
Abstract
Mycobacterium tuberculosis, the causative agent of tuberculosis, has infected humans for millennia. M. tuberculosis is well adapted to establish infection, persist in the face of the host immune response and be transmitted to uninfected individuals. Its ability to complete this infection cycle depends on it both evading and taking advantage of host immune responses. The outcome of M. tuberculosis infection is often a state of equilibrium characterized by immunological control and bacterial persistence. Recent data have highlighted the diverse cell populations that respond to M. tuberculosis infection and the dynamic changes in the cellular and intracellular niches of M. tuberculosis during the course of infection. M. tuberculosis possesses an arsenal of protein and lipid effectors that influence macrophage functions and inflammatory responses; however, our understanding of the role that specific bacterial virulence factors play in the context of diverse cellular reservoirs and distinct infection stages is limited. In this Review, we discuss immune evasion and provocation by M. tuberculosis during its infection cycle and describe how a more detailed molecular understanding is crucial to enable the development of novel host-directed therapies, disease biomarkers and effective vaccines.
Similar content being viewed by others

Introduction
Mycobacterium tuberculosis is a respiratory pathogen that is estimated to infect one-quarter of the world’s population, and it has killed more people over the course of human history than any other microorganism. M. tuberculosis is thought to have originated from environmental mycobacteria that entered human populations in the Horn of Africa more than 70,000 years ago, and the global lineages of M. tuberculosis today mirror the subsequent routes of human migration1. Having evolved with humans for millennia, M. tuberculosis is exquisitely well adapted to navigate the human immune system. The life cycle of M. tuberculosis (Fig. 1) depends on its ability to interact with the immune system in seemingly distinct ways: it evades the innate immune response, persists in the face of an adaptive immune response without causing symptomatic disease, and elicits a robust inflammatory response to cause extensive tissue pathology for it to be transmitted. Understanding how M. tuberculosis orchestrates its life cycle is crucial for the design of preventive and therapeutic vaccines, as well as novel therapies and disease biomarkers.

Mycobacterium tuberculosis is transmitted by aerosol from an individual with active pulmonary infection. The first cells infected are alveolar macrophages. After infected alveolar macrophages migrate into the lung interstitium, the bacilli infect a variety of monocyte-derived and tissue-resident macrophages, dendritic cells and neutrophils. Whether the innate immune response clears infection in some individuals is not clear. Dendritic cells travel to the draining lymph node, where antigen-specific T cells are primed. T cells return to the site of infection and are crucial to establish control and prevent dissemination. With an effective adaptive immune response, most infected individuals develop latent infection, a spectrum of outcomes ranging from sterilized infection to subclinical disease. For reasons that are not well understood, ~5–10% of infected individuals will develop active tuberculosis (TB), most often cavitary disease in the lungs. Most transmission occurs from individuals with cavitary pulmonary disease.
A central feature of tuberculosis (TB) pathogenesis is the ability of the causative organism, M. tuberculosis, to survive in diverse intracellular environments within a variety of myeloid cell populations. The outcome of M. tuberculosis infection is shaped by host genetics, co-morbidities, environmental factors and microbial virulence determinants in ways we are just beginning to understand. To establish infection, M. tuberculosis resists and disarms macrophages and neutrophils in the lung, undermining lysosomal trafficking pathways to survive intracellularly. M. tuberculosis infects tissue-resident alveolar macrophages (AMs), which arise during embryogenesis, as well as a variety of phenotypically distinct macrophage populations of haematological origin2,3,4,5,6,7. Infected dendritic cells (DCs) travel to the draining lymph node and prime T cells, which then return to the infected lung. This takes several weeks, but once an effective adaptive immune response develops, T cells, B cells and activated macrophages form the characteristic granuloma, and bacterial control is established. Most often, bacterial replication is contained, and the inflammatory response subsides, leading to latent TB. Individuals with latent TB have an adaptive immune response to M. tuberculosis but no symptoms, culturable bacilli or disease manifestations. Latent TB likely encompasses a spectrum of outcomes that include bacterial elimination and subclinical disease8, although currently there is no way to distinguish individuals who sterilized infection from those that harbour viable bacilli. Approximately 5–10% of infected individuals will go on to develop active TB, due to either progressive primary infection or ‘reactivation’, which can occur long after initial infection9. In reactivation, TB develops in the setting of a previously ‘successful’ (although not sterilizing) adaptive immune response. A wide variety of disease manifestations are possible, from cavitary lung disease to focal infection involving nearly any organ system, to widely disseminated infection. Cavitary lung disease is most common, and, importantly, individuals with cavitary lesions are the most infectious. Thus, although disseminated infection can be devastating for the infected individual, it is not a successful outcome for the bacteria either, as it is much less likely to lead to transmission. In this Review, we discuss how M. tuberculosis evades immune-mediated clearance while capitalizing on the host inflammatory response at different phases of its life cycle. We focus on recent studies, highlight gaps in knowledge and consider how our current understanding will inform new therapies, vaccines and diagnostics.
How M. tuberculosis establishes infection
The cellular niche of M. tuberculosis
Understanding the earliest events of infection is crucial for the development of a preventive vaccine. The infectious dose of M. tuberculosis is remarkably low, estimated to be approximately three bacilli, highlighting how effective M. tuberculosis is at evading the innate immune response10. Even before uptake by phagocytic cells, M. tuberculosis encounters alveolar lining fluid (Fig. 2), a complex mixture of lipids and proteins secreted by alveolar epithelial cells, which includes surfactant proteins and hydrolases that interact with mycobacterial surface glycolipids. Alveolar lining fluid enhances pathogen uptake and killing by phagocytes and has a variable impact on interactions with alveolar epithelial cells11,12. Deficiencies in pulmonary surfactant due to inflammageing or smoking promote intracellular M. tuberculosis replication and increase the risk of developing TB12,13. In addition, antibody opsonization may promote innate immune control against M. tuberculosis14. Interestingly, recent data suggest that M. tuberculosis-specific IgM antibodies elicited by vaccination might be protective15. This indicates that it might be possible to develop vaccines that generate humoral responses that disrupt the earliest steps of infection, for example, by neutralizing secreted or cell envelope-associated virulence factors or by functionally altering subsequent macrophage interactions, as discussed further herein.

In the airways, Mycobacterium tuberculosis first encounters alveolar macrophages (AMs), which present a permissive niche for infection establishment. In addition, M. tuberculosis infects pulmonary epithelial cells and sheds virulence lipids such as phthiocerol dimycoserosate (PDIM) and sulfolipids into epithelial host cell membranes. Infected AMs migrate into the lung interstitium, in a manner that depends on the ESX-1 secretion system of M. tuberculosis and production of host IL-1β. When M. tuberculosis enters the lung interstitium, it infects additional macrophage populations. Neutrophils respond to M. tuberculosis infection by inducing reactive oxygen species (ROS) and neutrophil extracellular traps (NETs), which do little to control bacterial replication and exacerbate inflammation instead. Some macrophages appear to be better at controlling infection than others, using antimicrobial mechanisms such as phagolysosomal fusion, autophagy and oxidative stress to kill M. tuberculosis, and exhibit a pro-inflammatory metabolic shift. M. tuberculosis detoxifies reactive oxygen with the catalase–peroxidase KatG, and also inhibits ROS production in macrophages and neutrophils using NuoG. When infected macrophages undergo an apoptotic mode of cell death, they can be cleared by efferocytosis, which limits pathogen spread. M. tuberculosis uses virulence factors such as EsxA, CpnT and PDIM to induce necrosis and promote M. tuberculosis dissemination, extracellular replication and immunopathology. M. tuberculosis also induces a foamy macrophage phenotype by enhancing accumulation of host lipids, which support bacterial nutrition and persistence. Host cytokines, such as type I interferons and TNF, and leukotrienes contribute to tissue inflammation, which in turn recruits more cells. Further, M. tuberculosis EsxH suppresses antigen presentation by dendritic cells to delay the onset of adaptive immunity.
Much of our knowledge of the earliest events of infection come from the mouse model. In that model, epithelial cells are infected in the first 48 h after infection; however, M. tuberculosis does not appear to replicate or persist in these cells7. The mycobacterial lipid phthiocerol dimycocerosate (PDIM) can spread into epithelial membranes and modulate immune responses16. Microfold cells (M cells) may also be a portal of entry, allowing M. tuberculosis to gain access to underlying lymphoid tissue in the upper airways17,18. In mice, tissue-resident AMs are the predominant cell type infected during the first 2 weeks7. AMs are located in the air spaces, where they are continually exposed to environmental particulates. In the absence of a microbial challenge, AMs are poised to suppress inflammatory responses to foreign material to prevent lung injury19,20. M. tuberculosis infection of AMs initiates a nuclear factor erythroid 2-related factor 2 (NRF2)-driven antioxidant transcriptional response that correlates with impaired control of M. tuberculosis growth21,22. Ageing may also make AMs more permissive to M. tuberculosis replication23. After 2 weeks, infected AMs migrate out of the alveolar space into the lung interstitium in a process dependent on host IL-1β signalling and an M. tuberculosis type VII secretion system, ESX-1 (ESAT-6 secretion system 1; ESAT-6 is also known as EsxA), which is discussed later. After entering the lung interstitium, M. tuberculosis infects additional phagocytic cell types7. On the basis of its transcriptional responses, M. tuberculosis within AMs appears to be able to access host iron and fatty acids, experiences minimal oxidative and nitrosative stress, and has a high replicative capacity2,4. Moreover, selective depletion of AMs decreases lung M. tuberculosis burden in mice2,24, supporting the idea that AMs are a particularly permissive niche that facilitates the establishment of infection. However, by 3 weeks after infection, infected AMs can exhibit a pro-inflammatory response5. Recent single-cell RNA sequencing analyses revealed that there are multiple AM subpopulations after infection, some of which mount a pro-inflammatory response and impose stress on the bacilli3. In addition, another study implicated AMs as an M. tuberculosis-restrictive cell type following their migration out of the airways into granulomas25. Overall, the data support the idea that AMs exert little control over M. tuberculosis during the first 2 weeks of infection; however, some AM populations may be restrictive, particularly as the infection progresses and an adaptive immune response develops.
