
Abstract
The COVID-19 pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is an unprecedented global health crisis. However, therapeutic options for treatment are still very limited. The development of drugs that target vital proteins in the viral life cycle is a feasible approach for treating COVID-19. Belonging to the subfamily Orthocoronavirinae with the largest RNA genome, SARS-CoV-2 encodes a total of 29 proteins. These non-structural, structural and accessory proteins participate in entry into host cells, genome replication and transcription, and viral assembly and release. SARS-CoV-2 proteins can individually perform essential physiological roles, be components of the viral replication machinery or interact with numerous host cellular factors. In this Review, we delineate the structural features of SARS-CoV-2 from the whole viral particle to the individual viral proteins and discuss their functions as well as their potential as targets for therapeutic interventions.
Similar content being viewed by others

Introduction
Coronaviruses are enveloped viruses that possess a positive-sense single-stranded RNA genome 26–32 kb in length1. Coronaviruses belong to the Coronaviridae subfamily Orthocoronavirinae. According to variations in the genome sequence and serological reactions, coronavirus members in the subfamily are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus2. Among them, Betacoronavirus is classified into five subgenera. Although infectious bronchitis virus was the first coronavirus isolated in chicken embryos in 1937 (ref.3), it was not until the 1960s that these viruses, particularly the human respiratory coronaviruses4, were characterized by electron microscopy. This subfamily of viruses has a unique structural feature on their surfaces which resembles a solar corona. This feature arises due to the presence of spike proteins on the virion surface.
Coronaviruses are characterized by high genetic recombination and mutation rates, which result in their ecological diversity5. They are able to infect and readily adapt to a wide range of hosts, from birds to whales. Seven coronaviruses have been found to infect humans. Human coronaviruses 229E, OC43, NL63 and HKU1 are responsible for 10–30% of upper respiratory tract infections annually, characterized by mild respiratory illnesses, such as the common cold6. By contrast, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus7 and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are able to cause severe human respiratory diseases, potentially resulting in high mortality. In 2002–2003, SARS-CoV resulted in 8,096 reported cases and 774 deaths (case–fatality rate of ~10%)7. By the end of January 2020, 2,500 cases of Middle East respiratory syndrome and more than 800 associated deaths (case–fatality rate ~34%) were reported worldwide8. In late December 2019, clustered cases of a severe pneumonia were reported, and the aetiological agent was isolated and identified as a novel betacoronavirus, named SARS-CoV-2, that shares ~80% similarity in genome sequence with SARS-CoV9. SARS-CoV-2 causes COVID-19, with symptoms including fever, cough, fatigue, nausea and shortness of breath10. To date, there have been more than 160 million confirmed COVID-19 cases and more than 3 million related deaths worldwide11.
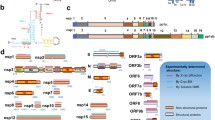
To date, there has been a lack of effective therapies to treat COVID-19. Due to the rampant and continuous spread of COVID-19, it is a matter of urgency to identify and characterize drug and vaccine targets for SARS-CoV-2. The genome of SARS-CoV-2 is close to 30 kb on size, contains 14 open reading frames (ORFs) and encodes 29 viral proteins. Approximately two thirds of the 5′ end of the SARS-CoV-2 genome encodes two overlapping polyproteins: pp1a and pp1ab12. These two polyproteins are digested by two viral proteases into 16 non-structural proteins (NSPs), which are essential for viral replication and transcription (Fig. 1a). Four ORFs at the 3′ terminus of the viral genome encode a canonical set of structural proteins that include the nucleocapsid (N), spike (S) protein, membrane (M) protein and envelope (E) protein, which are responsible for virion assembly and also participate in suppression of the host immune response. A series of accessory genes, which encode accessory proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8b, ORF9b and ORF14), lie between these structural genes. The accessory proteins are involved in regulating viral infection but may not be incorporated into the virion, except for the structural proteins ORF3a and ORF7a.

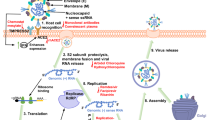
a | Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome organization, with functional domains shown in rectangles and the prime drug targets emphasized in the outlined box. The first part of the SARS-CoV-2 genomic RNA, encoding non-structural proteins (NSPs), can be directly translated into two polyproteins (a polyprotein is a single chain of polypeptides that are linked together by covalent peptide bonds), pp1a and pp1ab, in which a −1 frameshift between open reading fame 1a (ORF1a) and ORF1b leads to differences in translation. The two polyproteins are cleaved by viral proteases, a papain-like protease (PLpro) and a 3C-like protease (3CLpro), to generate 16 NSPs and to form the replication and transcription machinery. The domains that have an important function within each NSP are shown in the genome structure. The first three peptide cleavages are performed by PLpro. The remainders are cleaved by 3CLpro (also known as the main protease). The second part of the RNA genome encodes mainly four structural proteins: the spike (S) protein, the membrane (M) protein, the envelope (E) protein and the nucleocapsid (N) protein. In addition to these structural proteins, several accessory proteins are also encoded. b | The life cycle of SARS-CoV-2, including viral entry, replication and transcription, assembly and release. In the native SARS-CoV-2 structure, S proteins can have prefusion and postfusion conformations (Electron Microscopy Data Bank entries EMD-30426, EMD-30427 and EMD-30428). SARS-CoV-2 enters host cells through an endocytosis pathway mediated by S protein–angiotensin-converting enzyme 2 (ACE2) interactions. Viral RNA enters the cytoplasm after the entry step, and then ORF1a or ORF1ab is translated by the host ribosome. The viral polyproteins are cleaved into NSPs and assemble themselves into the replication and transcription complexes. Subgenomic viral mRNAs (after capping) act as templates for viral protein translation. Progeny virions are assembled in the endoplasmic reticulum and Golgi body. Afterwards, the virions are exocytosed to complete the life cycle. ERGIC, endoplasmic reticulum–Golgi intermediate compartment; ExoN, exonuclease; HEL, helicase; Mac1, macrodomain 1; NendoU, uridine-specific endoribonuclease; NiRAN, nidovirus RNA-dependent RNA polymerase-associated nucleotidyltransferase; NMT, guanine-N7-methyltransferase; OMT, 2′-O-methyltransferase; PL2, papain-like protease 2; RBD, receptor-binding domain; RdRp, RNA-dependent RNA polymerase; SUD, SARS-unique domain; +ss, positive-sense single-stranded; TM, transmembrane; Ubl1, ubiquitin-like domain 1; UTR, untranslated region.
Briefly, in the first step of the SARS-CoV-2 life cycle, the S protein on the outer surface of the virion is responsible for binding to the host receptor or receptors for attachment to the cell membrane, which is followed by viral and host cellular membrane fusion and the release of viral genomic RNA into the cells. Subsequently, host ribosomes are hijacked to produce the two viral replicase polyproteins, which can further be processed into 16 mature NSPs through two virus-encoding proteases: main protease (Mpro) and papain-like protease (PLpro). These NSPs are able to assemble into the replication and transcription complex (RTC) to initiate viral RNA replication and transcription. The genomic RNA and structural proteins then assemble into mature progeny virions, which are subsequently released through exocytosis to initiate another round of infection10 (Fig. 1b). Viral proteins can individually perform important physiological roles, constitute the viral protein machinery for specific essential events in the viral life cycle or extensively interplay with the cellular factors in the host immune response and pathogenesis13. In the following sections, we delineate the structural features of SARS-CoV-2 extending from the whole viral particle to individual proteins, including several antiviral drug targets, including the S protein, PLpro, Mpro and viral RNA-dependent RNA polymerase (RdRP)14.
Structural proteins in the viral life cycle
S protein in viral entry
The S protein is a homotrimer, which protrudes from the virion and extensively decorates the viral surface like a crown. It is heavily glycosylated, belongs to the type I membrane-protein family and is anchored in the viral membrane, where it mediates fusion of the viral membrane with the host cell membrane15. In the native state, prefusion and postfusion conformations of S proteins can be traced simultaneously on the reconstructed virions. The SARS-CoV-2 S protein comprises ~1,200 residues and can be cleaved by a furin-like protease into two functional subunits, S1 and S2, which are responsible for mediating attachment to host cells and membrane fusion, respectively16. After cleavage during viral entry into the host cells, S1 and S2 remain associated with each other through non-covalent interactions. As shown by cryogenic electron microscopy (cryo-EM) (Fig. 2a), the S1 subunit of the SARS-CoV-2 S protein wraps around a threefold axis, covering the S2 subunit underneath17. The S1 subunit contains a receptor-binding domain (RBD) and an amino-terminal (N-terminal) domain (NTD). The RBD has a five-stranded antiparallel β-sheet core, flanked on either side by a short helix. The receptor-binding motif (RBM) extends out of the core (connecting β4 and β5), taking on a cradle-like structure for receptor binding. The RBM, which is stabilized by a disulfide bond, does not possess a regular secondary structure except for two small β-sheets. The RBD can adopt two distinct conformational states: the closed ‘down’ state and the open ‘up’ state17. In the ‘down’ state, RBD angles are close to the central cavity of the trimer to shield the receptor-binding regions, while in the ‘up’ state, the RBD undergoes hinge-like conformational movement, exposing its determinant regions to recognize the human angiotensin-converting enzyme 2 (hACE2) receptor on the host cellular membrane, the state of which is considered to be less stable than in the ‘down’ state. The NTD of the S protein adopts a galectin-like fold with a sugar-binding pocket and contains a ceiling-like structure on top. The NTD may recognize sugar moieties upon initial attachment and play a significant role in the transition of the conformation of the S protein. The S2 subunit comprises four conserved structural regions: a fusion peptide, two heptad repeats (HR1 and HR2) and a transmembrane region. The HR1 region constitutes the main helical stalk of S2, whereas the HR2 region is temporarily flexible in the prefusion state. The fusion peptide forms a short hydrophobic segment.

