. et al. Gene expression in systemic lupus erythematosus: bone marrow analysis differentiates active from inactive disease and reveals apoptosis and granulopoiesis signatures. Arthritis Rheum. 58, 3541–3549 (2008).
Mishra, N., Reilly, C. M., Brown, D. R., Ruiz, P. & Gilkeson, G. S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111, 539–552 (2003).
Rother, N. et al. Hydroxychloroquine inhibits the trained innate immune response to interferons. Cell Rep. Med. 1, 100146 (2020).
Jeljeli, M. et al. Trained immunity modulates inflammation-induced fibrosis. Nat. Commun. 10, 5670 (2019).
Betjes, M. G. Immune cell dysfunction and inflammation in end-stage renal disease. Nat. Rev. Nephrol. 9, 255–265 (2013).
Kara, I. H., Yilmaz, M. E., Suner, A., Kadiroglu, A. K. & Isikoglu, B. The evaluation of immune responses that occur after HBV infection and HBV vaccination in hemodialysis patients. Vaccine 22, 3963–3967 (2004).
Garcia, P. et al. COVID-19 vaccine type and humoral immune response in patients receiving dialysis. J. Am. Soc. Nephrol. 33, 33–37 (2022).
Council, E.-E. & Group, E. W. Chronic kidney disease is a key risk factor for severe COVID-19: a call to action by the ERA-EDTA. Nephrol. Dial. Transpl. 36, 87–94 (2021).
Hilbrands, L. B. et al. COVID-19-related mortality in kidney transplant and dialysis patients: results of the ERACODA collaboration. Nephrol. Dial. Transpl. 35, 1973–1983 (2020).
Akar, H., Akar, G. C., Carrero, J. J., Stenvinkel, P. & Lindholm, B. Systemic consequences of poor oral health in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 6, 218–226 (2011).
Sarnak, M. J. & Jaber, B. L. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney Int. 58, 1758–1764 (2000).
Stewart, J. H. et al. The pattern of excess cancer in dialysis and transplantation. Nephrol. Dial. Transpl. 24, 3225–3231 (2009).
Yeun, J. Y., Levine, R. A., Mantadilok, V. & Kaysen, G. A. C-reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am. J. Kidney Dis. 35, 469–476 (2000).
Himmelfarb, J., Stenvinkel, P., Ikizler, T. A. & Hakim, R. M. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 62, 1524–1538 (2002).
Samouilidou, E. C. et al. Lipid abnormalities and oxidized LDL in chronic kidney disease patients on hemodialysis and peritoneal dialysis. Ren. Fail. 34, 160–164 (2012).
Diepeveen, S. H. et al. Oxidative stress in patients with end-stage renal disease prior to the start of renal replacement therapy. Nephron Clin. Pract. 98, c3–c7 (2004).
Bentzon, J. F., Otsuka, F., Virmani, R. & Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 114, 1852–1866 (2014).
Swirski, F. K. et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205 (2007).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Rogacev, K. S. et al. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 32, 84–92 (2011).
Pertosa, G., Grandaliano, G., Gesualdo, L. & Schena, F. P. Clinical relevance of cytokine production in hemodialysis. Kidney Int. Suppl. 76, S104–S111 (2000).
de Cal, M. et al. Oxidative stress and ‘monocyte reprogramming’ after kidney transplant: a longitudinal study. Blood Purif. 26, 105–110 (2008).
Sela, S. et al. Primed peripheral polymorphonuclear leukocyte: a culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J. Am. Soc. Nephrol. 16, 2431–2438 (2005).
Zawada, A. M. et al. SuperTAG methylation-specific digital karyotyping reveals uremia-induced epigenetic dysregulation of atherosclerosis-related genes. Circ. Cardiovasc. Genet. 5, 611–620 (2012).
Li, X. et al. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell 185, 1709–1727 e1718 (2022).
Bekkering, S. et al. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 34, 1731–1738 (2014).
Crisan, T. O. et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann. Rheum. Dis. 75, 755–762 (2016).
Cabau, G., Crisan, T. O., Kluck, V., Popp, R. A. & Joosten, L. A. B. Urate-induced immune programming: consequences for gouty arthritis and hyperuricemia. Immunol. Rev. 294, 92–105 (2020).