Over time, the bacilli diversify their niche by infecting polymorphonuclear neutrophils (PMNs), DCs and a variety of tissue-resident and recruited macrophage populations. Although PMNs eradicate a wide variety of microorganisms with reactive oxygen species (ROS) and neutrophil extracellular traps (NETs), M. tuberculosis resists ROS-mediated and NET-mediated killing26,27, and PMNs create a permissive niche for M. tuberculosis replication28,29,30. M. tuberculosis induces necrosis of infected PMNs, which promotes further M. tuberculosis growth following their phagocytosis by macrophages, which in turn recruits even more PMNs31,32. In humans, PMNs are implicated in lung immunopathology and the failed immune response that is characteristic of active TB33. On the other hand, during initial infection, PMNs promote CD4+ T cell priming in mice34. Thus, PMNs may mediate an indirect role in protection early in M. tuberculosis infection but overall appear to exert little direct antimycobacterial activity and are strongly associated with disease progression. Monocyte-derived DCs have been found to play a key role in transporting M. tuberculosis antigens from the lung to the draining lymph node, where conventional DCs present antigens to naive T cells35. Conventional lung DCs can be separated into two major populations on the basis of the surface markers CD11b and CD103. Both DC populations become infected with M. tuberculosis and exhibit delayed migration to the draining lymph node, but they differ in their capacity to prime naive T cells36,37. More recently, eosinophils have been shown to be recruited to the M. tuberculosis granuloma and were found to be protective against M. tuberculosis infection in mice38.
After infected AMs migrate into the lung interstitium, the bacilli infect additional populations of macrophages. The myeloid cells that become infected have been difficult to classify as they are recruited, proliferate, respond to stimuli and differentiate into macrophages and DCs in the inflammatory environment of the M. tuberculosis-infected lung. Aside from AMs, there are additional CD11c+ populations (which have been classified as either DCs or macrophages), as well as CD11clow/int populations (called ‘interstitial macrophages’ or ‘recruited macrophages’2,5,6,7,36) that become infected. In mice, selective depletion of circulating monocytes using intravenous clodronate increases lung M. tuberculosis burden, suggesting the importance of monocyte-derived macrophages in M. tuberculosis control2. In addition, transcriptomic analysis of human monocyte-derived macrophages revealed that they generate a more robust inflammatory response to M. tuberculosis than AMs39. Thus, a prevailing idea is that monocyte-derived, recruited macrophages are more restrictive than AMs. However, recent data suggest a more complex picture. Single-cell RNA sequencing suggests there are at least four distinct M. tuberculosis-infected interstitial macrophage populations3. By 6 weeks after infection, a subset of CD11c+ monocyte-derived macrophage-like cells becomes the predominantly infected cell type, with up to 30% of this population in the lung infected5. The bacilli within this cell population also appear to experience less stress, suggesting that this CD11c+ macrophage population is more permissive than other monocyte-derived populations3. In a zebrafish larva model, Mycobacterium marinum selectively recruits permissive macrophages40. M. tuberculosis may also promote the recruitment of permissive macrophages as a consequence of manipulation of myelopoiesis and epigenetic reprogramming41 (Box 1). Thus, as suggested above for AMs, different interstitial and recruited macrophages may differ in the degree to which they are able to control M. tuberculosis replication. Interestingly, differences in the antimicrobial capacity of distinct macrophage populations are associated with different metabolic phenotypes (as discussed in more detail later), with AMs broadly appearing more committed to oxidative phosphorylation and interstitial macrophages broadly appearing more committed to glycolysis2. As macrophage control of M. tuberculosis appears to depend on direct interaction with CD4+ T cells42, some of the more restrictive macrophages may be those that have productively engaged with T cells. The extent to which the distinct myeloid populations are epigenetically preprogrammed is an area of investigation3. Whether some individuals are able to sterilize the infection during the innate immune phase is unknown. Interestingly, there is evidence that monocytes from some individuals who appear resistant to M. tuberculosis infection have altered immunometabolic responses43.
To conclude, M. tuberculosis is remarkably effective at disarming the innate immune response. It appears to grow readily in AMs, which deliver the bacilli to the lung interstitium, where additional phagocytes support expansion of the bacterial niche and dissemination from the site of infection. With the onset of adaptive immunity, some myeloid cell subsets appear to restrict bacterial growth, whereas others continue to provide a permissive niche. Interestingly, in mice that have been vaccinated with the attenuated vaccine (Mycobacterium bovis bacillus Calmette–Guérin (BCG)) before M. tuberculosis infection compared with unvaccinated mice, there is earlier transfer of bacilli from AMs into other macrophage populations and PMNs44. Strategies to shift the balance of infection towards restrictive macrophages or to enhance the antimycobacterial capacity of the permissive subsets might enable novel host-directed therapies (HDTs) to promote bacterial clearance. In addition, there is intense interest in understanding how these innate responses could be ‘trained’ to generate enhanced protection as part of vaccine strategies45 (Box 1).
Mechanisms of macrophage control
Macrophages detect a variety of M. tuberculosis pathogen-associated molecular patterns and respond by activating antimicrobial pathways. Cell surface and intracellular pattern-recognition receptors that respond to M. tuberculosis infection include Toll-like receptors (TLRs), C-type lectin receptors, NOD-like receptors and cyclic GMP–AMP synthase (cGAS)–STING, which drive macrophage transcriptional responses and modulate intracellular trafficking. Additionally, phagocytic receptors such as Fcγ receptors, complement receptors and scavenger receptors promote mycobacterial uptake (reviewed in ref.46). Detection of M. tuberculosis pathogen-associated molecular patterns by TLR2 and C-type lectin receptors activates NF-κB signalling, resulting in the production of pro-inflammatory cytokines, such as TNF and IL-6, as well as IL-1β and IL-18, which are processed to mature proteins by the NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome. Some host pattern-recognition receptors actually restrain pro-inflammatory responses, such as DC-SIGN, a major M. tuberculosis-binding C-type lectin receptor, which favours M. tuberculosis replication in macrophages47. These early host–pathogen interactions also influence downstream intracellular trafficking pathways. For instance, Fcγ receptor-mediated phagocytosis directs the bacteria to lysosomes, whereas mannose-receptor engagement inhibits this process48. In addition, in humans, M. tuberculosis-specific antibodies have been found that differ in their functional profiles and glycosylation patterns, and these antibodies impact lysosomal maturation and inflammasome activity49. How signals from these different pathways are integrated when individual bacilli engage multiple macrophage receptors, the degree to which different receptors operate in distinct myeloid cells and whether M. tuberculosis differentially regulates engagement of pattern-recognition receptors at different times of its life cycle are all open questions.
Once bacilli are internalized, macrophages have many ways to eliminate bacteria: restricting essential nutrients, such as iron; intoxication with heavy metals; antimicrobial peptide production; generation of reactive oxygen and nitrogen intermediates; and progressive phagosome acidification and lysosomal fusion50. Fighting off infection also induces a major overhaul of host metabolism to provide ATP and NADPH to fuel antimicrobial pathways. Macrophages switch to an inflammatory phenotype characterized by a Warburg-like shift to glycolysis, which contributes to M. tuberculosis growth restriction as shown in bone marrow-derived macrophages, mice and non-human primates (NHPs)2,51,52. The inflammatory metabolic phenotype is associated with remodelling of the tricarboxylic acid cycle, which enhances IL-1β production53,54, and supports production of itaconate55, which limits inflammatory responses and can inhibit enzymes of M. tuberculosis central carbon metabolism56,57. Although IL-1β is host-protective during early stages of TB, as the disease progresses, it can enhance pro-inflammatory eicosanoid production, resulting in an influx of neutrophils and exacerbating tissue pathology58,59. In the case of human pulmonary TB, a recent study of patients with untreated multidrug-resistant M. tuberculosis found that IL-1β and pro-inflammatory eicosanoids correlated with tricarboxylic acid cycle remodelling60. Mitochondria are central to host immunometabolism as they house multiple metabolic pathways, impact cell death and inflammasome activation, and produce ROS61. M. tuberculosis also induces the accumulation of fatty acids and cholesterol in lipid droplets by modulating the expression of host factors such as miR-33 and PPARα62,63,64. Host lipid droplets play a role in immune defence, and they are also metabolized by M. tuberculosis as a carbon source and used to build virulence lipids necessary for pathogenesis62,65,66. Thus, macrophages respond to infection by profoundly changing their metabolism, and M. tuberculosis appears to take advantage of the changes to its benefit.