a | Cartoon representation of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) trimeric spike protein. The receptor-binding domain (RBD), amino-terminal domain (NTD), SD1/SD2 and S2 are blue, green, pink and red, respectively. b,c | Cartoon representations of the RBD in complex with the host cell receptor angiotensin-converting enzyme 2 (ACE2). ACE2 is orange. The side chains of amino acids participating in the interactions between the spike protein RBD and ACE2 are shown as stick models. d | Cartoon representation of the spike protein NTD in complex with a neutralizing antibody. The antibody is gold. e–j | Cartoon representations of the spike protein RBD in complex with class I, II, III and IV RBD neutralizing antibodies. In part f, representative interactions between the spike protein RBD and the nanobody are shown as stick models. Antibodies can bind with the RBD despite conformational changes. Antibodies are in gold. Protein Data Bank accession codes are indicated in parentheses.
Undergoing a substantial structural rearrangement, from the metastable prefusion conformation to the postfusion conformation, the S protein fulfils its function in regulating the fusion of viral membrane with the host cell membrane18. Fusion is triggered when the S1 subunit binds to hACE2 (Fig. 2b,c). As observed in the complex structure, the N-terminal helix of hACE2 interacts with the outer surface of the RBM in the S1 subunit19,20,21,22. The interaction involves 16 residues in the RBD and 20 residues in hACE2, which forms a network consisting of 14 hydrogen bonds and one salt bridge19. The binding of hACE2 to the RBD can lock the RBD in the ‘up’ conformation and trigger S1 shedding, which is mediated by the proteolytic cleavage of host TMPRSS2 and cathepsin B or cathepsin L. Thus, three HR1 helices of trimeric S2 interact with the pairing HR2 helices and constitute a stable six-helix bundle23. In this unique helix bundle, three HR2 helices are packed into the hydrophobic grooves of the HR1-trimer core in an antiparallel manner. This conformational arrangement brings viral and host cell membranes into proximity and facilitates subsequent membrane fusion. Because of the indispensable function of the S protein, it is an attractive target for inhibition by neutralizing antibodies (nAbs), and characterization of the S protein structure provides atomic-level information for rational vaccine design.
S protein-neutralizing antibodies
nAbs targeting the SARS-CoV-2 S trimer have shown protection from viral infection in animal models and are being evaluated as therapeutics in humans. These antibodies comprise human monoclonal antibodies isolated from COVID-19 convalescent donors and single-domain antibodies (also known as nanobodies) which can bind novel epitopes, including buried cavities that are inaccessible to conventional antibodies. Determination of a number of structures of nAbs in complex with the S trimer has elucidated their modes of neutralization. Although some nAbs target the NTD or S2, most nAbs bind to the RBD, the latter of which can be further classified into four distinct classes (classes I, II, III and IV) on the basis of the nAb–RBD binding characteristics.
The nAbs in class I can bind to the RBD only in the ‘up’ state (Fig. 2e). They are expected to bind to the flat area on the top side of the cradle-like surface of the RBD, which extensively overlaps with the binding site for hACE2. Through direct competition with hACE2, nAbs in this class would produce steric hindrance when binding to RBD, blocking hACE2 attachment. CB6 (ref.24), C105 (ref.25), CV30 (ref.26), B38 (ref.27), CC12.1, CC12.3 (ref.28), PR1077 (ref.29) and P4A1 (ref.30) nAbs belong to this class. Most contain IGHV3-53- or IGHV3-66-encoded heavy chains and utilize residues in complementarity-determining regions 1, 2 and 3.
The nAbs in class II also bind to the RBD in the ‘up’ state, but exhibit no overlap with hACE2-binding sites (Fig. 2f,g). CR3022 (ref.31), EY6A32 and nanobody VHH-72 (ref.33) belong to this class. The binding region is located at the bottom of the RBD, and is spatially separated from the hACE2-binding sites. Structural analysis showed that the RBD undergoes a rotation that exposes the epitopes for these nAbs. Such a rearrangement is considered to cause a premature conversion of the S protein from the prefusion state to the postfusion state. The resulting unstable configuration of the S protein consequently inactivates SARS-CoV-2.
The nAbs in class III can bind to RBDs only in the ‘down’ conformation (Fig. 2h). They comprise Fab 2-4, Fab 2-43 (ref.34) and BD23 (ref.35). The heavy chains of the nAbs reach the RBD and interact with the cradle-like surface or the flexible ridge region. However, the binding pattern between these nAbs and the RBD is different from that for class I nAbs, according to the orientation change in the RBD, and the binding area becomes narrower. Notably, N-glycan chains are supposed to play a significant role in stabilizing the binding of class III nAbs to the ‘down’ RBD. Additionally, epitopes of some nAbs extend to the NTD, which may help to resist dynamic instability. Collectively, this binding mode would lock the RBD in the ‘down’ conformation, which also sterically hinders hACE2 access.
The nAbs in class IV can recognize both the ‘up’ RBD conformation and the ‘down’ RBD conformation (Fig. 2i,j). They comprise H11-D4, H11-H4 (ref.36), P2B-2F6 (ref.37), Ty1 (ref.38), S309 (ref.39), REGN10987 (ref.40) and P17 (ref.41). Structural studies show that these nAbs target different regions. P2B-2F6 and the nanobodies H11-D4 and H11-H4 can bind to the top cradle-like surface in a similar orientation as class III nAbs. Their binding can be further reinforced by a protruding loop on the RBD. These three nAb epitopes are largely located on the opposite side of the RBM compared with the epitopes of class I nAbs. By partially overlapping with the hACE2-binding site, these nAbs sterically block hACE2 binding to the RBD as well. S309 targets a region distinct from the RBM. Its epitope comprises the α1 helix, a section of the β1 strand and two loops formed by residues 358–361 and 333–335. RGEN10987 is another class VI nAb that binds distal to the hACE2-binding site. The binding of this nAb would spatially hinder hACE2 attachment.
4A8 (ref.42), COV57 (ref.25), 2–17, 5–24, 4–8 (ref.34) and FC05 (ref.43) are nAbs that target other parts of the S protein. Structural analysis reveals that 4A8, which shows a high level of neutralization of SARS-CoV-2, recognizes the NTD and does not sterically hinder the binding between hACE2 and the S protein (Fig. 2d). Regarding the S2 subunit, only a few targeted monoclonal antibodies have been reported. Antibody 1A9 (ref.44) has been found to interact with the S2 subunit but fails to neutralize SARS-CoV-2. In a recent report, the nAb CC40.8 was identified and found to neutralize SARS-CoV-2 and specifically recognize the S2 subunit45. The discovery of non-RBD-targeted nAbs may benefit the strategy of nAb cocktail therapeutics.
Since SARS-CoV and SARS-CoV-2 share the same host cell receptor, hACE2, development of cross-neutralizing antibodies to both coronaviruses seems feasible. H014 (ref.46) is a recently reported humanized antibody which efficiently neutralizes both SARS-CoV and SARS-CoV-2. It can recognize and interact with open RBDs, but the binding interface is located distinct from the RBM, and exhibits no competition with hACE2 attachment. Consistently, other cross-neutralizing antibodies (for example, VHH-72, ADI-56046 (ref.47), COV21 (ref.25) and CC6.33 (ref.48)) also avoid the RBM and prefer to recognize the core domain of the RBD.