Joosten, L. A. B., Crisan, T. O., Bjornstad, P. & Johnson, R. J. Asymptomatic hyperuricaemia: a silent activator of the innate immune system. Nat. Rev. Rheumatol. 16, 75–86 (2020).
Zawada, A. M. et al. Serum uric acid and mortality risk among hemodialysis patients. Kidney Int. Rep. 5, 1196–1206 (2020).
Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion–from mechanism to translation. Nat. Med. 17, 1391–1401 (2011).
Fernandez, A. R., Sanchez-Tarjuelo, R., Cravedi, P., Ochando, J. & Lopez-Hoyos, M. Review: ischemia reperfusion injury-a translational perspective in organ transplantation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21228549 (2020).
Iyer, S. S. et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl Acad. Sci. USA 106, 20388–20393 (2009).
McDonald, B. et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366 (2010).
Amores-Iniesta, J. et al. Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep. 21, 3414–3426 (2017).
Barbera-Cremades, M. et al. P2X7 receptor induces tumor necrosis factor-alpha converting enzyme activation and release to boost TNF-α production. Front. Immunol. 8, 862 (2017).
Vergani, A. et al. Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation 127, 463–475 (2013).
Isenberg, J. S., Frazier, W. A. & Roberts, D. D. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol. Life Sci. 65, 728–742 (2008).
Thakar, C. V. et al. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J. Clin. Invest. 115, 3451–3459 (2005).
Li, Y., Qi, X., Tong, X. & Wang, S. Thrombospondin 1 activates the macrophage Toll-like receptor 4 pathway. Cell Mol. Immunol. 10, 506–512 (2013).
Stein, E. V., Miller, T. W., Ivins-O’Keefe, K., Kaur, S. & Roberts, D. D. Secreted thrombospondin-1 regulates macrophage interleukin-1β production and activation through CD47. Sci. Rep. 6, 19684 (2016).
Yamauchi, Y. et al. Thrombospondin-1 differentially regulates release of IL-6 and IL-10 by human monocytic cell line U937. Biochem. Biophys. Res. Commun. 290, 1551–1557 (2002).
Batal, I. et al. The mechanisms of up-regulation of dendritic cell activity by oxidative stress. J. Leukoc. Biol. 96, 283–293 (2014).
Jong, W. M. et al. Reduced acute myocardial ischemia-reperfusion injury in IL-6-deficient mice employing a closed-chest model. Inflamm. Res. 65, 489–499 (2016).
Uehara, M. et al. Ischemia augments alloimmune injury through IL-6-driven CD4+ alloreactivity. Sci. Rep. 8, 2461 (2018).
Solhjou, Z. et al. Novel application of localized nanodelivery of anti-interleukin-6 protects organ transplant from ischemia-reperfusion injuries. Am. J. Transpl. 17, 2326–2337 (2017).
Zhao, H., Alam, A., Soo, A. P., George, A. J. T. & Ma, D. Ischemia-reperfusion injury reduces long term renal graft survival: mechanism and beyond. EBioMedicine 28, 31–42 (2018).
Fagenson, A. M. et al. Liver ischemia reperfusion injury, enhanced by trained immunity, is attenuated in caspase 1/caspase 11 double gene knockout mice. Pathogens https://doi.org/10.3390/pathogens9110879 (2020).
Tammaro, A., Kers, J., Scantlebery, A. M. L. & Florquin, S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: the fine balance between adaptive repair and tissue degeneration. Front. Immunol. 11, 1346 (2020).
Nankivell, B. J. & Alexander, S. I. Rejection of the kidney allograft. N. Engl. J. Med. 363, 1451–1462 (2010).
Ochando, J., Ordikhani, F., Boros, P. & Jordan, S. The innate immune response to allotransplants: mechanisms and therapeutic potentials. Cell Mol. Immunol. 16, 350–356 (2019).
Poggio, E. D., Augustine, J. J., Arrigain, S., Brennan, D. C. & Schold, J. D. Long-term kidney transplant graft survival-making progress when most needed. Am. J. Transpl. 21, 2824–2832 (2021).
Garcia, M. R. et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J. Clin. Invest. 120, 2486–2496 (2010).
Zecher, D., van Rooijen, N., Rothstein, D. M., Shlomchik, W. D. & Lakkis, F. G. An innate response to allogeneic nonself mediated by monocytes. J. Immunol. 183, 7810–7816 (2009).