Mycobacterial immune evasion strategies
Decades of work has been directed at defining how M. tuberculosis resists and impairs the myriad of macrophage defences. Its ability to impair phagolysosomal fusion was documented as early as 1971 (ref.67). We now appreciate that M. tuberculosis survives within diverse intracellular environments (Fig. 3). We also have an increasing understanding of the protein and lipid effectors that M. tuberculosis uses to undermine distinct host lysosomal trafficking pathways, as briefly discussed here and reviewed in detail recently68. NdkA prevents recruitment of endosomal markers such as RAB5 and RAB7, and SapM dephosphorylates phosphatidylinositol 3-phosphate69. PknG, a serine/threonine protein kinase, targets the RAB GTPase RAB7L1 to inhibit phagosome maturation, and more recently has been described to function as an unusual ubiquitin ligase that interferes with NF-κB signalling70,71. A variety of M. tuberculosis lipids, including phosphatidylinositol mannosides, lipoarabinomannan, diacyltrehaloses, polyacyltrehaloses, trehalose dimycolates and sulfoglycolipid 1, modulate inflammatory signalling and have also been implicated in arresting phagosome maturation, as recently reviewed68. The M. tuberculosis proteins NdkA, CpsA and PPE2 impair acquisition of NADPH oxidase on mycobacterial phagosomes, whereas M. tuberculosis catalase (KatG) detoxifies ROS72,73,74,75. By blocking NADPH oxidase, M. tuberculosis impairs an autophagy-related pathway called ‘LC3-associated phagocytosis’ which normally traffics microorganisms to the lysosome. M. tuberculosis NuoG can also detoxify phagosomal ROS and thereby limit subsequent TNF-mediated apoptosis76. M. tuberculosis encodes five type VII secretion systems (ESX-1–ESX-5) that export substrates out of the cell. ESX-1 and ESX-3 effectors play prominent roles in macrophage interactions and are required for virulence77. EsxA (exported by ESX-1) and PDIM (a cell envelope lipid) promote damage to the phagosomal membrane78,79,80,81,82, although exactly how remains unclear. Permeabilizing the phagosome enables M. tuberculosis to access nutrients and deliver effectors to the cytosol. In addition to modulating lysosomal trafficking, M. tuberculosis effectors inhibit the AIM2 and NLRP3 inflammasome and promote host cell necrosis83,84,85 (reviewed in ref.68). Phagosomal damage sets off a complex series of host–pathogen attacks and counterattacks. Macrophages can repair damage to the endolysosomal system, but the M. tuberculosis ESX-3 effector EsxH interferes with repair86. Macrophages recognize damaged phagosomes and attempt to route bacteria to the lysosome through selective autophagy. Damaged phagosomes expose glycan residues on intraluminal proteins, which are then bound by galectins, whereas ubiquilin 1 and the ubiquitin ligases parkin and SMURF1 promote ubiquitin deposition around the bacilli69,87,88. Galectin-tagged and ubiquitin-tagged mycobacteria are recognized by adaptor proteins such as p62, NDP52 and TAX1BP1 and are targeted for autolysosomal degradation. Bacterial and mitochondrial DNA that enter the cytoplasm activate the cGAS–STING pathway to promote production of type I interferons and activate autophagy89,90. Members of the M. tuberculosis PE-PGRS protein family block autophagy91,92, and, at least in human lymphatic endothelial cells, cytosolic mycobacteria aggregate to form cords, which are resistant to selective autophagy93. In addition, unlike infection with other bacilli, M. tuberculosis infection does not generate substantial mitochondrial ROS, which also appears to contribute to impaired NADPH oxidase activity and autophagy94. The ability of M. tuberculosis to block lysosomal trafficking pathways is overcome in part if macrophages are stimulated with interferon-γ before infection. Interferon-γ has a major impact on macrophage physiology and phagosome biology, as discussed further later. Finally, M. tuberculosis neutralizes the pH of the phagosome by producing an unusual terpene nucleoside (1-tuberculosinyladenosine) and also has mechanisms to resist killing by acidification95,96. As a final resort to eliminate intracellular infection, the host cell initiates apoptosis, a programmed cell death pathway, in which degraded cellular contents are retained inside membrane blebs (reviewed in ref.97). An apoptotic mode of cell death, combined with subsequent efferocytosis, is host protective98; however, M. tuberculosis induces necrosis using factors such as CpnT, PDIM and iron overload to favour bacterial dissemination68,81,83,99,100,101,102. Excessive tissue inflammation by type I interferons and TNF also contributes to M. tuberculosis-induced macrophage death61,103. Overall, M. tuberculosis takes a multifaceted approach to undermine macrophage antimicrobial responses and survive in diverse intracellular environments.

Mycobacterium tuberculosis is taken up in a single membrane-bound phagosome, which is targeted by the host to promote bacterial clearance. The interactions between host proteins (blue) and M. tuberculosis virulence factors (proteins in dark green; lipids in light green) shape infection outcomes. Sequential recruitment of molecular markers such as RAB5, RAB7 and phosphatidylinositol 3-phosphate (PtdIns3P) normally promote maturation of early to late phagosomes, but M. tuberculosis actively evades phagosomal maturation. In a process called ‘LC3-associated phagocytosis’ (LAP), NADPH oxidase assembles on the M. tuberculosis phagosome (LAPosome) and induces oxidative stress, but M. tuberculosis effectors impair the recruitment of the NADPH oxidase. M. tuberculosis can also be targeted to spacious phagosomes in a RAB20-dependent manner. In addition, the M. tuberculosis ESX-1 substrate EsxA and phthiocerol dimycoserosate (PDIM) promote phagosomal damage. M. tuberculosis EsxH inhibits the host endosomal sorting complex required for transport (ESCRT) machinery to prevent membrane repair. M. tuberculosis escapes into the cytosol and can replicate to form cords. The host attempts to recapture cytosol-exposed M. tuberculosis in double-membrane autophagosomes, which is inhibited by effectors, including PE-PGRS family members. M. tuberculosis contained within single-membrane or double-membrane compartments is targeted for lysosomal killing. M. tuberculosis has mechanisms to resist acidification, such as blocking vacuolar-type ATPase (V-ATPase) and producing the antacid 1-tuberculosinyladenosine (1-TbAd) to neutralize the pH. DAT, diacyltrehalose; LAM, lipoarabinomannan; PAT, polyacyltrehalose; PIM, phosphatidylinositol mannoside; ROS, reactive oxygen species; SL-1, sulfoglycolipid 1; TDM, trehalose dimycolate.
Our molecular understanding of how M. tuberculosis undermines macrophage functions comes largely from ex vivo studies, although there are efforts to develop more physiologically relevant systems12,104. As discussed earlier herein, in vivo macrophages differ on the basis of ontogeny, tissue residence and inflammatory environment. They are exposed to the alveolar lining fluid and undergo epithelioid transformations, form multinucleated giant cells and become lipid-laden and ‘foamy’. How ex vivo macrophages (most often bone marrow-derived or monocyte-derived macrophages) recapitulate the diverse macrophage subsets in vivo is poorly defined. Whether M. tuberculosis preferentially resides in particular intracellular compartments in particular macrophage populations is not known. M. tuberculosis could also selectively deploy effectors to differentially manipulate macrophages on the basis of the macrophage state. There are several notable examples in which the findings of the ex vivo studies and in vivo data appear discordant. For example, numerous autophagy proteins contribute to macrophage control of M. tuberculosis ex vivo, but they are dispensable in vivo28, and NLRP3 is essential for M. tuberculosis-induced inflammasome activity in macrophages ex vivo but is expendable for IL-1β production in vivo105. In addition to differences between macrophages, these discrepancies could reflect compensatory host pathways in vivo or differences in the physiology of bacilli grown in vivo compared with liquid culture. For example, in vivo M. tuberculosis has access to host cholesterol and fatty acids, which impact virulence lipids of M. tuberculosis, such as PDIM66. Moreover, in ex vivo studies, investigators routinely generate single-cell suspensions of M. tuberculosis before macrophage infections, which could impact macrophage interactions. Although future work will have to assess the relevance of individual effectors in distinct cell types and during different stages of the life cycle of M. tuberculosis, overall, these studies have yielded substantial insight into molecular mechanisms that enable M. tuberculosis to disrupt macrophage functions. Understanding these immune evasion strategies of M. tuberculosis lays the groundwork for HDTs aimed at enhancing mycobacterial clearance, as discussed in more detail later.
Withstanding adaptive immunity
Mechanisms of host control
An effective adaptive immune response is required to prevent progressive, disseminated TB. Granulomas are the histopathological hallmark of M. tuberculosis infection. Mature granulomas are an organized collection of macrophages, neutrophils and lymphocytes. When there is an effective adaptive immune response, granulomas control and even sterilize the infection, becoming sclerotic and calcified, whereas active TB granulomas are necrotic and have a caseating appearance (like cheese). CD4+ T cells and TNF are crucial for well-organized granuloma formation and host protection106,107. Individuals with CD4+ T cell dysfunction from HIV infection or those treated with TNF inhibitors fail to form well-organized granulomas, and they are at high risk of developing active TB and disseminated infection8,46. CD8+ T cells are also activated during M. tuberculosis infection, and are detectable in the blood of human patients with TB108. Recent data support the idea that CD8+ T cells and CD4+ T cells synergize to control M. tuberculosis infection109. The role of B cells and humoral immunity in TB is less well established than that of T cells. Historically, B cell depletion studies failed to definitively establish a role for B cells or antibodies in M. tuberculosis infection control, although recent studies have uncovered potentially protective antibodies in NHPs and humans after intravenous BCG vaccination15,110. Inducible bronchus-associated lymphoid tissue is ectopic lymphoid tissue that contains B cell follicles and is found in a variety of inflammatory lung diseases. The abundance of inducible bronchus-associated lymphoid tissue and its proximity to M. tuberculosis granulomas correlates with protection against TB111.
Interferon-γ has long been implicated as a key factor through which CD4+ T helper 1 (TH1) cells mediate protection112. Interferon-γ has a major impact on macrophage physiology and phagosome biology, including enhancing autophagy and promoting RAB20-dependent targeting to spacious phagosomes113,114. There are a number of differences in how interferon-γ enhances the antimicrobial activity of human macrophages as compared with murine macrophages. Interferon-γ induces the expression of immunity-related GTPases, which target intracellular pathogens for destruction115. In mice, this is a large family, whereas humans have only two immunity-related GTPases, and their expression is not interferon-γ dependent115. In mouse macrophages, but not human macrophages, interferon-γ increases nitric oxide production by enhancing expression of inducible nitric oxide synthase114. Nitric oxide has direct toxic effects on M. tuberculosis and reduces immunopathology in vivo30,58. In human cells, interferon-γ induces the expression of the antimicrobial peptide cathelicidin, which depends on vitamin D116. Despite these apparent species-specific differences in macrophage responses to interferon-γ, a deficient type II interferon response is associated with TB susceptibility in both mice and humans. In addition to potentiating the antimicrobial activity of macrophages, the susceptibility of interferon-γ-deficient hosts may also be explained by impaired myelopoiesis and a lack of trained, protective monocytes and macrophages117 (Box 1). Interferon-γ also represses the recruitment of M. tuberculosis-permissive PMNs by inhibiting IL-1 and 12/15-lipoxygenase30, and it limits the accumulation of terminally differentiated, non-protective CD4+ T cells in the lung vasculature118. Interferon-γ is particularly important to limit disseminated infection in the spleen, whereas excessive interferon-γ in the lung can be detrimental if it is not repressed by PD1119. Similarly, infection of mice lacking cyclophilin D, a mitochondrial matrix protein, led to increased T cell proliferation, with higher production of TNF and interferon-γ, with associated tissue damage and reduced survival120.
Interferon-γ-independent mechanisms also contribute to CD4+ T cell-mediated control in the lungs in ways that are not well understood121. Studies to define interferon-γ-independent mechanisms identified the TNF family member CD153, whose expression on M. tuberculosis-specific TH1 cells inversely correlates with bacterial burden within granulomas in mice and NHPs, and is required for the protective capacity of CD4+ T cells122. In humans, CD153 expression is higher in patients with latent TB than in patients with active TB123. Further defining protective T cell subsets on the basis of their effector capabilities, memory and activation status, and migratory potential may reveal T cell signatures that correlate with protection and can guide vaccine strategies124. In conclusion, CD4+ TH1 cells are crucial for the control of M. tuberculosis infection and to limit dissemination. Interferon-γ plays a key role in their function, but interferon-γ-independent mechanisms also contribute to M. tuberculosis infection control in ways that are not well defined.
