
Abstract
Traditionally, the viral replication cycle is envisioned as a single, well-defined loop with four major steps: attachment and entry into a target cell, replication of the viral genome, maturation of viral proteins and genome packaging into infectious progeny, and egress and dissemination to the next target cell. However, for many viruses, a growing body of evidence points towards extreme heterogeneity in each of these steps. In this Review, we reassess the major steps of the viral replication cycle by highlighting recent advances that show considerable variability during viral infection. First, we discuss heterogeneity in entry receptors, followed by a discussion on error-prone and low-fidelity polymerases and their impact on viral diversity. Next, we cover the implications of heterogeneity in genome packaging and assembly on virion morphology. Last, we explore alternative egress mechanisms, including tunnelling nanotubes and host microvesicles. In summary, we discuss the implications of viral phenotypic, morphological and genetic heterogeneity on pathogenesis and medicine. This Review highlights common themes and unique features that give nuance to the viral replication cycle.
Similar content being viewed by others
Introduction
The textbook depiction of the viral replication cycle1,2 contains four relatively uniform core steps: entry, replication, assembly and egress (Fig. 1a). Innovative studies have uncovered novel and heterogeneous strategies for accomplishing each of these steps. In this Review, we examine the phenotypic implications of both host and viral heterogeneity in shaping the viral replication cycle. We consider several aspects of heterogeneity in hosts, including availability and post-translational modifications (PTMs) of host receptors, and variability in antiviral defences. We also discuss heterogeneity within viruses, specifically how viral polymerase fidelity, virion morphology and egress mechanisms lead to a more nuanced view of viral infection (Fig. 1b).

a | Traditional depiction of the viral replication cycle with its series of defined, uniform steps, using human metapneumovirus (HMPV) as an example. HMPV virions bind to a target receptor and fuse with the host cell membrane (step 1). After fusion, viral RNA is replicated in cytoplasmic replication organelles (step 2) and viral proteins are produced and undergo assembly with the viral RNA (step 3). Homogeneous mature virus particles undergo egress through the cellular membrane (step 4). b | Evidence of substantial heterogeneity in the viral replication cycle is accumulating. Entry can be impacted by the presence of post-translational modification of receptors, including glycosylation (step 1). Upon entry, HMPV replication organelles can traffic along actin-containing nanotubes across tight junctions of the lung epithelium3 (step 2). Variation in assembly (step 3) and egress (step 4) lead to the maturation of heterogeneous virus particles from the cell. Similar heterogeneity has been observed in many different viruses.
Technological advances (Table 1) have been crucial for identifying how heterogeneity redefines viral replication. Human airway epithelial cell cultures3,4,5 are emerging as an important resource for studying the role of cellular heterogeneity during respiratory viral infections. These cultures maintain well-differentiated patient-derived primary airway bronchial surface epithelial cells at an air–liquid interface3,4,5. These cultures comprise ciliated, non-ciliated, goblet and basal cells, thus recapitulating the complex multicellular environment of the lung5. Single-cell RNA sequencing (scRNA-seq)6,7,8,9,10,11,12,13,14,15,16,17 techniques reveal cell-to-cell variability in viral infections and provide important insights into the heterogeneity of transcriptional responses and viral growth. Live-cell imaging and advances in single-particle tracking have also increased our understanding of heterogeneous viral infections of individual cells18. The study of pleomorphic viruses has also benefited from technological advances. Historically, cryoelectron microscopy has been used to resolve the structure of homogeneous virus particles through two-dimensional averaging and single-particle reconstruction. However, heterogeneous samples such as pleomorphic viruses cannot be resolved in this way. Cryoelectron tomography19,20,21 and subtomogram averaging eliminated this barrier in structural biology and now enable visualization of heterogeneous virus particles.
In this Review, we highlight the key findings of studies using the above techniques as well as other innovative studies as we dissect phenotypic heterogeneity at different steps of the viral replication cycle. Although we have restricted our discussion to recent examples relevant for human disease, we invite the reader to consider the implications of the heterogeneity discussed here on other viruses, such as environmental viruses and viruses with plant and bacterial hosts.
Entry
Heterogeneity in entry receptor usage is a well-established concept in virology. It has long been recognized that viruses may use several receptors to gain entry into different cell types1. In this section, we highlight recent studies that reveal the importance of viral heterogeneity for receptor avidity and the importance of host heterogeneity in PTMs of receptors. Heterogeneity in host receptor preference and PTMs extends our understanding beyond dogmatic claims of viral tropism based on the presence of specific receptors on host cells. Glycosylation is perhaps the best-studied PTM relevant for viral entry; accordingly, we focus on this PTM and its implications for viral pathogenesis and host range.
Viral heterogeneity in receptor avidity
Viruses exploit abundant host receptors to enter into target cells, and recent studies have shown that receptor glycosylation influences avidity. Haemagglutinin (HA) of human influenza virus binds ubiquitous α2,6-linked sialic acid glycan moieties on the cell surface to initiate entry into target cells, and several different cellular receptors then contribute to the entry process22. A recent study demonstrated that weak virion affinity for sialic acid can lead to multiple transient association and dissociation events along the cell surface, enabling the virus to travel to regions actively undergoing endocytosis and enhancing uptake into an infected cell23. Interestingly, the direction of virion motion on the cell surface depends on virion morphology: movement of spherical virus particles lacks directionality, but filamentous virions travel in a straight, directed manner24. As discussed in the Assembly section, neuraminidase (NA) from influenza virus, which cleaves sialic acids and is required for motility23, is highly polarized on the surface of filamentous virions25. Given these observations, it is tempting to speculate that the density and distribution of HA and NA molecules on a virion and the respective affinity for sialic acid binding and release may influence the movement of viruses along cell surfaces and affect virus entry. It is worth noting that receptor avidity is not static for influenza viruses, as evolution of the HA protein increases26 or decreases27 the affinity for α2,6-linked sialic acid. In recent years, H3N2 viruses and pandemic H1N1 viruses have caused more severe disease than previous strains28,29, which may be linked to changes in receptor avidity for long-chain branched glycans30. Further insight into the relationship between viral entry and virion morphology is needed to connect the role of pleomorphic viral populations in the infectivity and spread of viruses.
Host heterogeneity in receptor glycosylation
Receptor glycosylation varies between cell types and in different hosts, leading to differential receptor avidity during viral infection31,32,33,34,35,36,37,38. The arenavirus Lassa virus preferentially enters cells that express α-dystroglycan (α-DG) modified with long-chain matriglycans39. Like-glycosyltransferase (LARGE) glycosylates α-DG31. The extent to which LARGE modifies α-DG influences the avidity of Lassa virus for α-DG, and in the absence of these long-chain PTMs, Lassa virus alternatively uses TAM (Tyro3, Axl, Mer) receptors32,40. Population genetics data indicate positive selection of certain LARGE alleles in West Africa, a region where Lassa haemorrhagic fever is endemic31. Therefore, population heterogeneity in α-DG glycosylation by LARGE may influence the prevalence and severity of Lassa fever.