Dai, H. et al. Donor SIRPα polymorphism modulates the innate immune response to allogeneic grafts. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aam6202 (2017).
Dai, H. et al. PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science 368, 1122–1127 (2020).
Braza, M. S. et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity 49, 819–828 e816 (2018).
Thiagarajan, P. S. et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc. Res. 99, 494–504 (2013).
Yang, H. et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl Acad. Sci. USA 107, 11942–11947 (2010).
Azimzadeh, A. M. et al. Humoral immunity to vimentin is associated with cardiac allograft injury in nonhuman primates. Am. J. Transpl. 5, 2349–2359 (2005).
Huang, Y. et al. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am. J. Transpl. 7, 799–808 (2007).
Fishman, J. A. Infection in organ transplantation. Am. J. Transpl. 17, 856–879 (2017).
Salehi, S. & Reed, E. F. The divergent roles of macrophages in solid organ transplantation. Curr. Opin. Organ. Transpl. 20, 446–453 (2015).
Jiang, X., Tian, W., Sung, Y. K., Qian, J. & Nicolls, M. R. Macrophages in solid organ transplantation. Vasc. Cell 6, 5 (2014).
Ahmed, E. B., Wang, T., Daniels, M., Alegre, M. L. & Chong, A. S. IL-6 induced by Staphylococcus aureus infection prevents the induction of skin allograft acceptance in mice. Am. J. Transpl. 11, 936–946 (2011).
Kapetanovic, R. et al. Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect. Immun. 75, 830–837 (2007).
Wang, T. et al. Infection with the intracellular bacterium, Listeria monocytogenes, overrides established tolerance in a mouse cardiac allograft model. Am. J. Transpl. 10, 1524–1533 (2010).
Wang, T. et al. Prevention of allograft tolerance by bacterial infection with Listeria monocytogenes. J. Immunol. 180, 5991–5999 (2008).
Shen, H. & Goldstein, D. R. IL-6 and TNF-α synergistically inhibit allograft acceptance. J. Am. Soc. Nephrol. 20, 1032–1040 (2009).
Kaminski, H. & Fishman, J. A. The cell biology of cytomegalovirus: implications for transplantation. Am. J. Transpl. 16, 2254–2269 (2016).
Kapoor, A., Forman, M. & Arav-Boger, R. Activation of nucleotide oligomerization domain 2 (NOD2) by human cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS One 9, e92704 (2014).
Mitchell, B. M., Leung, A. & Stevens, J. G. Murine cytomegalovirus DNA in peripheral blood of latently infected mice is detectable only in monocytes and polymorphonuclear leukocytes. Virology 223, 198–207 (1996).
Pulliam, L., Moore, D. & West, D. C. Human cytomegalovirus induces IL-6 and TNF α from macrophages and microglial cells: possible role in neurotoxicity. J. Neurovirol. 1, 219–227 (1995).
Hargett, D. & Shenk, T. E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl Acad. Sci. USA 107, 20039–20044 (2010).
Smith, P. D. et al. Cytomegalovirus enhances macrophage TLR expression and MyD88-mediated signal transduction to potentiate inducible inflammatory responses. J. Immunol. 193, 5604–5612 (2014).
Sen, P. et al. Linking indirect effects of cytomegalovirus in transplantation to modulation of monocyte innate immune function. Sci. Adv. 6, eaax9856 (2020).
Cook, C. H. et al. Disruption of murine cardiac allograft acceptance by latent cytomegalovirus. Am. J. Transpl. 9, 42–53 (2009).
Zuhair, M. et al. Estimation of the worldwide seroprevalence of cytomegalovirus: a systematic review and meta-analysis. Rev. Med. Virol. 29, e2034 (2019).
Brown, G. D. et al. Dectin-1 mediates the biological effects of β-glucans. J. Exp. Med. 197, 1119–1124 (2003).
Tang, C. et al. Inhibition of Dectin-1 signaling ameliorates colitis by inducing lactobacillus-mediated regulatory T cell expansion in the intestine. Cell Host Microbe 18, 183–197 (2015).
Xie, J. et al. Laminarin-mediated targeting to Dectin-1 enhances antigen-specific immune responses. Biochem. Biophys. Res. Commun. 391, 958–962 (2010).