How M. tuberculosis undermines adaptive immunity
Although a robust adaptive immune response to M. tuberculosis develops, it is delayed and fails to sterilize the infection. In animal models, it takes several weeks before antigen-specific T cells are detected in the lungs. Similarly, in humans, it takes 2–8 weeks before TB-specific T cell responses are detectable. M. tuberculosis delays T cell priming by impairing DC maturation and interfering with efficient antigen presentation through a variety of mechanisms, as recently reviewed125 (Fig. 4). For example, infected DCs export M. tuberculosis antigens through kinesin 2-dependent vesicular transport to divert them from major histocompatibility complex class II presentation, making them less effective at activating T cells than uninfected DCs that take up M. tuberculosis antigens126. The cell envelope-associated serine protease Hip1 cleaves the chaperone GroEL2, which is strongly immunogenic in its full-length form, to prevent its presentation by DCs127. Several effectors that undermine phagosome integrity and phagosome maturation have also been shown to impair T cell priming. For example, EsxH impairs antigen processing by inhibiting the endosomal sorting complex required for transport (ESCRT) machinery128, PE_PGRS47 impairs antigen presentation by inhibiting autophagy92, and PDIM inhibits CD86 and IL-12p40 expression in infected DCs129. In addition, as mentioned earlier, NuoG, which inhibits apoptosis of infected neutrophils, delays antigen acquisition by DCs and trafficking to the lymph nodes130. It is possible to experimentally overcome the delay in T cell priming by direct pulmonary installation of M. tuberculosis antigen-treated DCs at the time of infection131. This leads to earlier recruitment of antigen-specific T cells to the lungs and more immediate control of M. tuberculosis replication. Overall, the delay in the adaptive immune response allows M. tuberculosis to establish infection, spread from the initial site of infection and grow relatively unhindered for weeks.

During Mycobacterium tuberculosis infection, there is a delay in migration of dendritic cells (DCs) to the lungs. NuoG contributes to this delay by inhibiting apoptosis of infected polymorphonuclear neutrophils (PMNs) and antigen uptake by DCs. Once infected DCs arrive in the lymph node, M. tuberculosis impairs their ability to prime CD4+ T cells by degrading antigens (for example, Hip1 degrades GroEL2), exporting antigens out of the cell, and inhibiting antigen processing (mediated by EsxH). Exported M. tuberculosis lipoglycans such as lipoarabinomannan also interfere directly with T cell responses. Phthiocerol dimycoserosate inhibits CD4+ T cell priming and differentiation by inhibiting expression of CD86 and IL-12. M. tuberculosis also preferentially elicits CD4+ and CD8+ responses to decoy antigens (for example, Ag85B and TB10.4), whose expression is downregulated after T cell priming or whose targeting by T cells is not protective. T cells in the infected lung are physically separated from infected macrophages. High levels of IL-10 and transforming growth factor-β (TGFβ) in granulomas also inhibit T cell effector functions. Finally, by inhibiting antigen presentation in infected macrophages, M. tuberculosis also prevents recognition of infected cells. TCR, T cell receptor.
Even when antigen-specific T cells are rapidly recruited to the lungs experimentally, they do not sterilize the infection131. Thus, the adaptive immune response is not just late but is also inadequate. Direct interaction between M. tuberculosis-specific CD4+ T cells and infected macrophages seems to be crucial for control of M. tuberculosis infection42. Effectors discussed above that impair T cell priming, such as EsxH, can impair recognition of infected macrophages by CD4+ T cells128. In addition, M. tuberculosis-infected macrophages release M. tuberculosis cell wall components, such as lipoarabinomannan, in extracellular vesicles. Lipoarabinomannan and other mycobacterial lipoglycans inhibit T cell receptor-mediated activation of naive and effector CD4+ T cells132. CD8+ T cell responses to M. tuberculosis are defined largely by immunodominant antigens, and T cells that are specific for these antigens have a poor ability to recognize infected macrophages133,134. Similarly, CD4+ T cells that recognize epitopes of Ag85B are rendered ineffective by the downregulation of Ag85B during chronic infection135,136. Furthermore, the MVA85A candidate vaccine was found to confer no more protection than BCG despite inducing a robust TH1 cell response to Ag85A137. Thus, immunodominant antigens may act as decoys, priming T cell populations that fail to recognize the directly infected cells because the antigen they recognize is exported from or not expressed in infected cells. The idea of decoy antigens is supported by the identification of a strain of M. tuberculosis containing a polymorphism in esxH, which encodes TB10.4, disrupting its immunodominance through increased proteolytic cleavage of the immunodominant epitope, thereby allowing other clonal populations to expand138. On the other hand, persistently expressed M. tuberculosis antigens can lead to CD4+ T cell and CD8+ T cell exhaustion through T cell receptor downregulation139,140,141. This phenomenon has been observed in EsxA-specific CD4+ T cells in humans136. Interestingly, CD4+ T cells specific for the non-dominant, or cryptic, epitopes of EsxA resist terminal differentiation and confer more protection than the CD4+ T cells specific for the dominant epitopes142. However, one study reported low levels of T cell exhaustion markers in TB granulomas in NHPs, suggesting that exhaustion does not fully explain the defects in T cell-mediated protection143. Another study found that CD4+ T cell responses against the immunodominant proteins encoded in the ESX-1, ESX-3 and ESX-5 loci are associated with protection in latent TB144.
Although granulomas are important in host protection and for preventing disseminated infection, they also facilitate persistent infection. To form a granuloma, macrophages undergo a programme of epithelialization that is driven by T helper 2 (TH2) cell immunity145,146. The immunosuppressive environment of the TB granuloma exhibits a PDL1 signature similar to that seen in tumours147. Granulomas also appear to keep CD4+ T cells peripherally located, away from the more central, infected macrophages148,149. Recruitment of nonspecific T cells may also limit antigen-specific T cell–macrophage interactions150. Granulomas are an immunosuppressive environment in which IL-10 impairs TH1 cell immunity and lysis of infected macrophages by CD8+ T cells151,152,153. Transforming growth factor-β (TGFβ) also inhibits CD4+ T cell function and survival in the TB granuloma154. Moreover, the lipid-rich caseum of necrotic granulomas provides fatty acids and cholesterol for M. tuberculosis metabolism. Even within an infected individual, different granulomas establish different degrees of immune control155,156, and the basis for this heterogeneity is not well understood. In summary, both the ability of M. tuberculosis to inhibit T cell function and the nature of the host granulomatous response contribute to a maladaptive immune response that fails to reliably sterilize the infection.
Reactivation and transmission
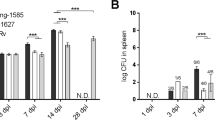
Why M. tuberculosis reactivates in seemingly immunocompetent hosts and how it promotes transmission are questions of intense interest. It has been difficult to approach these questions because mouse models do not have latent infection, and there are no well-established systems for studying transmission. Host responses that are important in maintaining the latent state were recently reviewed8. Polyfunctional CD4+ T cells producing interferon-γ, TNF and IL-2 appear to be important, and treatment with anti-TNF agents or co-infection with simian immunodeficiency virus promotes reactivation in NHP models8. CD8+ T cells and donor-unrestricted T cells may also play a role, and antibodies, natural killer cells and type 3 innate lymphoid cells are also correlated with the latent TB state8. In the face of this immune pressure, M. tuberculosis is thought to survive in a dormant state, although the physiology of the bacilli during latent infection is not well understood. Hypoxic conditions, as found inside solid granulomas, promote a non-replicating, persistent phenotype and activate the bacterial DosR regulon, which is required for persistence in NHPs157,158.
Type I interferon signalling accompanies reactivation, and accumulating evidence suggests it plays a causative role in reactivation. To identify host signatures associated with reactivation, a cohort of adolescents with latent TB were followed up prospectively. A neutrophil-driven blood transcriptional signature of type I interferon signalling preceded clinical diagnosis of active TB33,159. This fits with the idea discussed above that neutrophils provide a replicative niche for M. tuberculosis, and they may also play an immunoregulatory role30. In addition, a genetic mutation in interferon-α/β receptor subunit 1 that is associated with increased risk of hepatitis C virus infection in humans is associated with decreased susceptibility to M. tuberculosis160. M. tuberculosis resuscitation-promoting factors are cell wall active enzymes that play a role in the transition from dormancy to active replication161, but the involvement of other bacterial factors that influence reactivation is not clear. As ESX-1 function and PDIM abundance impact type I interferon production, as discussed earlier, we speculate that they might be important bacterial drivers of reactivation, but there are no well-established, facile systems for studying this. The lack of small animal models of reactivation disease makes it difficult to establish whether responses that are correlated with disease progression are causally linked to reactivation. The large knowledge gap regarding the relevant host and bacterial determinants hampers strategies to develop therapeutic vaccines that prevent disease progression in infected individuals. However, substantial efforts have made use of signatures of the host response to M. tuberculosis to try to develop new biomarkers and diagnostics (Box 2).
For transmission, M. tuberculosis gains access to the airways, grows to a high number and is aerosolized by cough. M. tuberculosis takes advantage of the adaptive immune response to promote transmission. Cavities play a central role; they are immune privileged sites with few blood vessels and a fibrotic cuff that limits immune cell access. When growing extracellularly in cavities, M. tuberculosis reaches burdens of 107–109 bacilli162. The development of cavities requires a tissue-damaging immune response directed against viable M. tuberculosis or M. tuberculosis antigen. Individuals with HIV infection are less likely to develop cavitary disease or to transmit infection163. There is a correlation between the number of circulating CD4+ T cells and the frequency of cavitary disease163, and rabbits are more likely to develop cavitary disease after repeated exposure164. The association between T cell responses and cavitary disease might explain why CD4+ T cell antigens are hyperconserved165. In addition, high levels of IL-1β and TNF, which are usually considered host protective, are associated with cavitary disease166. TNF can drive necrotic cell death and expression of matrix metalloproteinases to promote tissue destruction61. Interestingly, when tested in C3HeB/FeJ mice, M. tuberculosis strains with different propensities for transmission caused strikingly different pathology, with high-transmission strains causing more caseating granulomas, and low-transmission strains resulting in more extensive and diffuse pneumonia167. How the genetic differences between these strains leads to differences in transmission potential is not yet established168. It is possible that M. tuberculosis could manipulate its cell envelope glycolipids to alter the balance between masking and eliciting host inflammation169. It is also plausible that virulence factors that promote macrophage necrosis, such as CpnT and ESX-1, promote cavity formation and transmission, but this has not been directly tested. Interestingly, neutrophils may promote transmission for reasons beyond their association with tissue damage, as a recent study demonstrated that M. tuberculosis aerosolized within infected PMNs had enhanced viability compared with extracellular bacilli170. Finally, direct evidence for cell envelope lipids in transmission comes from recent data showing that sulfolipid in the mycobacterial cell envelope promotes cough in guinea pigs171.