Similar findings have been reported for hepatitis C virus (HCV)34 (Fig. 2a). Glycosylation of human scavenger receptor class B type I (SR-BI), an HCV co-receptor41,42, is thought to be mediated by UDP-glucose:glycoprotein glucosyltransferase 1 (UGGT1)34. Silencing of UGGT1 or inhibition of N-linked glycosylation of SR-BI reduces its expression, and this diminishes HCV entry34. SR-BI is also a receptor for high-density lipoprotein (HDL) and some individuals with high HDL levels carry the SR-BI variant T175A33, which not only interferes with removal of HDL but also disrupts SR-BI glycosylation and does not support SR-BI-mediated HCV entry43. Therefore, allelic variants that change receptor PTMs may profoundly impact receptor avidity. Future studies will be necessary to determine whether such allelic variants influence disease outcomes.

a | Differences between individual hosts can affect viral entry, such as heterogeneous host populations (left) that carry differentially modified viral receptors. In the example shown, hepatitis C virus (HCV) has different affinities for variants of its co-receptor, scavenger receptor class B type I (SR-BI). The glycosylated wild-type (WT) T175 variant (top) binds HCV, and the unmodified T175A variant found in human SR-BI (bottom) does not. b | Furthermore, species-level determinants of viral entry exist. Middle East respiratory syndrome coronavirus (MERS-CoV) differs in its affinity for different variants of the receptor dipeptidyl peptidase 4 (DPP4). Humans, camels and bats carry unmodified DPP4 (left), which binds MERS-CoV, whereas the glycosylated DPP4 variants found in mice, ferrets and guinea pigs (right) do not support infection. *Additional determinants exist.
Consequences of receptor heterogeneity for host range
Receptor glycosylation can also influence the host range of a virus (Fig. 2b), as is the case for many pathogens associated with human pandemics, including influenza virus and at least two coronaviruses: severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). SARS-CoV and the newly emerged SARS-CoV-2, the causative agent of coronavirus disease 2019 (COVID-19), both use angiotensin-converting enzyme 2 (ACE2) as a receptor44,45, whereas MERS-CoV uses dipeptidyl peptidase 4 (DPP4) for entry into host cells46. For both MERS-CoV and SARS-CoV, receptor glycosylation leads to a species barrier. Bat, camel and human DPP4 orthologues, which are not glycosylated, support infection of MERS-CoV, but glycosylated DPP4 orthologues from other species do not36,37. Glycosylated DPP4 does not support MERS-CoV entry in cells expressing mouse, ferret, hamster and guinea pig DPP4 orthologs36,37. Removal of DPP4 glycosylation fully restores permissivity of cells expressing mouse DPP4 and partially restores permissivity of cells expressing hamster and guinea pig DPP4 to MERS-CoV infection36,37. SARS-CoV efficiently replicates in a wide range of animals, most prominently humans, bats and palm civets (reviewed elsewhere38). A species barrier exists in mice and rats, however38. This barrier has been mapped to two mutations in the ACE2 receptor, one of which introduces a glycosylation site in rat ACE2 that sterically hinders SARS-CoV infection. The newly emerged SARS-CoV-2 also uses ACE2 as its receptor45 and considerable interest in the receptor avidity of SARS-CoV-2 is already apparent47. Whether glycosylation or other PTMs in ACE2 or in the receptor-binding protein, spike (S), have influenced the emergence of this pandemic virus should be explored.
Host range is heavily influenced by the glycan specificities of influenza A viruses (IAVs) in different hosts. Most epithelial cells in the human respiratory tract preferentially express long-chain α2,6-linked sialic acid glycans, whereas avian gut cells express α2,3-linked sialic acids35. Correspondingly, human-origin IAV strains have higher affinity for long-chain α2,6-linked sialic acids, which predominate in the human respiratory tract, and avian-origin IAV strains have higher affinity for α2,3-linked sialic acids35,48,49. As we have already seen, affinity for specific sialic acid linkages can change over time30, with consequences for disease severity. As we continue to study and learn from past pandemics, the impact of PTMs on receptor avidity and host range must be explored.
Replication
There is an exquisite balance between viruses and hosts during viral replication, and clinical outcomes are further influenced by interactions between viruses and the host immune response. Accordingly, we see extensive viral and host phenotypic heterogeneity during this step. Error-prone viral polymerases50,51,52 and genetic reassortment contribute to viral phenotypic heterogeneity. Relative to DNA viruses, RNA viruses show exceptionally high replication error rates that provide genetic versatility in different fitness landscapes53. In addition to the accumulation of random mutations, low-fidelity viral polymerases are prone to ‘slipping’ during replication, increasing heterogeneity in viral transcripts54. Host heterogeneity can also alter viral replication. Here, we examine how viral and host heterogeneity manifest during viral replication, specifically the diversity of host immune responses in individual cells, defective viral genomes (DVGs) and viral population diversity.
Inverse relationship between viral replication and host immunity within a cell
scRNA-seq has been used to examine the viral RNA burden across single cells. This methodology revealed cell-to-cell variation in viral RNA abundance for IAVs, dengue virus, poliovirus, West Nile virus and Zika virus6,9,11,12 ranging from <0.1 to 50% of the cellular transcriptome12. Viral infections trigger antiviral defences upon activation of interferon and downstream interferon-stimulated genes55. However, scRNA-seq demonstrated that robust activation of interferon-β (IFNβ) mRNA transcripts is found in only a few cells infected with IAVs or West Nile virus6,8,12. Defective IAV particles activate interferon expression in bulk cells56, yet individual infected cells rarely induce interferon production16,57. West Nile virus-infected cells exhibit several expression patterns of individual interferon-stimulated genes across single cells6. Interferon-stimulated gene expression ranged from strongly negatively correlated to weakly positively correlated with viral RNA abundance6. It is likely that use of scRNA-seq in a wide range of cell types will spur the detection of novel antiviral gene candidates with therapeutic potential6,11. Whether these observations indicate active suppression of interferon or a failure to activate interferon should be explored further.
The scRNA-seq methodology has been indispensable in the study of incomplete or semi-infectious influenza virus particles. The IAV genome consists of eight viral RNA segments that are not always expressed in an infected cell13,58,59. Failure to express all eight segments impairs viral mRNA transcription and the productivity of infection12,13,17,60,61. More recently, one study coupled single-cell transcriptomics with long-read PacBio sequencing, determining that two-thirds of cells were infected with IAV variants with mutations and deletions that lead to higher levels of immune activation than the wild-type variant8. Sequencing viral genes in individual cells captures heterogeneity that has so far been overlooked and provides a unique perspective on viral species present during infection.