Wang, J. E. et al. Peptidoglycan primes for LPS-induced release of proinflammatory cytokines in whole human blood. Shock 16, 178–182 (2001).
Wray, G. M., Foster, S. J., Hinds, C. J. & Thiemermann, C. A cell wall component from pathogenic and non-pathogenic gram-positive bacteria (peptidoglycan) synergises with endotoxin to cause the release of tumour necrosis factor-α, nitric oxide production, shock, and multiple organ injury/dysfunction in the rat. Shock 15, 135–142 (2001).
Juarez-Verdayes, M. A., Rodriguez-Martinez, S., Cancino-Diaz, M. E. & Cancino-Diaz, J. C. Peptidoglycan and muramyl dipeptide from Staphylococcus aureus induce the expression of VEGF-A in human limbal fibroblasts with the participation of TLR2-NFB and NOD2-EGFR. Graefes Arch. Clin. Exp. Ophthalmol. 251, 53–62 (2013).
Ogawa, C., Liu, Y. J. & Kobayashi, K. S. Muramyl dipeptide and its derivatives: peptide adjuvant in immunological disorders and cancer therapy. Curr. Bioact. Compd. 7, 180–197 (2011).
Rickard, D. J. et al. Identification of benzimidazole diamides as selective inhibitors of the nucleotide-binding oligomerization domain 2 (NOD2) signaling pathway. PLoS One 8, e69619 (2013).
Lenis, A. T., Lec, P. M., Chamie, K. & Mshs, M. D. Bladder cancer: a review. JAMA 324, 1980–1991 (2020).
Lobo, N. et al. 100 years of Bacillus Calmette-Guerin immunotherapy: from cattle to COVID-19. Nat. Rev. Urol. 18, 611–622 (2021).
Bisiaux, A. et al. Molecular analyte profiling of the early events and tissue conditioning following intravesical bacillus Calmette-Guerin therapy in patients with superficial bladder cancer. J. Urol. 181, 1571–1580 (2009).
Priem, B. et al. Trained immunity-promoting nanobiologic therapy suppresses tumor growth and potentiates checkpoint inhibition. Cell 183, 786–801 e719 (2020).
Morales, A., Eidinger, D. & Bruce, A. W. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol. 116, 180–183 (1976).
Patard, J. J. et al. Intravesical Bacillus Calmette-Guerin treatment improves patient survival in T1G3 bladder tumours. Eur. Urol. 41, 635–641 (2002).
Moorlag, S., Arts, R. J. W., van Crevel, R. & Netea, M. G. Non-specific effects of BCG vaccine on viral infections. Clin. Microbiol. Infect. 25, 1473–1478 (2019).
Teufel, L. U., Arts, R. J. W., Netea, M. G., Dinarello, C. A. & Joosten, L. A. B. IL-1 family cytokines as drivers and inhibitors of trained immunity. Cytokine 150, 155773 (2022).
Dinarello, C. A., Simon, A. & van der Meer, J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652 (2012).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Dinarello, C. A., Koch, K. M. & Shaldon, S. Interleukin-1 and its relevance in patients treated with hemodialysis. Kidney Int. Suppl. 24, S21–S26 (1988).
Hung, A. M., Ellis, C. D., Shintani, A., Booker, C. & Ikizler, T. A. IL-1β receptor antagonist reduces inflammation in hemodialysis patients. J. Am. Soc. Nephrol. 22, 437–442 (2011).
Nowak, K. L. et al. IL-1 inhibition and vascular function in CKD. J. Am. Soc. Nephrol. 28, 971–980 (2017).
Mulder, W. J. M., Ochando, J., Joosten, L. A. B., Fayad, Z. A. & Netea, M. G. Therapeutic targeting of trained immunity. Nat. Rev. Drug. Discov. 18, 553–566 (2019).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Bekkering, S. et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell 172, 135–146 e139 (2018).
Sohrabi, Y. et al. LXR activation induces a proinflammatory trained innate immunity-phenotype in human monocytes. Front. Immunol. 11, 353 (2020).
van der Heijden, C. et al. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc. Res. 116, 317–328 (2020).
Kiefer, J. et al. An unbiased flow cytometry-based approach to assess subset-specific circulating monocyte activation and cytokine profile in whole blood. Front. Immunol. 12, 641224 (2021).