Implications for therapies and vaccines
How can a better understanding of TB pathogenesis inform new approaches for vaccines and therapies? Because TB investigators demonstrated that interferon-γ, TNF and CD4+ TH1 cells are crucial determinants of host immunity, most vaccine efforts have focused on augmenting these immune responses. However, although they are crucial for controlling TB, particularly to prevent dissemination, ‘more’ does not promote sterilizing immunity and instead can exacerbate tissue damage and pathology. Given the plethora of strategies that M. tuberculosis has to undermine both the efficacy of CD4+ T cells and the antimicrobial capacity of macrophages, it is not surprising that this approach has been largely unsuccessful. Prior infection does not prevent reinfection in humans, and NHPs can be secondarily infected even while they have ongoing active infection143, suggesting that an immune response that prevents infection is going to be different from what M. tuberculosis normally elicits in most people. New vaccine strategies should take into account the immune evasion mechanisms of the bacilli (Fig. 5). A protective host response will require overcoming of the immune evasion mechanisms of M. tuberculosis, restoration of the antimicrobial capacity of myeloid cells and restoration of their functional interactions with the adaptive immune system. Understanding how M. tuberculosis undermines host immunity can define its Achilles heel. Protection could be achieved at the level of phagocyte function by enhancing autophagy, LC3-associated phagocytosis and phagosome–lysosome fusion; production of ROS; nutritional immunity; optimal metabolic responses; or antigen presentation and communication with innate and adaptive lymphocytes.

a | Possible strategies for generating a protective vaccine. A vaccine could generate antibodies that neutralize a panel of essential cell surface or secreted virulence factors that are required for Mycobacterium tuberculosis to establish infection. Opsonizing antibodies could promote bacterial uptake specifically into protective macrophages or direct the bacilli to a bactericidal path intracellularly. By generating epigenetic changes, macrophages could be trained to a more protective phenotype. It may be possible to identify antigens that generate a protective T cell response, as opposed to the decoy antigens that are dominant during natural infection. Finally, generating lymphocytes that are poised in the lung, ready to respond to infection, would overcome the delay in T cell priming. b | The approaches proposed in part a could protect against M. tuberculosis infection by restoring macrophages’ effector functions, such as lysosomal trafficking, reactive oxygen species (ROS) production and signalling, optimal metabolic responses and protective cell death pathways. Antibodies or training could also skew cellular recruitment towards protective myeloid cells and away from permissive macrophages and neutrophils. Finally, restoring functional interactions with the adaptive immune system could enhance the antimicrobial capacity of myeloid cells and their protective inflammatory responses. LAM, lipoarabinomannan; LAP, LC3-associated phagocytosis; Me1, 1-methylation.
How could we overcome immune evasion mechanisms in the context of a vaccine? As we learn more, we may be able to harness trained immunity to increase the barrier to primary infection (Box 1 and Fig. 5). There is also increasing evidence suggesting that antibodies can be protective against TB. For example, certain antibodies targeting arabinomannan, an immunomodulatory component of the mycobacterial cell wall, and those targeting the PstS1 transporter of the bacilli can be protective110,172. There has been minimal investigation into identifying protective antigens or exploring antigens that are not normally generated in most people during natural infection. For example, generating vaccine-induced antibodies that neutralize a panel of essential cell surface or secreted virulence factors, such as ESX effectors or CpsA, which are required for the earliest events of infection, could be one approach. Humoral or trained responses may also promote bacterial uptake into protective macrophages or direct the bacilli to a bactericidal path intracellularly. Along the same lines, skewing responses away from decoy antigens might confer protective adaptive immune responses. Finally, creative approaches to overcome the delay in T cell priming would also be protective131. Given the myriad of approaches M. tuberculosis uses to resist clearance, a combination of these strategies to overcome immune evasion by M. tuberculosis is likely to be necessary to generate substantial protection.
Although these strategies may seem fanciful or out of reach currently, the idea that a protective vaccine is going to need to overcome the macrophage and T cell dysfunction imposed by M. tuberculosis is based on a wealth of pathogenesis studies and provides an important framework for future approaches. Recent work demonstrating that BCG administered by an intravenous route provides remarkable protection from infection offers the possibility to define heretofore elusive correlates of protection173 and to elucidate how they overcome M. tuberculosis immune evasion. Protective immunity might also be exhibited by a small fraction of individuals who resist M. tuberculosis infection despite heavy exposure43,174.
Understanding molecular mechanisms of immune evasion by M. tuberculosis may also lead to HDTs, which could be used as an adjunctive therapy along with direct-acting antimicrobials to shorten treatment duration and circumvent the problem of antibiotic resistance. HDTs might be able to clear persistent and drug-tolerant M. tuberculosis reservoirs, thereby reducing relapses. A rational approach towards HDT is to reverse the M. tuberculosis-imposed deficiencies in immune cells. For example, AMs may be particularly permissive to infection because they rely predominantly on fatty acid metabolism rather than glycolysis2. Inhibiting macrophage fatty acid oxidation induces mitochondrial ROS, enhances NADPH oxidase activity and promotes autophagy2,94, thereby overcoming key immune evasion strategies of M. tuberculosis. Similarly, metformin enhances mitochondrial ROS production and autophagy in infected cells, decreasing M. tuberculosis burden in macrophages and mice. Metformin can also educate CD8+ T cells and enhance BCG vaccine responses, and is currently in clinical trials as an adjunct for TB therapy175. As an alternative to small molecules, targeting host microRNAs that are induced by M. tuberculosis may also have the potential to enhance antimicrobial immunity63,176. Although a handful of HDTs have been shown to have antimycobacterial activity in mice, their ability to reduce bacterial burden has generally been modest59,94,177,178. This may be because the cells infected in vivo are not as responsive to the treatment as macrophages infected ex vivo. In the hope of translating findings to humans, an animal model that better recapitulates human pathology, such as NHPs, ultra-low-dose infections and C3HeB/FeJ mice, as well as capturing more genetic diversity, such as the use of collaborative cross and diversity outbred mice, may be important179,180,181,182. In addition, many of the HDTs that have been evaluated are premised on restoring macrophage antimicrobial responses; restoring T cell functionality would address the other side of the equation. As granulomas promote M. tuberculosis persistence and support an immunosuppressive environment, another strategy is to reverse the immunosuppressive environment or disrupt the granuloma architecture to promote lymphocyte infiltration and access to the inner region harbouring infected macrophages and free bacteria, an approach that has shown promise in mice, NHPs and zebrafish145,148,154. Approaches to develop rational combinations of HDTs that synergize are needed. Finally, it may seem paradoxical to treat infection by reducing inflammation, but in combination with antibiotics such an approach might improve immune function, reduce tissue injury and enhance drug penetration into granulomatous tissue183. Given their role as a permissive niche for M. tuberculosis and their role in immunopathology, neutrophils have also been evaluated as targets of HDTs to reduce lung pathology and M. tuberculosis burden184.
Conclusions
To conclude, TB research in the past decade has made huge strides. We have an increasing understanding of how the tubercle bacilli undermine macrophage and T cell functions. The bacilli also take advantage of the host immune response to establish a cellular niche, generate extensive tissue damage and drive transmission. We have gained substantial mechanistic insight into how M. tuberculosis modulates intracellular trafficking, undermines macrophage effector functions, controls cell death pathways, impairs antigen presentation and survives in diverse intracellular environments. Recent work has highlighted the complexity of the cellular response to M. tuberculosis, demonstrating that there are a variety of distinct myeloid cell types that are infected and that these cells differ in the degree to which they can restrict M. tuberculosis growth. How virulence strategies of the bacilli are deployed in vivo in the different types of infected myeloid cells and during different phases of the infection cycle remains to be established. Understanding the immune evasion strategies of M. tuberculosis is crucial for the development of HDTs and an effective vaccine. For the host to clear the bacilli, therapies and vaccines will have to overcome myeloid cell dysfunction and the failure of the innate and adaptive immune systems to communicate effectively. In the context of preventive vaccines, recent work suggests that trained immunity, unique antibody responses and novel T cell epitopes are promising paths to pursue. Although there are many gaps remaining, the fundamental understanding of host–pathogen interactions should enable well-reasoned, new approaches to develop better biomarkers, therapies and vaccines.
References
Comas, I. et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182 (2013).
Huang, L., Nazarova, E. V., Tan, S., Liu, Y. & Russell, D. G. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 215, 1135–1152 (2018). This study demonstrates that macrophage populations have differential abilities to restrict M. tuberculosis growth during in vivo infection.
Pisu, D. et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med. https://doi.org/10.1084/jem.20210615 (2021).
Pisu, D., Huang, L., Grenier, J. K. & Russell, D. G. Dual RNA-Seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30, 335–350.e4 (2020).
Lee, J. et al. CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. 16, e1008621 (2020).
Norris, B. A. & Ernst, J. D. Mononuclear cell dynamics in M. tuberculosis infection provide opportunities for therapeutic intervention. PLoS Pathog. 14, e1007154 (2018).
Cohen, S. B. et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24, 439–446.e4 (2018). This is the first study to demonstrate that AMs, although long suspected, are the initial cells infected during pulmonary M. tuberculosis infection and that they initiate bacterial dissemination into the lung interstitium.
Boom, W. H., Schaible, U. E. & Achkar, J. M. The knowns and unknowns of latent Mycobacterium tuberculosis infection. J. Clin. Invest. https://doi.org/10.1172/JCI136222 (2021).
Pai, M. et al. Tuberculosis. Nat. Rev. Dis. Primers 2, 16076–16076 (2016).
Donald, P. R. et al. Droplets, dust and guinea pigs: an historical review of tuberculosis transmission research, 1878–1940. Int. J. Tuberc. Lung Dis. 22, 972–982 (2018).
Scordo, J. M. et al. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal Immunol. 12, 795–804 (2019).