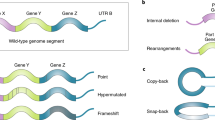
Defective viral genomes are drivers of replicative heterogeneity
Most RNA viruses produce DVGs upon replication at high multiplicity of infection7. DVGs are truncated forms of the viral genome that lack essential genes and cannot replicate in the absence of a helper virus with complementing components. Two main types of DVGs exist, deletion and copy-back DVGs7,62. Deletion DVGs lack parts of the genome and hundreds of different variants of these have been identified from a single infection7. By contrast, copy-back DVGs are regulated by sequence-specific ‘break points’ and ‘rejoin points’ at which the polymerase detaches and rejoins the template, and thus are not stochastic in origin62. For unknown reasons, Sendai virus DVGs are unable to interact with host factors involved in viral packaging and budding, limiting replication of these variant genomes in the absence of full-length genome complementation63,64. Evading or counteracting the host antiviral response is critical for productive infection and efficient replication. Yet DVG production decreases infectivity by competing with full-length genomes and robustly triggering the host antiviral response. DVGs also affect temporal variation of the host cell transcriptome61. The role of DVGs during infection remains unclear but may lie in providing diversity to viral populations with high recombination or reassortment rates, or in offering a link to the development of persistent infections.
Viral population diversity and implications for host fitness
Cross-species transmission is a major driver of viral evolution and diversity, with greater genetic heterogeneity maximizing the likelihood of host jumping. RNA virus families such as Rhabdoviridae and Picornaviridae that undergo frequent host switching are substantially less co-evolved with their hosts than double-stranded DNA viruses, which infrequently switch hosts65. This suggests that the genetic plasticity of RNA viruses allows host switching with the potential cost of reduced fitness in the original host; however, there are noteworthy exceptions to this theory, including the HCV and HIV-1 examples described at the end of this section. Here, we highlight recent examples of genetic heterogeneity in different viruses and examine how they impact host fitness and host jumping.
Population heterogeneity facilitates host jumping in different ways. In the arthropod-borne chikungunya virus, acquisition of the A226V mutation in the envelope protein E1 expands vector competence from the tropical mosquito vector Aedes aegypti to the Asian tiger mosquito Aedes albopictus, an invasive mosquito tolerant of both tropical and temperate climates66,67. However, the E1-A226V mutation requires lineage-specific epistatic mutations, with the Asian chikungunya virus lineage unable to adapt to Ae. albopictus68. Different rabies virus (RABV) lineages also exhibit differential propensities for host jumping69. Dog-adapted and fox-adapted RABV lineages both show comparable rates of population heterogeneity during serial passage, yet only dog-adapted RABV efficiently replicates in both the domestic dog and the European red fox69. These observations suggest that adaptation within certain host species can increase viral variants capable of host switching. In addition, the acquisition of certain genetic changes through viral evolution can influence the success of a given strain in a new host.
The importance of host-adaptive mutations on cross-species jumping has been examined most often for zoonotic viruses that successfully jump into human hosts, often spurring severe outbreaks that occasionally lead to widespread pandemics. Wild birds are the primary reservoir for zoonotic IAVs70, and the virus can spread from birds to a wide range of hosts, which can promote viral heterogeneity and occasional spillover events to humans, as observed for H5N1, H7N9, H9N2, H6N1 and H10Nx strains. Bats are believed to harbour more zoonotic viruses than any other animal, including RABV, Hendra virus, Nipah virus, Ebola virus, Marburg virus, hantaviruses and coronaviruses (reviewed elsewhere71,72). Spillover events of these viruses to human hosts tends to cause severe morbidity and mortality71. However, spillover of bat viruses to humans often involves an intermediate host70,73,74. The exact reasons for the involvement of intermediate hosts remain unclear, but include barriers to infection71 such as receptor specificities described in the Entry section. Nevertheless, viral families with a wide host-tropism can accommodate viral diversity to facilitate host jumping from animal reservoirs to humans.
Whereas population heterogeneity widens the host range of some viruses, viral diversity in a single host is associated with persistent infections in a single host. Considerable within-host diversity and population structure are observed for HCV and HIV-1 (refs75,76,77,78). Viral diversity in HCV infection can enhance liver cirrhosis necessitating transplantation79, with minor variants persisting in the new liver following transplantation76. A hallmark of HIV-1 infection is compartmentalization in different anatomical sites, triggering diversification into distinct, independently evolving subpopulations75,78,80,81,82,83. HIV-1 env gene compartmentalization in cerebrospinal fluid is linked to HIV-associated dementia84,85, suggesting that genetic diversification of HIV-1 and pathogenesis and disease progression might be related.
From these examples, it is clear that population heterogeneity can have vastly different outcomes for viral fitness in different hosts. The examples illustrated here include a wide range of genetically plastic RNA viruses that nonetheless exhibit very different propensities for host jumping. It is clear that population heterogeneity affords extreme flexibility in a virus to either co-adapt to a specific host or to host switch.
Assembly
Heterogeneity in virion structure, composition and morphology impact biological processes86,87,88,89,90. For viruses with segmented genomes, a full-length genome must be packaged with high accuracy by properly folded virion components to ensure formation of fully infectious virions. For many viruses, highly regular particles form with readily defined morphology and identifiable patterns of symmetry that cannot accommodate more than one copy of the viral genome. Recent reviews have discussed heterogeneity in structural protein maturation, such as the high proportion of immature virions produced during flavivirus infection91,92,93. Here, we focus our discussion on newly described and historically difficult to resolve structures of pleomorphic viruses and variations in virion glycoprotein levels. Pleomorphic virus particles are more flexible than viruses with regular morphology, producing a highly heterogeneous mixture of virion particle sizes. These heterogeneous structures have relaxed constraints on genomic packaging and, in some cases, do package more than one genome copy94,95. Accordingly, we also discuss low-fidelity packaging of segmented viral genomes.
Semi-infectious particles
Many viruses exhibit a particle to plaque-forming unit (PFU) ratio greater than 1, suggesting that not all viral particles are equally infectious. Particle to PFU ratios vary among virus family and type, ranging from 1 or 2 for Semliki Forest virus to as high as 107 for some HIV-1 variants96. Segmented viruses have complex packaging requirements in which a single copy of each genomic segment must be packaged to produce an infectious virion. Packaging of the segmented bunyavirus, Rift Valley fever virus, is thought to be random, producing virions lacking one or more segments and leading to infected cells failing to express all viral proteins97. Meanwhile, packaging of IAVs is tightly coordinated to incorporate one copy of each segment into the majority of virions98. Despite this quality control, approximately 20% of IAV particles package fewer than eight genomic segments99, and a large number of infected cells fail to express IAV proteins from all eight segments or to replicate each segment equally58. Viruses either lacking gene segments or unable to fully translate or replicate all eight viral gene segments are referred to broadly as ‘semi-infectious particles’58. The origin of naturally occurring semi-infectious particles is unknown and could be a consequence of incomplete genome packaging, failed nuclear import of viral RNA segments, abortive replication or epigenetic silencing of viral RNA segments. Recently, one study demonstrated that the fitness cost of infection with semi-infectious IAV particles is reduced through complementation by incoming virions from neighbouring infected cells87. Semi-infectious particles are observed in IAV populations in vitro and in vivo87, and may drive viral heterogeneity within a host and, potentially, between hosts. Therefore, the possibility of complementation of IAV genomic segments in humans and its relationship to IAV transmission should be explored.