It is noteworthy that SARS-CoV-2 has a high mutation rate, and numerous mutant strains (variants) have been reported. Mutations in the S protein, especially the epitopes for nAbs, would attenuate the potency of nAbs. The D614G mutation is the most commonly reported mutation in the S protein49, and results in increased infectivity and morbidity. The cryo-EM structure of the trimeric S protein with D614G demonstrated a conformational shift towards the hACE2-binding fusion-competent state49 and exhibited attenuation of efficacy in nAb binding. N501Y is a mutant variant emerging from the United Kingdom, South Africa and Brazil50. The mutation site is located at the RBD–hACE2 interface and has been experimentally shown to cause an increase in hACE2 affinity51. Other mutations worth noting include K417N and K417T, which appear in the epitopes of class I nAbs and are considered to affect the binding of class I antibodies. Mutations at residues in the NTD were also found in the new variants of concern, such as ΔY144 and Δ242–244. They were shown to abrogate neutralization of NTD-specific nAbs52,53,54. Additionally, SARS-CoV-2 with the naturally occurring mutations to E484, F490, Q493 or S494 of the S protein was found to escape from potential therapeutic antibodies such as C121 and C144 (ref.55). Combination treatment with two or more nAbs targeting distinct epitopes would be a strategy to suppress nAb escape variants.
E protein
After a coronavirus enters host cells, the E protein regulates viral lysis and the subsequent viral genome release. The E protein was found to be involved in viral assembly and budding by localizing to endoplasmic reticulum (ER) and Golgi body membranes2. Moreover, the E protein has been shown to participate in activating the host inflammasome56.
The structure of the SARS-CoV-2 E protein57 solved by nuclear magnetic resonance spectroscopy shows that it is composed of a five-helix bundle ~35 Å in length (Fig. 3b). As the E protein can function as an ion channel, the pore inside the transmembrane region is predominantly occupied by hydrophobic residues except for the N-terminal pore. Owing to non-specific interhelical interactions, the entrance site at the N terminus is a drug target for inhibitor binding. The E protein is recognized topologically to be Nlumen–Ccyto (N-terminal ER–Golgi intermediate compartment lumen and carboxy-terminal (C-terminal) cytoplasm) and involved in regulation of pumping Ca2+ out of the ER, which may lead to activation of the cellular inflammasome, thereby enhancing the host antiviral response.

a | Structures of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nucleocapsid protein. The upper panel shows a schematic representation of the domains of the nucleocapsid protein. The lower-left panel shows a cartoon representation and the electrostatic surface of the amino-terminal (N-terminal) domain. It has the shape of a right-handed fist and contains a four-stranded antiparallel β-sheet as a core subdomain. The loops protruding out of the core are positively charged. These facilitate RNA binding. The lower-right panel shows a cartoon representation and the electrostatic surface of the dimeric carboxy-terminal (C-terminal) domain. Overall, it has a rectangular shape, but each protomer displays a crescent shape. Two β-hairpin structures from each protomer form four antiparallel β-strands by inserting themselves into each cavity. A positively charged groove is found in the helix face of the dimer, which also facilitates RNA binding. b | The structure of the transmembrane domain of the envelope protein. It comprises a five-helix bundle and folds like a pipe with a channel in the middle. Protein Data Bank accession codes are indicated in parentheses.
N protein
The N protein serves as the only structural protein inside the virion. It is a crucial component that protects the viral RNA genome and packages it into a ribonucleoprotein complex. A native reconstruction of SARS-CoV-2 using electron cryotomography suggests that a significant number of ribonucleoproteins may be membrane proximal. The N protein also plays a role in antagonizing the host immune response58 and has been identified to counter cellular RNAi-mediated antiviral activities through its binding with double-stranded RNA ‘strings’59, and can be regarded as a viral suppressor of RNA silencing. The N protein has potential as a target for vaccine development because it induces a severe immune responses during infection.
The N protein has two conserved structural domains, the NTD (N-NTD) and the CTD (N-CTD), each of which is independently folded60. In the crystal structures of the N protein61, the N‐NTD exists as a monomer, whereas the N‐CTD exists as a dimer (Fig. 3a). The N‐NTD has the shape of a right‐handed fist and contains a four‐stranded antiparallel β‐sheet as a core subdomain. The loops protruding out of the core are positively charged, putatively to allow RNA binding. The N‐CTD homodimer forms a rectangular shape, with each protomer displaying a crescent shape. To stabilize the dimer interface, two β‐hairpin structures from each protomer can form four antiparallel β-strands by inserting themselves into each cavity. Compared with other coronaviruses, the N protein from SARS-CoV-2 displays different charge distributions in the N-terminal loop, the RNA protruding tip, the bottom of the N-NTD core and the N-CTD β-strand face. Hence, the variations in RNA binding to the N protein may further guide inhibitor optimization.
NSPs and inhibitors
Host translation shutdown by nsp1
nsp1 originates from the N-terminal cleavage of polypeptides pp1a and pp1ab by PLpro. The biological functions of nsp1 manifest themselves mainly in virus–host interactions to suppress host translation62,63, and thus nsp1 can be regarded as a canonical virulence factor. To hinder the host translation process, nsp1 is proposed to function by two mechanisms: the first is to bind the ribosomal 40S subunit during the initiation stage64 and the second is to induce host mRNA degradation65. Importantly, nsp1 does not impede viral protein expression while it binds to the mRNA 5′ untranslated region, leading to efficient viral translation and replication. The structure of nsp1 and the ribosomal 40S subunit has been determined to show the interactions between them and to explain the potential inhibition mechanism66. In this cryo-EM structure, the C-terminal domain of nsp1 possesses a short α-helix which is connected to a longer α-helix through a short loop (Fig. 4a). Thus, the host mRNA entry channel is blocked by nsp1 insertion. This hypothesis is corroborated by the loss of host translation inhibition in the K164A–H165A double mutant. The long α-helix also contributes to the interactions between nsp1 and the ribosome. Through the shutdown of host translation, especially antiviral factors, nsp1 assists in evading immune defences, which suggests that disrupting nsp1–ribosome interactions is a plausible approach for SARS-CoV-2 drug discovery.

a | Surface representation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nsp1 in complex with the host ribosomal 40S subunit. A cartoon representation of the interaction pattern between nsp1 and the ribosomal 40S subunit is shown in the inset. nsp1, ribosomal proteins us3 and us5, and h18 ribosomal RNA (rRNA) are coloured violet, blue, green and yellow, respectively. b | Schematic representation of functional domains of nsp3. Cartoon representations of the three solved structures are shown below. c | Cartoon representation of papain-like protease (PLpro) in complex with different inhibitors. The left, middle and right panels show the inhibitors VIR250, GRL0617, and YM155, respectively. All inhibitors are presented as stick models. d | Cartoon representation of PLpro in complex with interferon-stimulated gene product 15 (ISG15). PLpro and ISG15 are coloured blue and yellow, respectively. Protein Data Bank accession codes are indicated in parentheses. Ac, acidic domain; HVR, hypervariable region; MR, marker domain; RBD, RNA-binding domain. SUD, severe acute respiratory syndrome-unique domain; TM, transmembrane.
Multidomain protein nsp3
nsp3 consists of 10–16 domains depending on the coronavirus genus. Eight are present in all coronaviruses, including ubiquitin-like domain 1 (Ubl1), a hypervariable region, a macrodomain, ubiquitin-like domain 2 (Ubl2), a PLpro, a zinc-finger domain, a Y1 domain and a CoV-Y domain67. Most of the conserved domains perform essential functions in the life cycle of the virus. The macrodomains possess highly conserved structures and similar functions. Macrodomain Mac1 can cleave the phosphate group of ADP-ribose 1-phosphate and reverse protein ADP-ribosylation by hydrolysis. The core structure of Mac1 contains seven β-strands flanked by six α-helices (Fig. 4b). ADP-ribose interacts with the Mac1 hydrophobic cleft through conserved hydrogen bonds68. This indicates that compounds targeting Mac1 may have broad-spectrum antiviral activities.
The ‘SARS-unique domain’ (SUD) participates in virus–host interactions. SUD has three subdomains: SUD-N (Mac2), SUD-M (Mac3) and SUD-C (DPUP). SUD-N and SUD-M adopt a macrodomain fold, whereas SUD-C has a frataxin‐like fold. Deletion of Mac2 decreases the viral replication rate to 65–70%, whereas Mac3 is indispensable for replication activity69. PAIP1, which is a component of the eukaryotic translation machinery, has been identified to interact with SUD. The structure of the Mac2–PAIP1M (middle domain of PAIP1) complex shows that Mac2 displays a typical α/β/α macrodomain fold, whereas PAIP1M adopts a HEAT repeat fold70. Strong complementarity which enhances complex stability is observed at the interface. This structure also supports the suggestion that Mac2–PAIP1M participates in regulating viral mRNA translation and is thus a good antiviral drug target.