Thacker, V. V. et al. A lung-on-chip model of early. Elife https://doi.org/10.7554/eLife.59961 (2020).
Moliva, J. I. et al. The lung mucosa environment in the elderly increases host susceptibility to Mycobacterium tuberculosis infection. J. Infect. Dis. 220, 514–523 (2019).
Li, H. & Javid, B. Antibodies and tuberculosis: finally coming of age? Nat. Rev. Immunol. 18, 591–596 (2018).
Irvine, E. B. et al. Robust IgM responses following intravenous vaccination with bacille Calmette-Guérin associate with prevention of Mycobacterium tuberculosis infection in macaques. Nat. Immunol. 22, 1515–1523 (2021). This article suggests that intravenous BCG mediates protection against M. tuberculosis through humoral immunity, with IgM titres correlating with reduced bacterial burden.
Cambier, C. J., Banik, S. M., Buonomo, J. A. & Bertozzi, C. R. Spreading of a mycobacterial cell-surface lipid into host epithelial membranes promotes infectivity. Elife https://doi.org/10.7554/eLife.60648 (2020).
Khan, H. S. et al. Identification of scavenger receptor B1 as the airway microfold cell receptor for Mycobacterium tuberculosis. Elife https://doi.org/10.7554/eLife.52551 (2020).
Nair, V. R. et al. Microfold cells actively translocate Mycobacterium tuberculosis to initiate infection. Cell Rep. 16, 1253–1258 (2016).
Hu, G. & Christman, J. W. Editorial: alveolar macrophages in lung inflammation and resolution. Front. Immunol. 10, 2275 (2019).
Hussell, T. & Bell, T. J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14, 81–93 (2014).
Lavalett, L. et al. Alveolar macrophages from tuberculosis patients display an altered inflammatory gene expression profile. Tuberculosis 107, 156–167 (2017).
Rothchild, A. C. et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aaw6693 (2019).
Lafuse, W. P. et al. Identification of an increased alveolar macrophage subpopulation in old mice that displays unique inflammatory characteristics and is permissive to Mycobacterium tuberculosis infection. J. Immunol. 203, 2252–2264 (2019).
Leemans, J. C. et al. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166, 4604–4611 (2001).
Dunlap, M. D. et al. A novel role for C-C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. 11, 1727–1742 (2018).
Corleis, B. et al. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell. Microbiol. 14, 1109–1121 (2012).
Filio-Rodríguez, G. et al. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a guinea pig model. Innate Immun. 23, 625–637 (2017).
Kimmey, J. M. et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569 (2015).
Lovewell, R. R., Baer, C. E., Mishra, B. B., Smith, C. M. & Sassetti, C. M. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. 14, 229–241 (2021).
Mishra, B. B. et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2, 17072 (2017).
Repasy, T. et al. Bacillary replication and macrophage necrosis are determinants of neutrophil recruitment in tuberculosis. Microbes Infect. 17, 564–574 (2015).
Dallenga, T. et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 22, 519–530.e3 (2017).
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010). The authors show that the transcriptional signature of active TB in humans is dominated by a neutrophil-driven type I interferon response.
Blomgran, R. & Ernst, J. D. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 186, 7110–7119 (2011).
Samstein, M. et al. Essential yet limited role for CCR2+ inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. Elife 2, e01086 (2013).
Wolf, A. J. et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179, 2509–2519 (2007).
Lai, R. et al. CD11b+ dendritic cell–mediated anti–Mycobacterium tuberculosis Th1 activation is counterregulated by CD103+ dendritic cells via IL-10. J. Immunol. 200, 1746–1760 (2018).
Bohrer, A. C. et al. Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J. Exp. Med. https://doi.org/10.1084/jem.20210469 (2021).
Papp, A. C. et al. AmpliSeq transcriptome analysis of human alveolar and monocyte-derived macrophages over time in response to Mycobacterium tuberculosis infection. PLoS ONE 13, e0198221 (2018).
Cambier, C. J. et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222 (2014).
Khan, N. et al. M. tuberculosis reprograms hematopoietic stem cells to limit myelopoiesis and impair trained immunity. Cell 183, 752–770.e22 (2020). This study demonstrates that M. tuberculosis undermines a protective trained immune response, reprograming haematopoietic stem cells by eliciting type I interferon.
Srivastava, S. & Ernst, J. D. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191, 1016–1020 (2013). This study illustrates the requirement for direct contact between infected macrophages and M. tuberculosis-specific CD4+ T cells for their effector functions.
Simmons, J. D. et al. Monocyte metabolic transcriptional programs associate with resistance to tuberculin skin test/interferon-γ release assay conversion. J. Clin. Invest. https://doi.org/10.1172/JCI140073 (2021).
Delahaye, J. L. et al. Cutting edge: bacillus Calmette–Guérin–induced T cells shape Mycobacterium tuberculosis infection before reducing the bacterial burden. J. Immunol. 203, 807–812 (2019).
Khader, S. A. et al. Targeting innate immunity for tuberculosis vaccination. J. Clin. Invest. 129, 3482–3491 (2019).
Kinsella, R. L. et al. Perspectives and advances in the understanding of tuberculosis. Annu. Rev. Pathol. 16, 377–408 (2021).
Lugo-Villarino, G. et al. The C-type lectin receptor DC-SIGN has an anti-inflammatory role in human M(IL-4) macrophages in response to Mycobacterium tuberculosis. Front. Immunol. 9, 1123 (2018).
Rajaram, M. V. S. et al. M. tuberculosis-initiated human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcRγ-chain, Grb2, and SHP-1. Cell Rep. 21, 126–140 (2017).
Lu, L. L. et al. A functional role for antibodies in tuberculosis. Cell 167, 433–443.e14 (2016). This study compares antibodies from patients with active TB to antibodies from patients with latent TB and demonstrates that the antibodies have different glycosylation and functional profiles, suggesting their potential role in mediating infection control.
Weiss, G. & Schaible, U. E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 264, 182–203 (2015).
Shi, L. et al. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci. Rep. 5, 18176 (2015).
Marino, S. et al. Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect. Immun. 83, 324–338 (2015).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013).
Mills, E. L. et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 (2016).
Lampropoulou, V. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 (2016).
Nair, S. et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J. Exp. Med. 215, 1035–1045 (2018).
Ma, J. et al. The roles of inflammasomes in host defense against Mycobacterium tuberculosis. Pathogens https://doi.org/10.3390/pathogens10020120 (2021).
Mishra, B. B. et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat. Immunol. 14, 52–60 (2013).
Mayer-Barber, K. D. et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103 (2014).
Collins, J. M. et al. TCA cycle remodeling drives proinflammatory signaling in humans with pulmonary tuberculosis. PLoS Pathog. 17, e1009941 (2021).
Roca, F. J. & Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153, 521–534 (2013). This study demonstrates the mechanism by which TNF, a host- protective cytokine, can also induce inflammation and enhance TB susceptibility.
Peyron, P. et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4, e1000204 (2008).
Ouimet, M. et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 17, 677–686 (2016).
Kim, Y. S. et al. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 198, 3283–3295 (2017).
Knight, M., Braverman, J., Asfaha, K., Gronert, K. & Stanley, S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-γ/HIF-1α signaling and supports host defense. PLoS Pathog. 14, e1006874 (2018).
Wilburn, K. M., Fieweger, R. A. & VanderVen, B. C. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathog. Dis. https://doi.org/10.1093/femspd/fty021 (2018).
Armstrong, J. A. & Hart, P. D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134, 713–740 (1971).
Augenstreich, J. & Briken, V. Host cell targets of released lipid and secreted protein effectors of Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 10, 595029 (2020).
Upadhyay, S., Mittal, E. & Philips, J. A. Tuberculosis and the art of macrophage manipulation. Pathog. Dis. https://doi.org/10.1093/femspd/fty037 (2018).
Pradhan, G., Shrivastva, R. & Mukhopadhyay, S. Mycobacterial PknG targets the Rab7l1 signaling pathway to inhibit phagosome-lysosome fusion. J. Immunol. 201, 1421–1433 (2018).
Wang, J. et al. Mycobacterium tuberculosis protein kinase G acts as an unusual ubiquitinating enzyme to impair host immunity. EMBO Rep. 22, e52175 (2021).
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D. & McKinney, J. D. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302 (2004).
Köster, S. et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl Acad. Sci. USA 114, E8711–E8720 (2017).
Sun, J. et al. Mycobacterium tuberculosis nucleoside diphosphate kinase inactivates small GTPases leading to evasion of innate immunity. PLoS Pathog. 9, e1003499 (2013).
Srivastava, S., Battu, M. B., Khan, M. Z., Nandicoori, V. K. & Mukhopadhyay, S. Mycobacterium tuberculosis PPE2 protein interacts with p67 phox and inhibits reactive oxygen species production. J. Immunol. 203, 1218–1229 (2019).
Miller, J. L., Velmurugan, K., Cowan, M. J. & Briken, V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 6, e1000864 (2010).
Rivera-Calzada, A., Famelis, N., Llorca, O. & Geibel, S. Type VII secretion systems: structure, functions and transport models. Nat. Rev. Microbiol. 19, 567–584 (2021).
van der Wel, N. et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298 (2007).
Augenstreich, J. et al. ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol. https://doi.org/10.1111/cmi.12726 (2017).
Barczak, A. K. et al. Systematic, multiparametric analysis of Mycobacterium tuberculosis intracellular infection offers insight into coordinated virulence. PLoS Pathog. 13, e1006363 (2017).
Quigley, J. et al. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. mBio https://doi.org/10.1128/mBio.00148-17 (2017).
Lerner, T. R. et al. Phthiocerol dimycocerosates promote access to the cytosol and intracellular burden of Mycobacterium tuberculosis in lymphatic endothelial cells. BMC Biol. 16, 1 (2018).
Amaral, E. P. et al. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J. Exp. Med. 216, 556–570 (2019).
Shah, S. et al. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-β and AIM2 inflammasome-dependent IL-1β production via its ESX-1 secretion system. J. Immunol. 191, 3514–3518 (2013).
Rastogi, S., Ellinwood, S., Augenstreich, J., Mayer-Barber, K. D. & Briken, V. Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase PknF. PLoS Pathog. 17, e1009712 (2021).
Mittal, E. et al. Mycobacterium tuberculosis type VII secretion system effectors differentially impact the ESCRT endomembrane damage response. mBio https://doi.org/10.1128/mBio.01765-18 (2018).