Pleomorphic particles
Influenza virus, HIV, rubella virus, pneumoviruses and paramyxoviruses such as measles virus are all pleomorphic19,100,101,102,103. Rather than a strictly uniform virion structure, these viruses are highly variable in length and/or diameter and versatile in genomic packaging capacity94,104,105. Characterization of pleomorphic structures has been historically challenging, but these particles are now being resolved through cryo-electron tomography19,20,21,103. This method was recently used to resolve the helical glycoprotein arrangement of rubella virus19.
Low-fidelity assembly is intricately interwoven with host adaptation, escape from antibodies or antiviral drugs and pathogenesis. IAVs are highly heterogeneous in structure, with a single cell capable of producing a diverse array of particles, including spherical, ovoid and bacilliform particles and filaments of varying length and diameter102. The filamentous morphology of IAVs has been comprehensively reviewed recently102. Disruption of the actin cytoskeleton selectively impairs the budding of filamentous, but not spherical, virions102, implicating distinct mechanisms underlying the formation of each type of virion. Human clinical IAV isolates are typically filamentous in morphology, but many laboratory strains, which are usually produced in embryonated eggs, produce exclusively spherical particles102. Further investigation of the host and viral determinants of virion morphology will be necessary to determine the factors underlying these observations.
Virion morphology may have implications for pathogenesis. In influenza virus, HA is enriched over NA in longer filamentous particles86, shifting the ratio of HA to NA molecules per virion and potentially impacting entry kinetics, NA activity and cleavage of sialic acids during budding. Differences in the HA to NA ratio on influenza virus particles could also influence sensitivity to NA antivirals86, and possibly antibody epitope accessibility. Interestingly, efficient transmission of H1N1 2009 pandemic influenza viruses correlated with NA activity and filamentous virion morphology88,89,90. Further investigation of the mechanism underlying these observations is necessary. Alternatively, the glycoprotein arrangement on filamentous particles may explain differences in HA and NA activity. As described in the Entry section, NA is polarized on filamentous virions25. Whether these morphological differences impact pathogenesis remains an active area of investigation.
Measles virus assembly also exhibits phenotypic heterogeneity, which may have consequences for clinical disease progression. During assembly, the measles virus matrix (M) protein binds the inner surface of the plasma membrane and the cytoplasmic tails of the viral fusion (F) glycoprotein and the attachment glycoprotein haemagglutinin (H)106, and is believed to coordinate assembly107. In rare cases, mutation or deletion of the F or H glycoprotein cytoplasmic tails accelerates measles virus dissemination through cell monolayers by the formation of syncytia and through the brain of patients suffering from subacute sclerosing panencephalitis106,108,109, a highly lethal, progressive neurological disorder resulting from persistent measles infection. Interestingly, measles virus vaccine strains with mutations in the M protein have higher avidity for the H cytoplasmic tail, but are defective for syncytia formation. Further investigation of this apparent link between the avidity of M protein for the glycoprotein cytoplasmic tails and the role of syncytia formation in disease progression is needed.
Future studies on the role of low-fidelity assembly in the pathogenesis of other pleomorphic viruses is warranted, but lacking. It stands to reason that low-fidelity assembly may promote dissemination of other pleomorphic viruses.
Egress
Pathogens are faced with a litany of obstacles in the extracellular environment of a host. Viruses are routinely confronted with antibody neutralization, pattern recognition receptors and the downregulation of the cell surface receptors required for entry. Viruses have, in turn, developed strategies to improve cell-to-cell spread and the chance of entry. Here, we examine a few alternate modes of cell-to-cell spread that blur the lines between entry and egress.
Tunnelling nanotubes promote rapid entry and egress
A growing body of evidence suggests that viruses can bypass the extracellular environment altogether during subsequent rounds of infection. For many viruses, this process involves subversion of the actin–myosin cytoskeletal system through tunnelling nanotubes (TNTs). TNTs are actin-rich filamentous cellular projections that facilitate intercellular exchange of cargo such as organelles, proteins and ions110. TNTs are sometimes referred to as nanotubular bridges or immunological or virological synapses, depending on the context. Interestingly, major histocompatibility complex (MHC) class I molecules are among cargo transferred along recently dispersed immunological synapses, suggesting a role for TNTs in antigen presentation111. Two distinct classes of TNTs have been reported that differ in size, composition and function110,112. Thin nanotubes comprise filamentous actin (F-actin) and facilitate unidirectional cargo transfer110,112. Thick nanotubes have a diameter greater than 0.7 μm, comprise both F-actin and tubulin, and are capable of bidirectional cargo transfer110,112. The latter class, thus far reported only in macrophages, permits cargo exchange between cells tens to hundreds of microns apart111,113. TNTs form either through actin polymerization or upon disassembly of a synapse111 with lifetimes ranging from several minutes to over an hour110.
A broad swath of pathogenic viruses reportedly hijack TNTs, including retroviruses, herpesviruses, vaccinia virus, flaviviruses, human metapneumovirus (HMPV), measles virus and IAVs3,4,5,113,114,115,116,117,118,119,120,121,122,123. The mechanism through which each of these viruses traverses a nanotubular bridge appears to be distinct, although the use of the actin–myosin cytoskeletal machinery for trafficking along a TNT is common for many3,4,114,115. Retroviruses specifically traffic along the exterior surface of TNTs in a fashion that retains the classical egress step of budding from a host cell and receptor-mediated entry into the next cell. Murine leukaemia virus and HIV-1 bud from an infected cell and traffic unidirectionally along the outer surface of TNTs113,114,116. Receptor-mediated binding of the murine leukaemia virus envelope glycoprotein, Env, stimulates outgrowth of a long, stable bridge between infected and uninfected cells, triggering apparent invagination through endocytosis116. TNTs are intrinsically more stable in human T cells, however, and intercellular dissemination of HIV-1 to uninfected T cells through TNTs is more efficient than that of cell-free virions113,117,118.
In stark contrast to the use of TNTs by retroviruses, HMPV, IAVs and measles virus commandeer nanotubular bridges for direct transfer of viral genomic RNA across tight junctions in the host epithelium3,4,5,119. HMPV phosphoprotein actively induces nanotube formation through actin polymerization and restructuring to promote cell-to-cell spread3. Intriguingly, HMPV and measles virus viral RNA and proteins traverse TNTs in lung epithelial cell lines and differentiated airway epithelial cell cultures3,5,119, suggesting that direct delivery of viral RNA into target cells occurs in the absence of classically described modes of virion egress and receptor-mediated entry. This mode of viral dissemination protects viruses from antibody neutralization during infection3.
TNTs thus seem to be a viable and attractive alternative mode of entry and egress for numerous viruses (Fig. 1b). Given that a diverse set of viruses have evolved mechanisms to traffic along TNTs, it is presumed that TNTs are widely used during viral infection. However, studies of viral trafficking along TNTs remain restricted to a small subset of cell types, frequently immortalized cell lines, some of which may not be representative of clinical disease. Given the considerable diversity of TNTs reported across cell types, further studies of TNT formation and use in relevant models of viral pathogenesis are warranted.