PLpro is located in nsp3 between SUD and a nucleic acid-binding domain. It cleaves the viral polyprotein precursors pp1a and pp1ab at three sites to produce NSPs nsp1, nsp2 and nsp3 (ref.71). Apart from viral polyproteins, PLpro can also cleave host proteins to antagonize the innate immune response72. It preferentially recognizes and cleaves interferon-stimulated gene product 15 (ISG15) from interferon regulatory factor 3 (IRF3) and attenuates type I interferon responses, facilitating escape of the virus from the immune system73. PLpro is a 36-kDa cysteine protease with a catalytic triad71. It contains an N-terminal ubiquitin-like domain and a catalytic core domain74. The catalytic core domain comprises three subdomains, the thumb, palm and fingers, which together fold like an open right hand. The thumb subdomain is composed of four α-helices, whereas the palm is formed by a six-stranded β-sheet. A four-stranded, twisted, antiparallel β-sheet makes up the finger subdomain. In the fingertip region, four cysteine residues constitute a zinc-finger motif, which coordinates a zinc ion with tetrahedral geometry. This zinc-finger is essential for structural integrity and protease activity.
The substrate-binding site is located in the solvent-exposed cleft between the thumb subdomain and the palm subdomain, which possess a catalytic triad composed of C111, H272 and D286. The substrate-binding site recognizes the consensus sequence LXGG↓X (the amino acid residues of the substrate are numbered P4–P3–P2–P1↓P1′–P2′ around the cleavage site, denoted by the downwards arrow). Subsites S1–S4 provide the binding sites for P1–P4, respectively75. The S1 and S2 subsites are rather narrow, and can accommodate only glycine residues. The S3 subsite is partially solvent exposed but prefers positively charged and hydrophobic residues. The S4 subsite is relatively large and accommodates only hydrophobic residues. A flexible β-hairpin BL2 loop, which contains an unusual β-turn at Y268 and Q269, is involved in controlling substrate access to the active site. Consideration of the conformation of the BL2 loop may be important for rational drug design.
Besides the catalytic site, PLpro harbours two distinct binding subsites (SUb1 and SUb2) for recognizing diubiquitin chains and ISG15. SUb1 recognizes one ubiquitin molecule of diubiquitin chains and the C-terminal ubiquitin-like domain of ISG15. SUb2 recognizes the other (K48-linked) uniquitin molecule and the N-terminal ubiquitin-like domain of ISG15 (refs74,76) (Fig. 4d). As shown in the complex structures of PLpro–ubiquitin and PLpro–ISG15, SUb1 of SARS-CoV-2 PLpro preferentially binds ISG15 through a different binding mode compared with uniquitin. Moreover, PLpro SUb2 provides exquisite specificity for K48-linked diubiquitin chains, which makes diubiquitin a suitable substrate compared with monoubiquitin.
Inhibitors targeting PLpro
Owing to the substantial role in mediating viral replication and suppressing the host immune response, PLpro is an attractive target for antiviral drug development. Thousands of compounds, including approved drugs and molecules in clinical trials, have been screened against this target, but the hit rate is extremely low compared with that of drug leads that target Mpro, another viral protease encoded by SARS-CoV-2. The peptidomimetic inhibitors VIR250 and VIR251 were the first identified covalent inhibitors of PLpro (ref.77) (Fig. 4c). A catalytic residue, C111, of PLpro engages in a Michael addition reaction with the β-carbon of the vinyl group of the vinylmethyl ester warheads from VIR250 and VIR251, resulting in the formation of a covalent thioether linkage. Residues at P2, P3 and P4 participate in an extensive network of hydrogen bonds and van der Waals interactions with their corresponding subsites. Similar substrate preferences and catalytic efficiencies are observed for SARS-CoV-2 and SARS-CoV PLpro, suggesting that inhibitors of SARS-CoV PLpro are a good starting point for lead compound optimization against SARS-CoV-2. GRL0617, an inhibitor of SARS-CoV PLpro, also inhibits SARS-CoV-2 PLpro (ref.78). Structural studies show that GRL0617 fits in the substrate cleft which was formed between the BL2 loop and the loop connecting α3 and α4, where it occupies the S3 and S4 subsites. The aromatic ring of GRL0617 fits into the S3 subsite, while the naphthalene group fills the S4 subsite. Thus, the binding of GRL0617 blocks the substrate from gaining access to the active site. Inspired by the success of GRL0617, several naphthalene-based compounds were synthesized and also show good inhibition of SARS-CoV-2 PLpro (ref.79). YM155, an anticancer drug candidate in clinical trials, has also been shown to inhibit SARS-CoV-2 PLpro and has potent antiviral activity (half-maximal effective concentration (EC50) of 170 nM)80. YM155 achieves such a strong inhibition by simultaneously recognizing three hotspots in PLpro. The first binding site is located at the entrance of the substrate-binding pocket and blocks substrate entry to the active site. The second is located on the thumb domain and hampers interactions between PLpro and ISG15. The third site is located on the zinc-finger motif, and the binding perturbs the stability of the zinc-finger motif and enzyme activity.
Mpro
Mpro is the major protease encoded by SARS-CoV-2. It cleaves replicase polyproteins at no fewer than 11 sites to release NSPs, allowing the assembly of the viral replication and transcription machinery. The pivotal role that Mpro plays in regulating viral replication and transcription makes it an attractive drug target. Crystal structures show that this 306 amino acid protease comprises three domains (domain I, residues 10–99; domain II, residues 100–182; and domain III, residues 198–303) and adopts a chymotrypsin-like fold81. Due to the similar substrate specificity and presence of a cysteine as a catalytic residue, Mpro is classified as a 3C-like protease82.
Since the first crystal structure of SARS-CoV-2 Mpro in complex with a Michael acceptor inhibitor N3 (Protein Data Bank accession code 6LU7) (Fig. 5a) was published81, many structures of Mpro in complex with inhibitors have been reported. SARS-CoV-2 Mpro functions as an active homodimer, in which the two protomers are nearly perpendicular to each other. The N-terminal finger (residues 1–7) of one protomer inserts itself between domains II and III of its neighbouring protomer, and promotes the formation of the dimer and the S1 subsite in the neighbouring protomer83. Dimerization is additionally regulated by domain III through a salt-bridge interaction between E290 of one protomer and R4 from its adjacent protomer. In each protomer, a deep cleft between domains I and II forms the substrate-binding site, with a catalytic dyad (H41 and C145) at its centre. Domain III contains five α-helices that arrange themselves into a large antiparallel globular cluster and exhibit a unique topology in coronaviruses. Domains II and III are connected by a long loop (residues 183–198).

a | Structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro) in complex with inhibitor N3. Protomer A is shown as a cartoon, and protomer B is shown as a surface representation. The surface representation of the substrate-binding site and N3 is shown in the right panel. Subsites S1′, S1, S2 and S4 are labelled. b | Interactions between N3 and SARS-CoV-2 Mpro. P1–P5 are labelled in N3. The residues interacting with P1′ are shown as cyan sticks, and the residues forming the S1, S2 and S4 subsites are shown as green sticks, white sticks and orange sticks, respectively. The residues interacting with P5 are shown as blue sticks. Intermolecular hydrogen bonds are shown as dashed lines. c | Surface representation of the substrate-binding site and with various inhibitors bound. GC376, boceprevir, carmofur and 11a are coloured yellow, blue, green and orange, respectively. All inhibitors are presented in stick form. Protein Data Bank accession codes are indicated in parentheses.
Coronavirus Mpros recognize the P4–P1′ positions of the substrate84,85,86 (Fig. 5b). The S1 subsite has an absolute preference for glutamine at P1. P2 is usually a bulky side chain that can be accommodated by the deep hydrophobic S2 subsite. The P3 side chain is solvent exposed, and the corresponding S3 subsite also shows tolerance to a wide range of functional groups. The hydrophobic S4 subsite is smaller than S2 and thus accommodates residues with small side chains. This binding pocket is highly conserved among coronavirus Mpros, suggesting that antiviral inhibitors targeting this pocket should have broad-spectrum activity against coronaviruses in general87.
Inhibitors of SARS-CoV-2 Mpro
Recently, numerous inhibitors of Mpro have been identified exhibiting a range of binding mechanisms (Fig. 5c). N3 is the representative peptidomimetic inhibitor, and harbours a Michael acceptor as a warhead and substituents spanning all substrate-binding subsites. The Michael acceptor forms a covalent bond with the active site residue, C145. N3 bears a lactam ring, an aliphatic isobutyl group, an isopropyl group, a methyl group and an isoxazole as the side chain for the P1–P5 sites, respectively. The lactam ring, which replaces glutamine at the P1 site, exhibits favourable binding at the S1 subsite81,88. Studies have shown that N3 displays strong inhibition of Mpros from different coronaviruses, and it could inhibit SARS-CoV-2 with EC50 of 16.77 μM in a Vero cell-based assay. This value may not be truly representative of activity as it is not clear whether the high levels of expression of the efflux transporter P-glycoprotein in Vero cells affected the evaluation of its antiviral efficacy88.