Sakowski, E. T. et al. Ubiquilin 1 promotes IFN-γ-induced xenophagy of Mycobacterium tuberculosis. PLoS Pathog. 11, e1005076 (2015).
Bell, S. L., Lopez, K. L., Cox, J. S., Patrick, K. L. & Watson, R. O. Galectin-8 senses phagosomal damage and recruits selective autophagy adapter TAX1BP1 to control. mBio 12, e0187120 (2021).
Watson, R. O., Manzanillo, P. S. & Cox, J. S. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815 (2012).
Wiens, K. E. & Ernst, J. D. The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. 12, e1005809 (2016).
Strong, E. J. et al. Identification of autophagy-inhibiting factors of Mycobacterium tuberculosis by high-throughput loss-of-function screening. Infect. Immun. https://doi.org/10.1128/IAI.00269-20 (2020).
Saini, N. K. et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 1, 16133 (2016).
Lerner, T. R. et al. Mycobacterium tuberculosis cords within lymphatic endothelial cells to evade host immunity. JCI Insight https://doi.org/10.1172/jci.insight.136937 (2020).
Chandra, P. et al. Inhibition of fatty acid oxidation promotes macrophage control of Mycobacterium tuberculosis. mBio https://doi.org/10.1128/mBio.01139-20 (2020).
Baker, J. J., Dechow, S. J. & Abramovitch, R. B. Acid fasting: modulation of Mycobacterium tuberculosis metabolism at acidic pH. Trends Microbiol. 27, 942–953 (2019).
Buter, J. et al. Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat. Chem. Biol. 15, 889–899 (2019).
Behar, S. M. et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 4, 279–287 (2011).
Martin, C. J. et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12, 289–300 (2012).
Srinivasan, L. et al. Identification of a transcription factor that regulates host cell exit and virulence of Mycobacterium tuberculosis. PLoS Pathog. 12, e1005652 (2016).
Sun, J. et al. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678 (2015).
Pajuelo, D. et al. Toxin secretion and trafficking by Mycobacterium tuberculosis. Nat. Commun. 12, 6592 (2021).
Izquierdo Lafuente, B., Ummels, R., Kuijl, C., Bitter, W. & Speer, A. Mycobacterium tuberculosis toxin CpnT is an ESX-5 substrate and requires three type VII secretion systems for intracellular secretion. mBio https://doi.org/10.1128/mBio.02983-20 (2021).
Zhang, L., Jiang, X., Pfau, D., Ling, Y. & Nathan, C. F. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J. Exp. Med. https://doi.org/10.1084/jem.20200887 (2021).
Elkington, P. et al. In vitro granuloma models of tuberculosis: potential and challenges. J. Infect. Dis. 219, 1858–1866 (2019).
Dorhoi, A. et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur. J. Immunol. 42, 374–384 (2012).
Caruso, A. M. et al. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162, 5407–5416 (1999).
Flynn, J. L. et al. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995).
Chávez-Galán, L. et al. Tuberculosis patients display a high proportion of CD8+ T cells with a high cytotoxic potential. Microbiol. Immunol. 63, 316–327 (2019).
Lu, Y. J. et al. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 36, 109696–109696 (2021).
Watson, A. et al. Human antibodies targeting a Mycobacterium transporter protein mediate protection against tuberculosis. Nat. Commun. 12, 602 (2021).
Marin, N. D., Dunlap, M. D., Kaushal, D. & Khader, S. A. Friend or foe: the protective and pathological roles of inducible bronchus-associated lymphoid tissue in pulmonary diseases. J. Immunol. 202, 2519–2526 (2019).
Flynn, J. L. et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254 (1993).
Schnettger, L. et al. A Rab20-dependent membrane trafficking pathway controls M. tuberculosis replication by regulating phagosome spaciousness and integrity. Cell Host Microbe 21, 619–628.e5 (2017).
MacMicking, J. D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12, 367–382 (2012).
Mitchell, G. & Isberg, R. R. Innate immunity to intracellular pathogens: balancing microbial elimination and inflammation. Cell Host Microbe 22, 166–175 (2017).
Fabri, M. et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci. Transl. Med. 3, 104ra102 (2011).
Kaufmann, E. et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176–190.e19 (2018). This study shows that BCG in the bone marrow reprogrammes haematopoietic stem cells, generating trained immunity of macrophages that can be harnessed to improve vaccine-elicited protection against TB.
Sallin, M. A. et al. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 18, 3091–3104 (2017).
Sakai, S. et al. CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog. 12, e1005667 (2016).
Tzelepis, F. et al. Mitochondrial cyclophilin D regulates T cell metabolic responses and disease tolerance to tuberculosis. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aar4135 (2018).
Gallegos, A. M. et al. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS Pathog. 7, e1002052 (2011). This study challenges the long-held belief that CD4+ T cell effector function is entirely mediated by interferon-γ in TB.
Sallin, M. A. et al. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol. 3, 1198–1205 (2018). This study identifies CD153 as a marker of protective interstitial CD4+ T cells and demonstrates that it is required for M. tuberculosis control in the lungs.
Du Bruyn, E. et al. Mycobacterium tuberculosis-specific CD4 T cells expressing CD153 inversely associate with bacterial load and disease severity in human tuberculosis. Mucosal Immunol. 14, 491–499 (2021).
Morgan, J. et al. Classical CD4 T cells as the cornerstone of antimycobacterial immunity. Immunol. Rev. 301, 10–29 (2021).
Ankley, L., Thomas, S. & Olive, A. J. Fighting persistence: how chronic infections with mycobacterium tuberculosis evade T cell-mediated clearance and new strategies to defeat them. Infect. Immun. https://doi.org/10.1128/IAI.00916-19 (2020).
Srivastava, S., Grace, P. S. & Ernst, J. D. Antigen export reduces antigen presentation and limits T cell control of M. tuberculosis. Cell Host Microbe 19, 44–54 (2016).
Georgieva, M., Sia, J. K., Bizzell, E., Madan-Lala, R. & Rengarajan, J. Mycobacterium tuberculosis GroEL2 modulates dendritic cell responses. Infect. Immun. https://doi.org/10.1128/IAI.00387-17 (2018).
Portal-Celhay, C. et al. Mycobacterium tuberculosis EsxH inhibits ESCRT-dependent CD4+ T-cell activation. Nat. Microbiol. 2, 16232 (2016).
Pinto, R. et al. Influence of phthiocerol dimycocerosate on CD4+ T cell priming and persistence during Mycobacterium tuberculosis infection. Tuberculosis 99, 25–30 (2016).
Blomgran, R., Desvignes, L., Briken, V. & Ernst, J. D. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11, 81–90 (2012).
Griffiths, K. L. et al. Targeting dendritic cells to accelerate T-cell activation overcomes a bottleneck in tuberculosis vaccine efficacy. Nat. Commun. 7, 13894 (2016).
Athman, J. J. et al. Membrane vesicles inhibit T cell activation. J. Immunol. 198, 2028–2037 (2017).
Woodworth, J. S. et al. Mycobacterium tuberculosis directs immunofocusing of CD8+ T cell responses despite vaccination. J. Immunol. 186, 1627–1637 (2011).
Yang, J. D. et al. Mycobacterium tuberculosis-specific CD4+ and CD8+ T cells differ in their capacity to recognize infected macrophages. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1007060 (2018).
Bold, T. D., Banaei, N., Wolf, A. J. & Ernst, J. D. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 7, e1002063 (2011).
Moguche, A. O. et al. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe 21, 695–706.e5 (2017).
Tameris, M. D. et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet 381, 1021–1028 (2013).
Sutiwisesak, R. et al. A natural polymorphism of Mycobacterium tuberculosis in the esxH gene disrupts immunodomination by the TB10.4-specific CD8 T cell response. PLoS Pathog. 16, e1009000 (2020). This study illustrates the immunofocusing of CD8+ T cells by M. tuberculosis antigens, supporting the idea that immunodominant antigens may serve as decoys.
Carpenter, S. M., Nunes-Alves, C., Booty, M. G., Way, S. S. & Behar, S. M. A higher activation threshold of memory CD8+ T cells has a fitness cost that is modified by TCR affinity during tuberculosis. PLoS Pathog. 12, e1005380 (2016).
Ganchua, S. K. C. et al. Lymph nodes are sites of prolonged bacterial persistence during Mycobacterium tuberculosis infection in macaques. PLoS Pathog. 14, e1007337 (2018).
Gallegos, A. M. et al. Control of T cell antigen reactivity via programmed TCR downregulation. Nat. Immunol. 17, 379–386 (2016).
Woodworth, J. S. et al. Protective CD4 T cells targeting cryptic epitopes of Mycobacterium tuberculosis resist infection-driven terminal differentiation. J. Immunol. 192, 3247–3258 (2014).
Cadena, A. M. et al. Concurrent infection with Mycobacterium tuberculosis confers robust protection against secondary infection in macaques. PLoS Pathog. 14, e1007305 (2018).
Lindestam Arlehamn, C. S. et al. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog. 9, e1003130 (2013).
Cronan, M. R. et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity 45, 861–876 (2016).
Cronan, M. R. et al. A non-canonical type 2 immune response coordinates tuberculous granuloma formation and epithelialization. Cell 184, 1757–1774 e1714 (2021).
McCaffrey, E. F. et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat. Immunol. 23, 318–329 (2022).
Gautam, U. S. et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 115, E62–E71 (2018). This study reports that host tryptophan metabolites generated by indoleamine 2,3-dioxygenase contribute to the immunosuppressive role of granulomas; by inhibiting indoleamine 2,3-dioxygenase, it is possible to alter granuloma architecture and enhance immune control of M. tuberculosis in NHPs.
Gern, B. H. et al. TGFβ restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe 29, 594–606.e6 (2021). This study identifies TGFβ as a potent suppressor of antigen sensing and interferon-γ production within granulomas, the inhibition of which reduces bacterial burdens.
Millar, J. A. et al. Spatial organization and recruitment of non-specific T cells may limit T cell-macrophage interactions within Mycobacterium tuberculosis granulomas. Front. Immunol. 11, 613638 (2021).
Moreira-Teixeira, L. et al. T cell-derived IL-10 impairs host resistance to Mycobacterium tuberculosis infection. J. Immunol. 199, 613–623 (2017).
de la Barrera, S. et al. IL-10 down-regulates costimulatory molecules on Mycobacterium tuberculosis-pulsed macrophages and impairs the lytic activity of CD4 and CD8 CTL in tuberculosis patients. Clin. Exp. Immunol. 138, 128–138 (2004).