The use of TNTs for delivery of viral RNA to neighbouring cells by HMPV and measles virus has enormous implications for viral dissemination and suggests that TNT cargo is more diverse than previously appreciated. Do most viruses traffic along the exterior of TNTs, as HIV-1 does, or are intercellular TNTs widely used to evade circulating antibodies and pattern recognition receptors on cell surfaces? The answers to these questions will undoubtedly reflect the heterogeneous nature of TNTs.
Cloaking of viruses in host microvesicles
Given the vast heterogeneity in viral replication, transmission of communities of many virus particles en masse may complement defective or incomplete genomes. Recent work has revealed that picornaviruses package multiple viral particles into a cellular microvesicle to promote the spread of many heterogeneous virions together124. Aggregation of enveloped viruses, such as vesicular stomatitis virus, can also facilitate the transmission of viral communities125. Intriguingly, for many RNA viruses, the minimal infectious unit is the viral genome rather than a fully encapsidated and/or enveloped virion. Although they may improve replication efficiency, proteins packaged by an RNA virus into a virion are often not essential for replication, with the exception of nucleoprotein complexes for negative-sense RNA viruses. Historically, viruses have been classified as one of two binary types, enveloped or non-enveloped, but recent studies have indicated surprising overlap in this classification.
Many enteroviruses, such as hepatitis A virus (HAV), were historically believed to only encapsidate their genome and undergo lytic egress as non-enveloped particles. Although this mode of egress may still predominate, recent work describes a duality in viral particle composition in which quasi-enveloped HAV particles acquire an envelope from extracellular vesicles and shed non-lytically from the host cell126. Enveloped HAV biogenesis resembles that of exosomes in multivesicular bodies as they are decorated with host exosomal markers, including CD9 and DPP4127. Assembly of enveloped HAV also depends on at least two components of the endosomal sorting complex (ESCRT), VPS4B and ALIX126.
Microvesicle cloaking has emerged in recent studies as a common egress strategy for many viruses. Other members of the Picornaviridae family exhibit particle duality as well, but the mechanism of envelopment during enterovirus assembly is distinct from that of hepatoviruses128. Poliovirus and Coxsackievirus B3 (CVB3) are shed non-lytically through the autophagy pathway128,129. Poliovirus, CVB3 and rhinovirus are all found in phosphatidylserine-containing lipid vesicles, and the infectivity of these enveloped particles is higher than that of canonical non-enveloped particles124. The authors of this study used innovative imaging approaches, including total-internal reflection fluorescence with super-resolution microscopy and direct stochastic optical reconstruction microscopy, to demonstrate that viral particles are packaged in microvesicles in clusters, thereby artificially increasing the multiplicity of infection124. Thus, it seems that the classically non-enveloped viruses of the Picornaviridae family can subvert different host processes to achieve non-lytic egress. In addition to HAV, the classically non-enveloped enteroviruses, norovirus and rotavirus, both use microvesicle cloaking for similarly enhanced infectivity130.
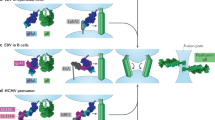
Enveloped viruses such as IAVs may also hijack microvesicle assembly131. The protein composition of the IAV virion includes over 300 host proteins, and many of these are known exosome markers, such as the tetraspanin CD9 (ref.131) identified in enveloped HAV127 (Fig. 3). Microvesicle assembly has therefore emerged as a common mode of egress for many virus families, from the canonically non-enveloped picornaviruses to enveloped viruses such as influenza.

a | Traditionally, all pleomorphic influenza virus particles were thought to comprise viral components only. This traditional influenza virus particle is shown, displaying spherical morphology and consisting of haemagglutinin (HA), neuraminidase (NA), matrix (M1) and viral genomic material. b | Careful studies of virions have revealed a revised architecture consisting of an influenza virion that encases several components of host exosomes131. c | Influenza virus also assumes a filamentous morphology of variable length with polarized NA proteins. ANXA, annexin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; UBB, ubiquitin.
Given that enveloped picornaviruses show higher infectivity than their non-enveloped counterparts, cloaking a virus in a microvesicle may have huge implications for entry and egress beyond particle morphology and infectivity. These viral vesicles can be passively taken up by neighbouring cells through processes such as clathrin-mediated endocytosis, bypassing the requirement for a cellular receptor for entry. In the case of picornaviruses, non-lytic egress presents a mechanism for rapid dissemination through tissues without alerting nearby cells to the presence of a pathogen upon lysis. Continued investigation of such possibilities remains paramount to our understanding of viral entry and egress and the development of antivirals to target these processes.
Implications for medicine
Viral heterogeneity poses a difficult challenge for medicine. Adequate medical interventions are still lacking for many viral infections, particularly the ones discussed in this Review. Vaccines are available for several of the pathogens described here, but the emergence of novel viruses such as SARS-CoV-2 or zoonotic IAV strains is also a major public health concern. Viral heterogeneity influences many facets of disease, so here we highlight some elements worthy of deeper contemplation.
Antivirals and vaccines in the face of heterogeneous entry mechanisms
Current technologies make it easier than ever before to screen thousands of compounds for efficacy against viral infection and rapidly identify potential new therapeutic candidates. Nevertheless, these results should be interpreted with caution. A given virus may exhibit extraordinary diversity in genomic content and particle morphology, so candidate therapeutics must be pan-protective against a heterogeneous viral population. Consideration should be given to the cell type and virion morphology as well as the diversity in entry and egress processes. Many antiviral therapies identified to date are entry inhibitors, yet we have seen how diverse mechanisms of entry and egress enable rapid dissemination through tissues. The use of TNTs for delivery of viral genomes to a neighbouring cell bypasses receptor-mediated entry altogether. Therefore, entry inhibitors may prove ineffective against such infections. TNTs may also facilitate viral escape from antibody neutralization, thus weakening the effectiveness of vaccination. Such infections may progress faster than a classical infection would, as rapid dissemination to cells hundreds of microns apart can readily occur111,113. Post-entry therapeutics or combination therapies with nanotube inhibitors might prove to be more effective against such viral diseases.
Dissemination of viral genomic material
A diverse set of viruses are cloaked in host microvesicles. Cloaked picornaviruses have higher infectivity than traditionally described non-enveloped particles and may disseminate more efficiently. Cloaked particles may have other functions in addition to spreading virus particles; in the case of influenza virus, they may complement semi-infectious virus particles lacking one or more gene segments. Given that picornaviruses reportedly package multiple viral particles per microvesicle to achieve dissemination of many virions en masse124, one could envision that microvesicles promote co-infection. Interestingly, microvesicle cloaking does not increase genetic complementation or population diversity during co-infection with multiple CVB3 variants132. This finding suggests that packaging of CVB3 into host microvesicles is highly selective. Considering that genetic reassortment is commonly reported at high multiplicity of infection during influenza virus co-infection133, further investigation will be necessary to determine whether the selectivity observed in CVB3 microvesicles applies to other viruses. The mechanism of such high-fidelity packaging in microvesicles is nonetheless an exciting new area of research.