A recent study reported a series of α-ketoamides that inhibit SARS-CoV-2 Mpro (ref.89). Distinct from the previously designed α-ketoamides, the P2–P3 amide bond is replaced with a pyridone ring, which increases the half-life in plasma. Replacement of the P2 cyclohexyl moiety with smaller cyclopropyl increases the antiviral activity against betacoronaviruses. Approved hepatitis C virus drugs, such as boceprevir, telaprevir and narlaprevir, are α-ketoamide inhibitors and also exhibit inhibition of SARS-CoV-2 Mpro. The ketone group undergoes a nucleophilic attack by the C145 thiolate to form a hemithioketal. Because boceprevir, telaprevir and narlaprevir are peptidomimetic inhibitors with similar structures, they form very similar interactions with the S1′–S4 subsites90. Another ketone-based potent inhibitor was discovered in the hydroxymethylketone class91. One of the hydroxymethylketone derivatives demonstrated inhibition of SARS-CoV-2 Mpro and also possesses antiviral activity with EC50 of 4.8 μM.
Another study presented two peptidomimetic aldehydes (named ‘11a’ and ‘11b’) which bear an indole moiety at the N terminus (P3 site) and an aldehyde warhead at the C terminus92. The complex structures show that the aldehyde groups covalently bind to C145 of the catalytic dyad to inhibit Mpro activity. Both inhibitors exhibited excellent inhibition of SARS-CoV-2 Mpro with half-maximal inhibitory concentrations of 0.053 μM and 0.040 μM, respectively. The inhibitors also exhibited strong anti-SARS-CoV-2 infection activity in Vero cell-based assays and good pharmacokinetic and toxicity properties. A recent study reported another series of aldehyde derivatives with EC50 ranging from 7.6 to 748.5 nM in cell-based assays. In a transgenic mouse model of SARS-CoV-2 infection, oral or intraperitoneal treatment with two compounds, MI-09 or MI-30, significantly reduced lung viral loads and lung lesions. Both also displayed good pharmacokinetic properties and safety in rats93. GC376, an inhibitor of feline infectious peritonitis virus in preclinical studies, has been found to efficaciously inhibit SARS-CoV-2 in Vero cells by targeting Mpro. It utilizes an aldehyde bisulfite to covalently bind to C145 (refs94,95). Based on 11a, 11b and GC376, a number of aldehyde-based dipeptidyl and tripeptidyl inhibitors of Mpro were designed, and the organocatalyst-mediated protein aldol ligation to C145 of the protease occurs96. A series of Mpro inhibitors that possess an aldehyde group for covalent inhibition have been reported97. Among them, two compounds inhibited SARS-CoV-2 replication in cultured primary human airway epithelial cells.
The repurposing of approved drugs, drug candidates and pharmacologically active compounds provides an alternative approach to identify potential drug leads that could rapidly be approved as clinical treatments for COVID-19. Through high-throughput screening, one study identified multiple drug leads that target Mpro, including ebselen, disulfiram and carmofur81. Ebselen exhibited antiviral activity in a plaque-reduction assay (EC50 = 4.67 μM). As an organoselenium compound, ebselen was previously investigated for treatment of bipolar disorders and hearing loss98. It has been shown to have low cytotoxicity in humans in clinical trials99. Ebselen has been approved by the US Food and Drug Administration to enter phase II clinical trials (NCT04484025 and NCT04483973) for COVID-19 treatment. Carmofur, which also exhibited antiviral activity in vitro, is a derivative of 5-fluorouracil. It is an approved antineoplastic agent, and has been investigated as a cancer treatment100. As observed in the complex structure of Mpro and carmofur, the catalytic C145 residue is covalently bound to the carbonyl reactive group of carmofur and its fatty acid tail extends into the hydrophobic S2 subsite101. Such a novel inhibitory mode makes carmofur a good lead compound for rational drug design. GRL-1720 and 5h were also identified as covalent inhibitors targeting Mpro through high-throughput screening. Crystal structures show that both GRL-1720 and 5h form extensive interactions with C145 and other residues in the Mpro active site102.
A recent study performed large-scale fragment screening against Mpro by combining mass spectrometry and X-ray approaches103. Seventy-one hits were identified to bind at the substrate-binding site, and three hits were found to bind near the dimer interface. These structures provide a starting point to design more elaborate and potent drug leads that target SARS-CoV-2 Mpro. Another study performed a high-throughput X-ray crystallographic screening of two drug repurposing libraries (the Fraunhofer IME Repurposing Collection and the Safe-in-Man library from Dompé Farmaceutici) against the SARS-CoV-2 Mpro (ref.104); the study authors identified 37 compounds that bind to Mpro. In subsequent cell-based assays, one peptidomimetic compound (calpeptin) and six non-peptidic compounds showed antiviral activity at non-toxic concentrations. Additionally, two allosteric binding sites representing potential targets against SARS-CoV-2 were identified. The first allosteric site is in the immediate vicinity of the S1 pocket of the adjacent protomer within the native dimer. The second allosteric site is formed by the deep groove between the catalytic domain and the dimerization domain.
Baicalin and baicalein, which are natural products derived from the flowering plant Scutellaria baicalensis, have been shown to inhibit SARS-CoV-2 Mpro with half-maximal inhibitory concentrations of 6.41 μM and 0.94 μM, respectively105. The structure of Mpro in complex with baicalein shows that the phenyl ring with three hydroxy groups forms π–S and π–π interactions with C145 and H41 of the catalytic dyad, while the hydroxy groups form multiple hydrogen bonds with the S1 subsite. The distal phenyl ring occupied the S2 subsite. Another example is shikonin106. The complex structure shows that shikonin forms a hydrogen bond network with the catalytic dyad C145 and H164 located in the S1 subsite. The aromatic head groups of shikonin form a π–π interaction with H41 on the S2 subsite. The hydroxy and methyl groups of the isohexenyl side chain of the shikonin tail form hydrogen bonds with R188 and Q189, respectively, in the S3 subsite. Such a unique mode of action expands our knowledge of Mpro inhibition.
Replication and transcription complex
Replication mechanism of the central RTC
In coronavirus infection, replication and transcription is regulated through a multisubunit mechanism107, where the RdRP nsp12 catalyses viral RNA synthesis and thus acts as the key component of the RTC108. In addition, the primase nsp8 (ref.109) and an auxiliary factor, nsp7, contribute to the activation and continuous production of viral RNA110. nsp12 along with nsp7 and nsp8 makes up the complete RdRP complex.
SARS-CoV-2 nsp12 is composed of three major domains, a nidovirus RdRP-associated nucleotidyltransferase (NiRAN) domain, an interface domain and a right-handed RdRP domain (finger, palm and thumb)111 (Fig. 6a). The active site of SARS-CoV-2 RdRP is located in the palm subdomain, which has a shape like other RNA polymerases, such as those from hepatitis C virus ns5b112 and poliovirus 3Dpol113. The architecture of the central cavity is shared by other conserved polymerases involving the primer-template entry, nucleoside triphosphate (NTP) entry and nascent strand exit paths. Residues D760 and D761 are involved in the coordination of two Mg2+ ions essential for polymerase activity. One Mg2+ ion coordinates motif C and binds at the 3′ end (‘i’ site) of the RNA primer, facilitating the condensation reaction in RNA chain synthesis, while the second Mg2+ positions the incoming NTP and stabilizes the charge environment. Separate from conserved motifs A–E at the active site, motif F and motif G inside the fingers subdomain are conducive to guiding the RNA template. During viral RNA synthesis, notable structural rearrangements occur in this complex to accommodate the RNA114. Along with the product chain synthesis, the protruding RNA template–product duplex exits through the active site without steric hindrance and extends to two positively charged ‘sliding poles’ formed by two nsp8 N-terminal helices115 (Fig. 6b). Consistent with SARS-CoV nsp8 adopting variable conformations116,117, N-terminal extensions of nsp8-2 (the second copy of nsp8) have two different orientations at the early replicating stage. In one orientation, it is adjacent to the finger subdomain, whereas in the other orientation, it interacts with the RNA duplex, suggesting that nsp8 may have regulatory functions in replication initiation. The complex consisting of nsp12, nsp7, nsp8 and RNA duplex reflects the replicating state in RdRP activity; therefore, it is referred to as the central RTC (C-RTC).