Arts, R. J. W. et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23, 89–100 e105 (2018).
Dos Santos, J. C. et al. β-Glucan-induced trained immunity protects against Leishmania braziliensis infection: a crucial role for IL-32. Cell Rep. 28, 2659–2672 e2656 (2019).
Arts, R. J. et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 24, 807–819 (2016).
Bulua, A. C. et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 (2011).
Chacko, B. K. et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab. Invest. 93, 690–700 (2013).
Nicholls, D. G. et al. Bioenergetic profile experiment using C2C12 myoblast cells. J. Vis. Exp. https://doi.org/10.3791/2511 (2010).
Mourits, V. P. et al. Lysine methyltransferase G9a is an important modulator of trained immunity. Clin. Transl. Immunol. 10, e1253 (2021).
van der Heijden, C. et al. Catecholamines induce trained immunity in monocytes in vitro and in vivo. Circ. Res. 127, 269–283 (2020).
Rodriguez-Ubreva, J. & Ballestar, E. Chromatin immunoprecipitation. Methods Mol. Biol. 1094, 309–318 (2014).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018).
de Almeida, M. C., Silva, A. C., Barral, A. & Barral Netto, M. A simple method for human peripheral blood monocyte isolation. Mem. Inst. Oswaldo Cruz 95, 221–223 (2000).
Meital, L. T. et al. A simple and effective method for the isolation and culture of human monocytes from small volumes of peripheral blood. J. Immunol. Methods 472, 75–78 (2019).
Ziegler-Heitbrock, L. et al. Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–e80 (2010).
Llitjos, J. F. et al. Assessing the functional heterogeneity of monocytes in human septic shock: a proof-of-concept microfluidic assay of TNFα secretion. Front. Immunol. 12, 686111 (2021).
Marimuthu, R. et al. Characterization of human monocyte subsets by whole blood flow cytometry analysis. J. Vis. Exp. https://doi.org/10.3791/57941 (2018).
Ong, S. M. et al. A novel, five-marker alternative to CD16-CD14 gating to identify the three human monocyte subsets. Front. Immunol. 10, 1761 (2019).
Garaude, J. et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 17, 1037–1045 (2016).
Monteiro, L. B., Davanzo, G. G., de Aguiar, C. F. & Moraes-Vieira, P. M. M. Using flow cytometry for mitochondrial assays. MethodsX 7, 100938 (2020).
Geng, J. et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 16, 1142–1152 (2015).
Kauffman, M. E. et al. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species 2, 361–370 (2016).
Acknowledgements
The authors’ work is supported by National Institutes of Health grants R01 AI139623AI (J.O.); Vici grant from the Dutch Research Council (NWO) and European Research Council Advanced Grant (TOLERANCE) (W.J.M.M.); European Research Council Consolidator Grant (310372) and Spinoza Grant of the Netherlands Organization for Scientific Research (M.G.N.); P01 HL158504 (J.C.M.); and Hypatia grant by the Radboud University Medical Center and Senior Kolff grant by the Dutch Kidney Foundation (R.D.).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, made substantial contributions to discussions of the content and wrote, reviewed or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
J.O., W.J.M.M., and M.G.N. declare that they are scientific founders of Trained Therapeutics Discovery. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Nephrology thanks C. Kurts, X. Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ochando, J., Mulder, W.J.M., Madsen, J.C. et al. Trained immunity — basic concepts and contributions to immunopathology. Nat Rev Nephrol 19, 23–37 (2023). https://doi.org/10.1038/s41581-022-00633-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-022-00633-5
This article is cited by
-
Bi-directional neuro-immune dysfunction after chronic experimental brain injury
Journal of Neuroinflammation (2024)
-
Vaccination with a combination of STING agonist-loaded lipid nanoparticles and CpG-ODNs protects against lung metastasis via the induction of CD11bhighCD27low memory-like NK cells
Experimental Hematology & Oncology (2024)
-
Evolution of innate immunity: lessons from mammalian models shaping our current view of insect immunity
Journal of Comparative Physiology B (2024)
-
Functional roles of sphingolipids in immunity and their implication in disease
Experimental & Molecular Medicine (2023)
-
Activation of immune signals during organ transplantation
Signal Transduction and Targeted Therapy (2023)