Heterogeneity in virion assembly may promote severe disease
Pleomorphic particles have been reported for decades, but the effect of heterogeneous assembly mechanisms on viral pathogenesis remains understudied. An intriguing function in mucus layer penetration and clearance has been proposed for long-filament IAV particles, which are defective for genomic packaging86. Such a role for low-fidelity assembly of IAV particles would have clear implications for pathogenesis and may be a viable target in the development of future therapeutics. Alternatively, the production of pleomorphic virions in a host can result in viruses with different HA to NA ratios (see Fig. 3 for an illustration). Altered HA to NA levels on a virion could impact antibody binding and may, therefore, influence vaccine efficacy.
Synergy among polymicrobial communities
Although outside the scope of this Review, the contribution of diverse polymicrobial communities to the morbidity and mortality of heterogeneous viruses is a public health dilemma in need of deeper investigation. Recently, a complex interplay between HIV-1 and Mycobacterium tuberculosis co-infection and TNT formation was described. Active tuberculosis in HIV-1-infected patients is associated with elevated HIV-1 infection of macrophages found in the lungs and pleural effusions. Active tuberculosis skews human monocyte differentiation towards an anti-inflammatory M2 macrophage pathway and blood monocytes treated with M. tuberculosis-conditioned medium are more permissive to HIV-1 infection134. M. tuberculosis infection stimulates TNT production in human macrophages, promoting HIV-1 dissemination into macrophages134. This particular phenotypic diversity likely contributes to M. tuberculosis-mediated exacerbation of HIV-1 morbidity and mortality. Further study of bacterial and viral co-infection will undoubtedly reveal additional layers of phenotypic heterogeneity in these complex systems.
Conclusions
As new technologies emerge, our appreciation of phenotypic heterogeneity in the viral replication cycle grows. From alternate egress and entry pathways to variations in genome assembly and virion morphology, these diverse replication mechanisms challenge how we classify viruses and what is considered an infectious particle. Here, we have highlighted the use of nanotubular bridges in cell-to-cell spread, exploitation of the microvesicle secretory pathway to cloak virions and promote infection, and how low-fidelity replication can drive viral heterogeneity. By redefining the canonical viral replication cycle, we will be better equipped to develop antiviral therapies against the many nuances of viral replication and address adaptive evasion strategies. Future studies examining the relationship between viral heterogeneity and disease severity are particularly needed to help refine specific antiviral targets and biosensors. Investigation of heterogeneity in infections with emerging pathogens such as SARS-CoV-2 are particularly essential for combating emerging viral threats. Studies already indicate heterogeneity in SARS-CoV-2 entry mechanisms in different cell types, which ultimately determines susceptibility to antivirals like chloroquine and its derivatives135. The gravity of the COVID-19 pandemic underscores the critical role of such studies in guiding global health policies.
References
Fields, B. N., Knipe, D. M., Howley, P. M. Fields Virology 6th edn (Lippincott Williams & Wilkins, 2013).
Shulla, A. & Randall, G. (+) RNA virus replication compartments: a safe home for (most) viral replication. Curr. Opin. Microbiol. 32, 82–88 (2016).
El Najjar, F. et al. Human metapneumovirus induces reorganization of the actin cytoskeleton for direct cell-to-cell spread. PLoS Pathog. 12, e1005922 (2016). This paper shows that HMPV viral proteins and genomic material use actin filaments to disseminate through the airway epithelium and evade neutralizing antibodies.
Roberts, K. L., Manicassamy, B. & Lamb, R. A. Influenza A virus uses intercellular connections to spread to neighboring cells. J. Virol. 89, 1537 (2015).
Singh, B. K., Pfaller, C. K., Cattaneo, R. & Sinn, P. L. Measles virus ribonucleoprotein complexes rapidly spread across well-differentiated primary human airway epithelial cells along F-actin rings. mBio 10, e02434-19 (2019).
O’Neal, J. T. et al. West Nile virus-inclusive single-cell RNA sequencing reveals heterogeneity in the type I interferon response within single cells. J. Virol. 93, e01778-18 (2019).
Vignuzzi, M. & López, C. B. Defective viral genomes are key drivers of the virus–host interaction. Nat. Microbiol. 4, 1075–1087 (2019).
Russell, A. B., Elshina, E., Kowalsky, J. R., Te Velthuis, A. J. W. & Bloom, J. D. Single-cell virus sequencing of influenza infections that trigger innate immunity. J. Virol. 93, e00500-19 (2019).
Schulte, M. B. & Andino, R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J. Virol. 88, 6205 (2014).
Patil, S. et al. Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-β response to viral infection. Sci. Signal. 8, ra16 (2015).
Zanini, F., Pu, S.-Y., Bekerman, E., Einav, S. & Quake, S. R. Single-cell transcriptional dynamics of flavivirus infection. eLife 7, e32942 (2018).
Russell, A. B., Trapnell, C. & Bloom, J. D. Extreme heterogeneity of influenza virus infection in single cells. eLife 7, e32303 (2018).
Heldt, F. S., Kupke, S. Y., Dorl, S., Reichl, U. & Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 6, 8938 (2015).
Wimmers, F. et al. Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-α production by plasmacytoid dendritic cells. Nat. Commun. 9, 3317 (2018).
Shalek, A. K. et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014).
Killip, M. J., Jackson, D., Pérez-Cidoncha, M., Fodor, E. & Randall, R. E. Single-cell studies of IFN-β promoter activation by wild-type and NS1-defective influenza A viruses. J. Gen. Virol. 98, 357–363 (2017).
Steuerman, Y. et al. Dissection of influenza infection in vivo by single-cell RNA sequencing. Cell Syst. 6, 679–691.e4 (2018).
Bhagwat, A. R. et al. Quantitative live cell imaging reveals influenza virus manipulation of Rab11A transport through reduced dynein association. Nat. Commun. 11, 23 (2020).
Mangala Prasad, V., Klose, T. & Rossmann, M. G. Assembly, maturation and three-dimensional helical structure of the teratogenic rubella virus. PLoS Pathog. 13, e1006377 (2017).
Liu, J., Bartesaghi, A., Borgnia, M. J., Sapiro, G. & Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 (2008).
White, T. A. et al. Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS Pathog. 6, e1001249 (2010).
Shaw M. L., Palese P. in Fields Virology Ch. 40 (eds Howley, P. & Knipe, D. M.) 1152–1181 (Lippincott Williams & Wilkins, 2013).
Sakai, T., Nishimura, S. I., Naito, T. & Saito, M. Influenza A virus hemagglutinin and neuraminidase act as novel motile machinery. Sci. Rep. 7, 45043 (2017). This paper shows that IAV HA and NA proteins coordinate iterative receptor binding and release, resulting in accelerated viral motility and endocytosis into target cells.