a–f | Cartoon representations of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) central replication and transcription complex (C-RTC), elongation RTC (E-RTC), cap(−1)′ RTC and backtracking RTC. The RNA-dependent RNA polymerase (RdRP) core complex contains nsp12 (nidovirus RdRP-associated nucleotidyltransferase (NiRAN), interface, finger, palm and thumb domains are yellow, orange, blue, red and olive, respectively), nsp7 (purple), nsp8-1 (individual; light grey), nsp8-2 (nsp7–nsp8 pair; aquamarine). C-RTC is composed of the RdRP core complex and RNA template–product chains (in wheat and deep sky blue). E-RTC is composed of the C-RTC along with two coupled nsp13 helicases, nsp13-1 (lime) and nsp13-2 (deep pink), and RNA strand fragments can be traced in nsp13-2. Cap(−1)′ RTC consists of E-RTC and nsp9 (claret; bound to nsp12 NiRAN domain). Backtracking RTC includes the C-RTC coupled with the proofreading-stimulating nsp13. g–i | Cartoon representations of SARS-CoV-2 C-RTC with bound inhibitors. Remdesivir can be inserted into the RNA product chain (−1 site), whereas favipiravir occupies the polymerase active centre. Two suramin molecules can bind with the RdRP core complex and suramin-1 occupies the space of positions −1 to −3 of the RNA template strand, whereas suramin-2 occupies the space of the primer strand. The inhibitors are all shown as pink stick models. Protein Data Bank accession codes are indicated in parentheses. FTP, favipiravir triphosphate; RMP, remdesivir monophosphate.
RNA elongation, capping and backtracking
The RTC needs to guarantee processive RNA duplex elongation without template–product dissociation so that viral genome or subgenome synthesis can be rapidly completed inside the host cell118. For coronaviruses, which have the largest known positive-sense RNA genomes, both replication efficiency and replication fidelity are essential for maintaining genetic integrity. The former relies on the functional elongation RTC (E-RTC), whereas the latter depends on proofreading by nsp14. An E-RTC is composed of a C-RTC and two coupled copies of the nsp13 helicase: nsp13-1 and nsp13-2 (ref.119) (Fig. 6c). nsp13 is believed to be crucial in viral replication and the mRNA capping process, which includes unwinding of the RNA duplex into single strands, 5′ to 3′ polarity formation and RNA 5′-triphosphatase activity120,121. The unique domains of coronavirus nsp13, such as the zinc-binding domain, the stalk and the 1B domain, are all important for helicase activity122. In the structure of E-RTC, two nsp13 zinc-binding domains form extensive interactions with two nsp8 N-terminal helices. In particular, the zinc-binding domain from nsp13-2 forms additional interactions with the nsp12 thumb subdomain, stabilizing the overall structure during elongation119,123. Before entering the nsp12 active site, the template RNA strand undergoes disruption of RNA secondary structure and guidance between the nsp13-2 RecA domain and the 1B domain to ensure the 5′ to 3′ translocation direction124. Structural characterization of E-RTC not only helps elucidate the RNA elongation mechanisms but also suggests different functional roles that nsp13 may play in this event. In nsp13-2, residues N361 in the domain 1A, S468, T532 and D534 in the domain 2A and R178 and H230 in the domain 1B collectively contribute to template RNA recognition and elongation, demonstrating that nsp13-2 is directly involved in positioning downstream template RNA. Interestingly, the interactions between the nsp13-1 1B domain and the nsp13-2 1B domain have been shown to play a pivotal role in E-RTC helicase activity, even though nsp13-1 is far from nsp13-2 (ref.123) (Fig. 6d). Therefore, nsp13-1 is indispensable for RNA elongation in that it is cooperatively coupled with nsp13-2 in the functioning E-RTC.
The capping modification of mRNA, which rigorously follows subgenomic mRNA synthesis, is essential for viral translation and propagation, mRNA protection and escape from host immune response125,126. Similarly to the RNA elongation process, multiple NSPs participate in RTC assembly during sequential stages of mRNA capping, which can be divided into four main steps: (1) removal of the γ-phosphate of 5′-pppA by nsp13 with RNA 5′-triphosphatase activity120; (2) transfer of GMP to 5′-ppA by the nsp12 NiRAN domain with guanylyltransferase (GTase) activity, leading to the generation of a GpppA cap structure127; (3) methylation of N7-guanine by nsp14, which has N7-methyltransferase activity128; and (4) methylation of the ribose 2′-O nucleotide into the final 7MeGpppA2′OMe cap structure by nsp1, which has 2′-O-methyltransferase activity129. Multiple NSPs are assembled into the RTC in order according to their functional roles, a process which is accompanied by structural conformational changes. On one hand, the nsp12 NiRAN domain is involved in the second step to catalyse the ppA to GpppA transfer through its newly identified GTase activity. On the other hand, an intermediate state which has been captured by cryo-EM, shows that nsp9 can inhibit the GTase activity by tight insertion into the NiRAN catalytic centre in order to terminate the reaction (Fig. 6e). nsp9 is an RNA-binding protein, which is characterized by a positively charged groove130. This groove, together with a β-hairpin at the nsp12 N terminus, provides an exit path for postcatalytic GpppA-RNA. Several hydrophobic interactions and hydrogen bonds enhance nsp9 binding to nsp12, suggesting that nsp9 plays a substantial role in the viral life cycle. Because it has been shown that disruption of the nsp9–nsp10 cleavage site is not lethal131 and nsp10 is able to tightly bind to nsp14 or nsp16 (refs132,133), nsp9 may serve as a core regulator in recruiting the nsp10–nsp14 or nsp10–nsp16 complex for the following capping RTC assembly with N7-methyltransferase activity and 2′-O-methyltransferase activity.
Another important aspect relating to the RTC is its proofreading mechanism. Most RNA viruses replicate with estimated error rates between 10−3 and 10−5, which results in approximately one mutation per genome per round of replication for a typical ∼10-kb genome134, a much higher mutation rate than occurs in cellular DNA replication135. The lower fidelity may largely be due to the lack of proofreading activity in these viruses. By contrast, SARS-CoV-2, which encodes nsp14 (an exonuclease with proofreading activity), can maintain high fidelity during replication of its large genome. Proofreading involves the backtracking of mismatched template–product RNA chains. The single-stranded 3′ segment of the product RNA generated by backtracking extrudes through the RdRP NTP entry tunnel. Then a mismatched nucleotide located at the 3′ end of product RNA enters the conserved NTP entry tunnel to initiate backtracking, and meanwhile, nsp13 stimulates RdRP backtracking. The structure of C-RTC in complex with the essential nsp13 helicase and RNA suggests that the helicase can facilitate the backtracking mechanism136 (Fig. 6f).
RdRP inhibitor discovery
The RdRP is a prime drug target for SARS-CoV-2 (Fig. 1a). Inhibition of RdRP activity will prevent viral replication and can potentially achieve clinical efficacy. Major efforts have been devoted to identify both nucleotide and non-nucleotide inhibitors, which have also been used as probes to understand the replication cycle of SARS-CoV-2 and to provide a basis for development of broad-spectrum antiviral drugs.
The prodrug remdesivir, which was initially developed for the treatment of Ebola virus infection, shows good activity against SARS-CoV-2 in in vitro assays137 but limited efficacy in clinical trials. In the cell, remdesivir is phosphorylated to remdesivir triphosphate, enabling it to act as an ATP analogue. The structure of pretranslocated catalytic C-RTC clearly demonstrates the incorporation mode of remdesivir and suggests its inhibition mechanism114 (Fig. 6g). Kinetic analysis shows remdesivir triphosphate is preferred as a substrate over ATP138 and terminates product chain elongation at a delayed position (i + 3). Once the inserted remdesivir monophosphate is transferred to the i + 3 position, the distance between the serine hydroxy oxygen from S861 and the 1′-cyano nitrogen from remdesivir monophosphate will be close to 2 Å, causing ‘delayed chain termination’. Further investigations indicate that an remdesivir-induced translocation barrier and RdRP stalling occur after the addition of three nucleotides upon incorporation of remdesivir into the product chain139. Favipiravir is another nucleoside analogue that has been approved as an anti-influenza virus drug in Japan. Favipiravir simulates the incorporation of ATP and GTP into the product RNA, yet it inhibits viral proliferation by increasing the mutation rate of the viral genome rather than causing product chain terminations140. The structure of the RdRP–favipiravir complex delineates a precatalytic state and identifies the conserved residues for favipiravir recognition (Fig. 6h).
Although nucleotide inhibitors can be inserted into RNA chains, they can later be cleaved by proofreading activity. Thus, non-nucleotide inhibitors have been considered as an alternative approach for drug development. Suramin, a century-old drug used to treat African sleeping sickness and river blindness, can effectively inhibit SARS-CoV-2 polymerase activity with at least 20-fold more activity than RDV-3Pi in biochemical assays and inhibits viral replication in vitro141. In the cryo-EM structure, two suramin molecules bind to the active sites of nsp12, with one occupying the template-binding site and the other occupying the primer catalytic active centre, implying that suramin may competitively inhibit protein–RNA binding due to its strong electronegativity (Fig. 6i). However, the highly negatively charged suramin has the potential to bind to many positively charged macromolecular surfaces, and thus its specific antiviral activity remains to be further investigated.