Carow, B. et al. Spatial and temporal localization of immune transcripts defines hallmarks and diversity in the tuberculosis granuloma. Nat. Commun. 10, 1823 (2019).
Gern, B. H. et al. TGFbeta restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe 29, 594–606.e6 (2021).
Martin, C. J. et al. Digitally barcoding mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. mBio https://doi.org/10.1128/mBio.00312-17 (2017).
Lin, P. L. et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med. 20, 75–79 (2014). This study demonstrates the heterogeneity of lung lesions in NHPs, with some generating sterilizing immunity, whereas other lesions support high bacterial burden within the same animal.
Sarathy, J. P. & Dartois, V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin. Microbiol. Rev. https://doi.org/10.1128/CMR.00159-19 (2020).
Veatch, A. V. & Kaushal, D. Opening Pandora’s box: mechanisms of Mycobacterium tuberculosis resuscitation. Trends Microbiol. 26, 145–157 (2018).
Zak, D. E. et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet 387, 2312–2322 (2016). When adolescents with latent TB were followed up prospectively, a transcriptional signature that correlated with risk of progression to active TB was apparent in whole blood in the 12 months preceding a clinical diagnosis of active TB.
Zhang, G. et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 9, 85 (2018).
Kana, B. D. & Mizrahi, V. Resuscitation-promoting factors as lytic enzymes for bacterial growth and signaling. FEMS Immunol. Med. Microbiol. 58, 39–50 (2010).
Kaplan, G. et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 71, 7099–7108 (2003).
Kwan, C. K. & Ernst, J. D. HIV and tuberculosis: a deadly human syndemic. Clin. Microbiol. Rev. 24, 351–376 (2011).
Urbanowski, M. E. et al. Repetitive aerosol exposure promotes cavitary tuberculosis and enables screening for targeted inhibitors of extensive lung destruction. J. Infect. Dis. 218, 53–63 (2018).
Comas, I. et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42, 498–503 (2010).
Sigal, G. B. et al. Biomarkers of tuberculosis severity and treatment effect: a directed screen of 70 host markers in a randomized clinical trial. EBioMedicine 25, 112–121 (2017).
Verma, S. et al. Transmission phenotype of Mycobacterium tuberculosis strains is mechanistically linked to induction of distinct pulmonary pathology. PLoS Pathog. 15, e1007613 (2019).
Nebenzahl-Guimaraes, H. et al. Transmissible Mycobacterium tuberculosis strains share genetic markers and immune phenotypes. Am. J. Respir. Crit. Care Med. 195, 1519–1527 (2017).
Garcia-Vilanova, A., Chan, J. & Torrelles, J. B. Underestimated manipulative roles of Mycobacterium tuberculosis cell envelope glycolipids during infection. Front. Immunol. 10, 2909 (2019).
Pfrommer, E. et al. Enhanced tenacity of mycobacterial aerosols from necrotic neutrophils. Sci. Rep. 10, 9159 (2020).
Ruhl, C. R. et al. Mycobacterium tuberculosis sulfolipid-1 activates nociceptive neurons and induces cough. Cell 181, 293–305.e11 (2020). This study is the first to show that an M. tuberculosis glycolipid directly activates neurons and stimulates cough in a guinea pig model to aid pathogen dissemination.
Chen, T. et al. Capsular glycan recognition provides antibody-mediated immunity against tuberculosis. J. Clin. Invest. 130, 1808–1822 (2020).
Darrah, P. A. et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 577, 95–102 (2020). This study explores the association between BCG vaccine efficacy and the route of administration, finding that intravenous BCG administration elicits unprecedented protection against M. tuberculosis infection.
Lu, L. L. et al. IFN-γ-independent immune markers of Mycobacterium tuberculosis exposure. Nat. Med. 25, 977–987 (2019).
Böhme, J. et al. Metformin enhances anti-mycobacterial responses by educating CD8+ T-cell immunometabolic circuits. Nat. Commun. 11, 5225 (2020).
Liu, F. et al. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat. Commun. 9, 4295 (2018).
Napier, R. J. et al. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 10, 635 (2011).
Parihar, S. P. et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 209, 754–763 (2014).
Plumlee, C. R. et al. Ultra-low dose aerosol infection of mice with Mycobacterium tuberculosis more closely models human tuberculosis. Cell Host Microbe 29, 68–82.e5 (2021).
Pichugin, A. V., Yan, B. S., Sloutsky, A., Kobzik, L. & Kramnik, I. Dominant role of the sst1 locus in pathogenesis of necrotizing lung granulomas during chronic tuberculosis infection and reactivation in genetically resistant hosts. Am. J. Pathol. 174, 2190–2201 (2009).
Smith, C. M. et al. Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife https://doi.org/10.7554/eLife.74419 (2022).
Kurtz, S. L. et al. The diversity outbred mouse population is an improved animal model of vaccination against tuberculosis that reflects heterogeneity of protection. mSphere https://doi.org/10.1128/mSphere.00097-20 (2020).
Guler, R. et al. Targeting molecular inflammatory pathways in granuloma as host-directed therapies for tuberculosis. Front. Immunol. 12, 733853 (2021).
Dallenga, T. et al. Targeting neutrophils for host-directed therapy to treat tuberculosis. Int. J. Med. Microbiol. 308, 142–147 (2018).
Divangahi, M. et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat. Immunol. 22, 2–6 (2021).
Fanucchi, S. et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet. 51, 138–150 (2019).
Arts, R. J. W. et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 17, 2562–2571 (2016).
Cheng, S. C. et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684 (2014).
van Puffelen, J. H. et al. Trained immunity as a molecular mechanism for BCG immunotherapy in bladder cancer. Nat. Rev. Urol. 17, 513–525 (2020).
O’Neill, L. A. J. & Netea, M. G. BCG-induced trained immunity: can it offer protection against COVID-19? Nat. Rev. Immunol. 20, 335–337 (2020).
Bekkering, S. et al. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 34, 1731–1738 (2014).
Kaufmann, E. et al. BCG vaccination provides protection against IAV but not SARS-CoV-2. Cell Rep. 38, 110502 (2022).
Cirovic, B. et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe 28, 322–334.e5 (2020).
World Health Organization. Global Tuberculosis Report 2018. (World Health Organization, 2018).
Goletti, D., Lee, M. R., Wang, J. Y., Walter, N. & Ottenhoff, T. H. M. Update on tuberculosis biomarkers: from correlates of risk, to correlates of active disease and of cure from disease. Respirology 23, 455–466 (2018).
Walzl, G. et al. Tuberculosis: advances and challenges in development of new diagnostics and biomarkers. Lancet Infect. Dis. 18, e199–e210 (2018).
Diray-Arce, J. et al. Integrative metabolomics to identify molecular signatures of responses to vaccines and infections. Metabolites https://doi.org/10.3390/metabo10120492 (2020).
Chandra, P., Coullon, H., Agarwal, M., Goss, C. W. & Philips, J. A. Macrophage global metabolomics identifies cholestenone as host/pathogen cometabolite present in human Mycobacterium tuberculosis infection. J. Clin. Invest. https://doi.org/10.1172/JCI152509 (2022).
Day, C. L. et al. PD-1 expression on Mycobacterium tuberculosis-specific CD4 T cells is associated with bacterial load in human tuberculosis. Front. Immunol. 9, 1995 (2018).
Roy Chowdhury, R. et al. A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature 560, 644–648 (2018).
Carvalho, A. C. C. et al. Pre-treatment neutrophil count as a predictor of antituberculosis therapy outcomes: a multicenter prospective cohort study. Front. Immunol. 12, 661934 (2021).
Acknowledgements
This work was funded by the US National Institutes of Health (R01 AI087682, R01 AI130454, R21 AI155380 and R21 AI160386 to J.A.P.; F31 AI152321 to S.J.G.). The authors thank members of the Philips laboratory for helpful discussions.
Author information
Authors and Affiliations
Contributions
J.A.P. conceived the Review; P.C. and S.J.G. contributed substantially to discussion of the content; P.C., S.J.G. and J.A.P. contributed equally to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Microbiology thanks Samuel Behar; Volker Briken, who co-reviewed with Shivangi Rastogi; and Maziar Divangahi for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Cavitary lung disease
-
A pathological process in the lungs that results in gas-filled spaces, which are typically thick-walled and arise in areas of consolidation, mass or nodule.
- Inflammageing
-
Age-associated chronic, sterile and low-grade inflammation that leads to disease exacerbation.
- Opsonization
-
The coating of microorganisms by components of the immune system such as antibodies and complement proteins to facilitate their uptake and elimination by phagocytes.
- Clodronate
-
A bisphosphonate that, when encapsulated in liposomes, is preferentially taken up by macrophages, where it accumulates and causes cell death.
- Myelopoiesis
-
The process in which mature myeloid cells such as macrophages and neutrophils differentiate from myeloid progenitors that arise from haematopoietic stem cells.
- Atherosclerosis
-
A disease of arteries that is characterized by deposition of plaque, which is composed of fats, cholesterol and lipid-laden macrophages, on the artery wall.
- Paucibacillary disease
-
Tuberculosis with such a low number of bacilli that they are not apparent on smear microscopy.
- Metformin
-
A medication that is used to treat type 2 diabetes mellitus and that is being evaluated as a host-directed therapy for tuberculosis.
Rights and permissions
About this article

Cite this article
Chandra, P., Grigsby, S.J. & Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol 20, 750–766 (2022). https://doi.org/10.1038/s41579-022-00763-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-022-00763-4
This article is cited by
-
circRNA_SLC8A1 promotes the survival of mycobacterium tuberculosis in macrophages by upregulating expression of autophagy-related protein SQSTM1/p62 to activate the NF-κB pathway
Scientific Reports (2024)
-
Interrogating the roles of lymph node metastasis in systemic immune surveillance
Clinical & Experimental Metastasis (2024)
-
Mycobacterium marinum mediates regulation of prostaglandin E2 expression on host immune response through cyclooxygenase pathway
Molecular Biology Reports (2024)
-
From immunology to artificial intelligence: revolutionizing latent tuberculosis infection diagnosis with machine learning
Military Medical Research (2023)
-
Mutational spectra are associated with bacterial niche
Nature Communications (2023)