Sakai, T., Takagi, H., Muraki, Y. & Saito, M. Unique directional motility of influenza C virus controlled by its filamentous morphology and short-range motions. J. Virol. 92, e01522-17 (2018).
Vahey, M. D. & Fletcher, D. A. Influenza A virus surface proteins are organized to help penetrate host mucus. eLife 8, e43764 (2019). This paper shows that HA and NA proteins are asymmetrically distributed on filamentous IAV particles, contributing to directional motility.
Hensley, S. E. et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 326, 734 (2009).
Nobusawa, E., Ishihara, H., Morishita, T., Sato, K. & Nakajima, K. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acid change in hemagglutinin altered its recognition of sialyloligosaccharides. Virology 278, 587–596 (2000).
Centers for Disease Control and Prevention. Summary of the 2017–2018 influenza season (CDC, 2019).
Jester, B. J., Uyeki, T. M. & Jernigan, D. B. Fifty years of influenza A (H3N2) following the pandemic of 1968. Am. J. Public Health 110, 669–676 (2020).
Peng, W. et al. Recent H3N2 viruses have evolved specificity for extended, branched human-type receptors, conferring potential for increased avidity. Cell Host Microbe 21, 23–34 (2017). This paper uses an influenza virus receptor glycan microarray to demonstrate that H3N2 viruses have evolved to retain specificity for human-type sialic acid but gain selectivity for elongated, branched glycans with greater avidity for HA.
Yoshida-Moriguchi, T. & Campbell, K. P. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 25, 702–713 (2015).
Fedeli, C. et al. Axl can serve as entry factor for Lassa virus depending on the functional glycosylation of dystroglycan. J. Virol. 92, e01613-17 (2018).
Chadwick, A. C. & Sahoo, D. Functional characterization of newly-discovered mutations in Human SR-BI. PLoS ONE 7, e45660 (2012).
Huang, J. et al. SR-BI interactome analysis reveals a proviral role for UGGT1 in hepatitis C virus entry. Front. Microbiol. 10, 2043 (2019).
de Graaf, M. & Fouchier, R. A. M. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 33, 823–841 (2014).
Peck, K. M. et al. Glycosylation of mouse DPP4 plays a role in inhibiting Middle East respiratory syndrome coronavirus infection. J. Virol. 89, 4696 (2015).
Peck, K. M. et al. Permissivity of dipeptidyl peptidase 4 orthologs to Middle East respiratory syndrome coronavirus is governed by glycosylation and other complex determinants. J. Virol. 91, e00534-17 (2017).
Li, F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 100, 246–254 (2013).
Hastie, K. M. et al. Structural basis for antibody-mediated neutralization of Lassa virus. Science 356, 923–928 (2017).
Shimojima, M., Ströher, U., Ebihara, H., Feldmann, H. & Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 86, 2067–2078 (2012).
Zeisel, M. B., Felmlee, D. J. & Baumert, T. F. in Hepatitis C Virus: From Molecular Virology to Antiviral Therapy (ed. Bartenschlager, R.) 87–112 (Springer, 2013).
Ding, Q., von Schaewen, M. & Ploss, A. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16, 562–568 (2014).
Westhaus, S. et al. Scavenger receptor class B member 1 (SCARB1) variants modulate hepatitis C virus replication cycle and viral load. J. Hepatol. 67, 237–245 (2017).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444 (2020).
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013).
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127-20 (2020).
Raman, R., Tharakaraman, K., Sasisekharan, V. & Sasisekharan, R. Glycan–protein interactions in viral pathogenesis. Curr. Opin. Struct. Biol. 40, 153–162 (2016).
Walther, T. et al. Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 9, e1003223 (2013).
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M. & Belshaw, R. Viral mutation rates. J. Virol. 84, 9733 (2010).
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Bordería, A. V. et al. Group selection and contribution of minority variants during virus adaptation determines virus fitness and phenotype. PLoS Pathog. 11, e1004838 (2015).
Lauring, A. S., Frydman, J. & Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 11, 327–336 (2013).
Sanjuán, R. & Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 73, 4433–4448 (2016).
Crosse, K. M., Monson, E. A., Beard, M. R. & Helbig, K. J. Interferon-stimulated genes as enhancers of antiviral innate immune signaling. J. Innate Immun. 10, 85–93 (2018).
Iwasaki, A. & Pillai, P. S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328 (2014).
Kallfass, C., Lienenklaus, S., Weiss, S. & Staeheli, P. Visualizing the β-interferon response in mice during infection with influenza A viruses expressing or lacking nonstructural protein 1. J. Virol. 87, 6925–6930 (2013).
Brooke, C. B. et al. Most influenza a virions fail to express at least one essential viral protein. J. Virol. 87, 3155–3162 (2013).
Dou, D. et al. Analysis of IAV replication and co-infection dynamics by a versatile RNA viral genome labeling method. Cell Rep. 20, 251–263 (2017).
Sjaastad, L. E. et al. Distinct antiviral signatures revealed by the magnitude and round of influenza virus replication in vivo. Proc. Natl Acad. Sci. USA 115, 9610–9615 (2018).
Wang, C. et al. Cell-to-cell variation in defective virus expression and effects on host responses during influenza virus infection. mBio 11, e02880-19 (2020).
Sun, Y. et al. A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog. 15, e1007707 (2019).
Genoyer, E. & López, C. B. Defective viral genomes alter how Sendai virus interacts with cellular trafficking machinery, leading to heterogeneity in the production of viral particles among infected cells. J. Virol. 93, e01579-18 (2019). This paper uses fluorescent in situ hybridization to reveal heterogeneity in the intracellular localization of DVGs compared with full-length genomes during Sendai virus infection.
Xu, J. et al. Replication defective viral genomes exploit a cellular pro-survival mechanism to establish paramyxovirus persistence. Nat. Commun. 8, 799 (2017). This paper uses single-cell analysis to reveal that DVGs promote a MAVS-dependent TNF pro-survival pathway, driving persistence of paramyxoviruses.
Geoghegan, J. L., Duchêne, S. & Holmes, E. C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 13, e1006215 (2017).
Tsetsarkin, K. A., Vanlandingham, D. L., McGee, C. E. & Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3, e201 (2007).
Tsetsarkin, K. A. et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 5, 4084 (2014).
Silva, L. A. & Dermody, T. S. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Invest. 127, 737–749 (2017).
Bonnaud, E. M. et al. Comparison of intra- and inter-host genetic diversity in rabies virus during experimental cross-species transmission. PLoS Pathog. 15, e1007799 (2019).
Parrish, C. R., Murcia, P. R. & Holmes, E. C. Influenza virus reservoirs and intermediate hosts: dogs, horses, and new possibilities for influenza virus exposure of humans. J. Virol. 89, 2990 (2015).
Wang, L.-F. & Anderson, D. E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 34, 79–89 (2019).