Accessory protein–host interactions
ORF3a, ORF9b, ORF7a and ORF8
ORF3a protein, encoded by ORF3a, is an ion channel membrane protein with 274 amino acids. It forms a potassium-sensitive channel and may promote virus release. The cryo-EM structure of SARS-CoV-2 ORF3a is the first viroporin family structure determined in coronaviruses142. The overall structure shows that ORF3a forms a dimer with the ion channel decorated with charged residues for cation conduction (Fig. 7a). It is noteworthy that ORF3a has a TRAF-binding domain at the N terminus that can activate NF-κB and the NLRP3 inflammasome143, suggesting an important role in the host immune response. As ion channels are important therapeutic targets and many ion-channel drugs have already been approved for clinical trials, ORF3a is another good antiviral drug target144.

a | Cryogenic electron microscopy structure of dimeric ORF3a. It forms a potassium-sensitive channel and may promote release of the virus. The two ORF3a proteins are shown as cartoon representations, and the domains are labelled. b | The structure of ORF9b in complex with human TOM70. ORF9b adopts a helical fold and is shown in cartoon representation. It interacts at the substrate binding site of TOM70, a subunit of the mitochondrial import receptor. TOM70 is shown in a surface representation. c | The structure of the ORF7a ectodomain. ORF7a is a type I transmembrane protein and is involved in virus–host interactions and protein trafficking within the endoplasmic reticulum and Golgi body. The ectodomain of ORF7a exhibits a seven-stranded β-sandwich fold and is shown in a cartoon representation. d | The structure of dimeric ORF8. ORF8 contains eight antiparallel β-strands and an immunoglobulin-like fold. The dimer is stabilized by surface hydrophobic interactions and a series of hydrogen bonds. The two ORF8 proteins are shown as cartoon representations. Protein Data Bank accession codes are indicated in parentheses.
ORF9b is encoded by an alternative ORF within the N protein gene. ORF9b suppresses the type I interferon immune response by interacting with the mitochondrial import receptor subunit TOM70. Targeting the interactions between ORF9b and TOM70 has been proposed as a therapeutic option for SARS-CoV-2. The structure of SARS-CoV-2 ORF9b shows that it is dimeric, with each protomer composed mainly of β-strands145 (Fig. 7b). The centre of the dimer has a hydrophobic environment for accommodating lipid molecules and membrane attachment.
ORF7a is a type I transmembrane protein and is also involved in virus–host interactions and protein trafficking within the ER and Golgi body. Its structure shows that it has a seven-stranded β-sandwich fold consistent with the immunoglobulin superfamily146 (Fig. 7c). A deep hydrophobic pocket has been identified for potential inhibitor binding.
ORF8 is an accessory protein that is composed of 121 amino acids. It has an N-terminal signal sequence and adopts an immunoglobulin-like fold147 (Fig. 7d). The structure of ORF8 shows that it can form a dimer, and each promoter of ORF8 contains eight antiparallel β-strands tied by three disulfide bonds. The covalently bonded dimer structure is stabilized by surface hydrophobic interactions and a series of hydrogen bonds. ORF8 is capable of assembling itself into large-scale homologous complexes; however, the oligomerization mechanism needs to be investigated further.
Conclusions
Coronaviruses have the largest genomes among all RNA viruses, encoding structural proteins and NSPs that achieve sustainability in a wide variety of ecological niches and hosts. Evolving viral proteins help coronaviruses to achieve host recognition and entry, genome replication, assembly and release of progeny viruses, and host immune surveillance evasion. In response to the COVID-19 pandemic, great efforts have been devoted to structural studies of SARS-CoV-2 proteins and viral–cellular protein complexes using X-ray crystallography and cryo-EM. Among them, the S protein, Mpro, PLpro and RdRP are the most widely studied drug targets. A multidisciplinary combination of structural virology, ’omics technologies, immunology and virology will produce a more effective approach to structure-aided design of vaccines and therapeutics that have the potential for clinical use.
References
Masters, P. S. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292 (2006).
Weiss, S. R. & Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69, 635–664 (2005).
Cavanagh, D. Coronaviruses in poultry and other birds. Avian Pathol. 34, 439–448 (2005).
Hamre, D. & Procknow, J. J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 121, 190–193 (1966).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Fung, T. S. & Liu, D. X. Human coronavirus: host-pathogen interaction. Annu. Rev. Microbiol. 73, 529–557 (2019).
de Wit, E., van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534 (2016).
World Health Organization. Middle East respiratory syndrome coronavirus. WHO https://www.who.int/health-topics/middle-east-respiratory-syndrome-coronavirus-mers#tab=tab_1 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Harrison, A. G., Lin, T. & Wang, P. Mechanisms of SARS-CoV-2 transmission and Pathogenesis. Trends Immunol. 41, 1100–1115 (2020).
Johns Hopkins Coronavirus Resource Center. COVID-19 dashboard. JHU https://coronavirus.jhu.edu/map.html (2021).
Chen, Y., Liu, Q. & Guo, D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92, 418–423 (2020).
Wang, M. Y. et al. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front. Cell Infect. Microbiol. 10, 587269 (2020).
Faheem et al. Druggable targets of SARS-CoV-2 and treatment opportunities for COVID-19. Bioorg Chem. 104, 104269 (2020).
Belouzard, S., Millet, J. K., Licitra, B. N. & Whittaker, G. R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4, 1011–1033 (2012).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904.e9 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Xia, S. et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020).
Shi, R. et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124 (2020).
Barnes, C. O. et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 182, 828–842.e16 (2020).
Hurlburt, N. K. et al. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat. Commun. 11, 5413–5413 (2020).
Wu, Y. et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020).
Yuan, M. et al. Structural basis of a shared antibody response to SARS-CoV-2. Science 369, 1119–1123 (2020).
Fu, D. et al. Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes. PLoS Biol. 19, e3001209 (2021).
Guo, Y. et al. A SARS-CoV-2 neutralizing antibody with extensive spike binding coverage and modified for optimal therapeutic outcomes. Nat. Commun. 12, 2623 (2021).
Yuan, M. et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020).
Zhou, D. et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat. Struct. Mol. Biol. 27, 950–958 (2020).
Wrapp, D. et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 181, 1004–1015.e15 (2020).
Liu, L. et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84.e16 (2020).
Huo, J. et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 27, 846–854 (2020).
Ju, B. et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 115–119 (2020).
Hanke, L. et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 11, 4420 (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014 (2020).
Yao, H. et al. Rational development of a human antibody cocktail that deploys multiple functions to confer pan-SARS-CoVs protection. Cell Res. 31, 25–36 (2021).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science 369, 650–655 (2020).
Wang, N. et al. Structure-based development of human antibody cocktails against SARS-CoV-2. Cell Res. 31, 101–103 (2021).
Zheng, Z. et al. Monoclonal antibodies for the S2 subunit of spike of SARS-CoV-1 cross-react with the newly-emerged SARS-CoV-2. Euro Surveill. 25, 2000291 (2020).
Song, G. et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat. Commun. 12, 2938 (2021).
Lv, Z. et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science 369, 1505–1509 (2020).
Wec, A. Z. et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 369, 731–736 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020).
Yurkovetskiy, L. et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 183, 739–751.e8 (2020).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Starr, T. N. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310.e20 (2020).
Collier, D. A. et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593, 136–141 (2021).
McCallum, M. et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 184, 2332–2347.e16 (2021).
McCarthy, K. R. et al. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 371, 1139–1142 (2021).
Weisblum, Y. et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 9, e61312 (2020).
Nieto-Torres, J. L. et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339 (2015).
Mandala, V. S. et al. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 27, 1202–1208 (2020).
Lu, X., Pan, J., Tao, J. & Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. 42, 37–45 (2011).
Mu, J. et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci. 63, 1413–1416 (2020).
Chang, C. K., Hou, M. H., Chang, C. F., Hsiao, C. D. & Huang, T. H. The SARS coronavirus nucleocapsid protein–forms and functions. Antivir. Res. 103, 39–50 (2014).
Peng, Y. et al. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 39, e105938 (2020).
Lei, L. et al. Attenuation of mouse hepatitis virus by deletion of the LLRKxGxKG region of Nsp1. PLoS ONE 8, e61166 (2013).
Narayanan, K. et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82, 4471–4479 (2008).
Kamitani, W., Huang, C., Narayanan, K., Lokugamage, K. G. & Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 16, 1134–1140 (2009).
Huang, C. et al. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 7, e1002433 (2011).
Thoms, M. et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369, 1134–1255 (2020).
Lei, J., Kusov, Y. & Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 149, 58–74 (2018).
Alhammad, Y. M. O. et al. The SARS-CoV-2 conserved macrodomain is a mono-ADP-ribosylhydrolase. J. Virol. 95, e01969-20 (2021).
Kusov, Y., Tan, J., Alvarez, E., Enjuanes, L. & Hilgenfeld, R. A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication-transcription complex. Virology 484, 313–322 (2015).
Lei, J. et al. The SARS-unique domain (SUD) of SARS-CoV and SARS-CoV-2 interacts with human Paip1 to enhance viral RNA translation. EMBO J. 40, e102277 (2021).
Harcourt, B. H. et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 78, 13600–13612 (2004).
Barretto, N. et al. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198 (2005).
Jiang, X. & Chen, Z. J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 12, 35–48 (2011).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Fu, Z. et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 12, 488–488 (2021).
Klemm, T. et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 39, e106275 (2020).
Rut, W. et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci. Adv. 6, eabd4596 (2020).
Gao, X. et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sinica. B 11, 237–245 (2021).
Osipiuk, J. et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 12, 743–743 (2021).
Zhao, Y. et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell https://doi.org/10.1007/s13238-021-00836-9 (2021).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Matthews, D. A. et al. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77, 761–771 (1994).
Anand, K. et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 21, 3213–3224 (2002).
Xue, X. et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 82, 2515–2527 (2008).
Yang, H. et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, e324 (2005).
Yang, H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA 100, 13190–13195 (2003).
Yang, H., Bartlam, M. & Rao, Z. Drug design targeting the main protease, the achilles heel of coronaviruses. Curr. Pharm. Des. 12, 4573–4590 (2006).
de Vries, M. et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 95, e01819-20 (2021).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
Kneller, D. W. et al. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 28, 1313–1320.e3 (2020).
Hoffman, R. L. et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 63, 12725–12747 (2020).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Qiao, J. et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378 (2021).
Fu, L. et al. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 11, 4417 (2020).
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020).
Yang, K. S. et al. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors*. ChemMedChem 16, 942–948 (2021).
Rathnayake, A. D. et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl Med. 12, eabc5332 (2020).
Kil, J., Pierce, C., Tran, H., Gu, R. & Lynch, E. D. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hearing Res. 226, 44–51 (2007).
Kil, J. et al. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 390, 969–979 (2017).
Dementiev, A. et al. Molecular mechanism of inhibition of acid ceramidase by carmofur. J. Med. Chem. 62, 987–992 (2019).
Jin, Z. et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 27, 529–532 (2020).
Hattori, S.-I. et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 12, 668 (2021).
Douangamath, A. et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 11, 5047 (2020).
Günther, S. et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372, 642 (2021).
Su, H.-x. et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 41, 1167–1177 (2020).
Li, J. et al. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 66, 661–663 (2021).
Ziebuhr, J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 287, 57–94 (2005).
Imbert, I. et al. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 25, 4933–4942 (2006).
te Velthuis, A. J., van den Worm, S. H. & Snijder, E. J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 40, 1737–1747 (2012).
Subissi, L. et al. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl Acad. Sci. USA 111, E3900–E3909 (2014).
Gao, Y. et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368, 779–782 (2020).
Appleby, T. C. et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 (2015).
Gong, P. & Peersen, O. B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl Acad. Sci. USA 107, 22505–22510 (2010).
Wang, Q. et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 182, 417–428.e3 (2020).
Hillen, H. S. et al. Structure of replicating SARS-CoV-2 polymerase. Nature 584, 154–156 (2020).
Li, S. et al. New nsp8 isoform suggests mechanism for tuning viral RNA synthesis. Protein Cell 1, 198–204 (2010).
Zhai, Y. et al. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat. Struct. Mol. Biol. 12, 980–986 (2005).
Choi, K. H. Viral polymerases. Adv. Exp. Med. Biol. 726, 267–304 (2012).
Chen, J. et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell 182, 1560–1573.e13 (2020).
Ivanov, K. A. et al. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 78, 5619–5632 (2004).
Ivanov, K. A. & Ziebuhr, J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 78, 7833–7838 (2004).
Jia, Z. et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 47, 6538–6550 (2019).
Yan, L. et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 11, 5874 (2020).
Saikrishnan, K., Powell, B., Cook, N. J., Webb, M. R. & Wigley, D. B. Mechanistic basis of 5′-3′ translocation in SF1B helicases. Cell 137, 849–859 (2009).
Daffis, S. et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456 (2010).
Ramanathan, A., Robb, G. B. & Chan, S. H. mRNA capping: biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016).
Yan, L. et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell 184, 184–193.e10 (2021).
Chen, Y. et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl Acad. Sci. USA 106, 3484–3489 (2009).
Bouvet, M. et al. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 6, e1000863 (2010).
Sutton, G. et al. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure 12, 341–353 (2004).
Deming, D. J., Graham, R. L., Denison, M. R. & Baric, R. S. Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 81, 10280–10291 (2007).
Ma, Y. et al. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl Acad. Sci. USA 112, 9436–9441 (2015).
Rosas-Lemus, M. et al. High-resolution structures of the SARS-CoV-2 2’-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 13, eabe1202 (2020).
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M. & Belshaw, R. Viral mutation rates. J. Virol. 84, 9733–9748 (2010).
Smith, E. C., Sexton, N. R. & Denison, M. R. Thinking outside the triangle: replication fidelity of the largest RNA viruses. Annu. Rev. Virol. 1, 111–132 (2014).
Malone, B. et al. Structural basis for backtracking by the SARS-CoV-2 replication-transcription complex. Proc Natl Acad Sci USA 118, e2102516118 (2021).
Williamson, B. N. et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 585, 273–276 (2020).
Gordon, C. J. et al. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 295, 6785–6797 (2020).
Kokic, G. et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 12, 279 (2021).
Peng, Q. et al. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovation 2, 100080 (2021).
Yin, W. et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 28, 319–325 (2021).
Kern, D. M. et al. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 28, 573–582 (2021).
Siu, K. L. et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 33, 8865–8877 (2019).
Zhong, X. et al. Amino terminus of the SARS coronavirus protein 3a elicits strong, potentially protective humoral responses in infected patients. J. Gen. Virol. 87, 369–373 (2006).
Gordon, D. E. et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, eabe9403 (2020).
Zhou, Z. et al. Structural insight reveals SARS-CoV-2 ORF7a as an immunomodulating factor for human CD14+ monocytes. iScience 24, 102187 (2021).
Flower, T. G. et al. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc Natl Acad Sci USA 118, e2021785118 (2021).
Acknowledgements
The authors thank L. Guddat, Y. Gao and W. Cui for discussions and technical support. This work was supported by the National Program on Key Research Project of China (2020YFA0707500 and 2017YFC0840300) and the National Natural Science Foundation of China (U20A20135).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Humanized antibody
-
Antibodies from non-human species whose protein sequence has been modified to increase their similarity to antibody variants produced naturally in humans. The aim of humanization is to make specific antibodies generated in non-human immune systems suitable for administration to humans.
- Michael addition reaction
-
The nucleophilic addition of a carbanion or another nucleophile to an α,β-unsaturated carbonyl compound containing an electron-withdrawing group. It belongs to the larger class of conjugate additions and results in the mild formation of C–C bonds.
- Naphthalene group
-
A chemical group which is composed of two aromatic rings sharing two adjacent carbon atoms.
- Half-maximal effective concentration
-
(EC50). A quantitative measure that indicates how much of a substance (for example, an antiviral agent) is effective in inducing a response (for example, eliminating a virus in cultured cells) halfway between the baseline level and the maximal level.
- Michael acceptor
-
The activated alkene in an α,β-unsaturated carbonyl compound, which is involved in the Michael addition reaction.
- Antineoplastic agent
-
A drug for cancer treatment. Such drugs interfere with the ability of a cancer cell to grow and spread.
- Prodrug
-
A pharmacologically inactive substance which can be converted into a pharmacologically active drug in vivo by metabolic reactions.
Rights and permissions
About this article

Cite this article
Yang, H., Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat Rev Microbiol 19, 685–700 (2021). https://doi.org/10.1038/s41579-021-00630-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-021-00630-8
This article is cited by
-
The gasdermin family: emerging therapeutic targets in diseases
Signal Transduction and Targeted Therapy (2024)
-
Non-coding RNAs in disease: from mechanisms to therapeutics
Nature Reviews Genetics (2024)
-
Targeting the receptor binding domain and heparan sulfate binding for antiviral drug development against SARS-CoV-2 variants
Scientific Reports (2024)
-
Immunized mice naturally process in silico-derived peptides from the nucleocapsid of SARS-CoV-2
BMC Microbiology (2023)
-
Immune profiling of SARS-CoV-2 epitopes in asymptomatic and symptomatic pediatric and adult patients
Journal of Translational Medicine (2023)