Calisher, C. H., Childs, J. E., Field, H. E., Holmes, K. V. & Schountz, T. Bats: important reservoir hosts of emerging viruses. Clin.l Microbiol. Rev. 19, 531 (2006).
Kan, B. et al. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 79, 11892–11900 (2005).
Gralinski, L. E. & Menachery, V. D. Return of the coronavirus: 2019-nCoV. Viruses 12, E135 (2020).
Raghwani, J. et al. Exceptional heterogeneity in viral evolutionary dynamics characterises chronic hepatitis C virus infection. PLoS Pathog. 12, e1005894 (2016).
Gray, R. R. et al. Unexpected maintenance of hepatitis C viral diversity following liver transplantation. J. Virol. 86, 8432 (2012).
Raghwani, J. et al. High-resolution evolutionary analysis of within-host hepatitis C virus infection. J. Infect. Dis. 219, 1722–1729 (2019).
Salemi, M. The intra-host evolutionary and population dynamics of human immunodeficiency virus type 1: a phylogenetic perspective. Infect. Dis. Rep. 5, e3 (2013).
Centers for Disease Control and Prevention. Hepatitis C questions and answers for health professionals (CDC, 2019).
Bednar, M. M. et al. Compartmentalization, viral evolution, and viral latency of HIV in the CNS. Curr. HIV/AIDS Rep. 12, 262–271 (2015).
Schnell, G., Price, R. W., Swanstrom, R. & Spudich, S. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J. Virol. 84, 2395–2407 (2010).
Anderson, J. A. et al. HIV-1 populations in semen arise through multiple mechanisms. PLoS Pathog. 6, e1001053 (2010).
Abrahams, M. R. et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J. Virol. 83, 3556–3567 (2009).
Ritola, K., Robertson, K., Fiscus, S. A., Hall, C. & Swanstrom, R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J. Virol. 79, 10830 (2005).
Harrington, P. R. et al. Cross-sectional characterization of HIV-1 env compartmentalization in cerebrospinal fluid over the full disease course. AIDS 23, 907–915 (2009).
Vahey, M. D. & Fletcher, D. A. Low-fidelity assembly of influenza a virus promotes escape from host cells. Cell 176, 281–294.e19 (2019). This paper uses site-specific labelling of IAV particles to reveal substantial heterogeneity in the size and composition of virions produced in individual cells.
Jacobs, N. T. et al. Incomplete influenza A virus genomes occur frequently but are readily complemented during localized viral spread. Nat. Commun. 10, 3526 (2019).
Campbell, P. J. et al. The M segment of the 2009 pandemic influenza virus confers increased neuraminidase activity, filamentous morphology, and efficient contact transmissibility to A/Puerto Rico/8/1934-based reassortant viruses. J. Virol. 88, 3802 (2014).
Seladi-Schulman, J., Steel, J. & Lowen, A. C. Spherical influenza viruses have a fitness advantage in embryonated eggs, while filament-producing strains are selected are selected in vivo. J. Virol. 87, 13343 (2013).
Lakdawala, S. S. et al. Eurasian-origin gene segments contribute to the transmissibility, aerosol release, and morphology of the 2009 pandemic H1N1 influenza virus. PLoS Pathog. 7, e1002443 (2011).
Kuhn, R. J., Dowd, K. A., Beth Post, C. & Pierson, T. C. Shake, rattle, and roll: impact of the dynamics of flavivirus particles on their interactions with the host. Virology 479–480, 508–517 (2015).
Sirohi, D. & Kuhn, R. J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 216, S935–S944 (2017).
Mukhopadhyay, S., Kuhn, R. J. & Rossmann, M. G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3, 13–22 (2005).
Rager, M., Vongpunsawad, S., Duprex, W. P. & Cattaneo, R. Polyploid measles virus with hexameric genome length. EMBO J. 21, 2364–2372 (2002).
Cattaneo, R., Donohue, R. C., Generous, A. R., Navaratnarajah, C. K. & Pfaller, C. K. Stronger together: multi-genome transmission of measles virus. Virus Res. 265, 74–79 (2019).
Klasse, P. J. Molecular determinants of the ratio of inert to infectious virus particles. Prog. Mol. Biol. Transl. Sci. 129, 285–326 (2015).
Wichgers Schreur, P. J. & Kortekaas, J. Single-molecule FISH reveals non-selective packaging of Rift Valley fever virus genome segments. PLoS Pathog. 12, e1005800 (2016).
Lakdawala, S. S., Fodor, E. & Subbarao, K. Moving on out: transport and packaging of influenza viral RNA into virions. Annu. Rev. Virol. 3, 411–427 (2016).
Nakatsu, S. et al. Complete and incomplete genome packaging of influenza A and B viruses. mBio 7, e01248-16 (2016).
Kuznetsov, Y. G., Victoria, J. G., Robinson, W. E. & McPherson, A. Atomic force microscopy investigation of human immunodeficiency virus (HIV) and HIV-infected lymphocytes. J. Virol. 77, 11896 (2003).
Cox, R. M. & Plemper, R. K. Structure and organization of paramyxovirus particles. Curr. Opin. Virol. 24, 105–114 (2017).
Dadonaite, B., Vijayakrishnan, S., Fodor, E., Bhella, D. & Hutchinson, E. C. Filamentous influenza viruses. J. Gen. Virol. 97, 1755–1764 (2016).
Ke, Z. et al. The morphology and assembly of respiratory syncytial virus revealed by cryo-electron tomography. Viruses 10, 446 (2018).
Dahlberg, J. E. & Simon, E. H. Physical and genetic studies of Newcastle disease virus: evidence for multiploid particles. Virology 38, 666–678 (1969).
Hausmann, S., Jacques, J. P. & Kolakofsky, D. Paramyxovirus RNA editing and the requirement for hexamer genome length. RNA 2, 1033–1045 (1996).
Tahara, M., Takeda, M. & Yanagi, Y. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell–cell fusion. J. Virol. 81, 6827–6836 (2007).
Ke, Z. et al. Promotion of virus assembly and organization by the measles virus matrix protein. Nat. Commun. 9, 1736 (2018).
Cathomen, T., Naim, H. Y. & Cattaneo, R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J. Virol. 72, 1224–1234 (1998).
Schmid, A. et al. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology 188, 910–915 (1992).
Gerdes, H.-H. & Carvalho, R. N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 20, 470–475 (2008).
Önfelt, B., Nedvetzki, S., Yanagi, K. & Davis, D. M. Cutting Edge: membrane nanotubes connect immune cells. J. Immunol. 173, 1511 (2004).
Önfelt, B. et al. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 177, 8476 (2006).
Sowinski, S. et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10, 211–219 (2008).
Lehmann, M. J., Sherer, N. M., Marks, C. B., Pypaert, M. & Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 170, 317–325 (2005).
Furnon, W. et al. Remodeling of the Actin network associated with the non-structural protein 1 (NS1) of West Nile virus and formation of NS1-containing tunneling nanotubes. Viruses 11, 901 (2019).
Sherer, N. M. et al. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 9, 310–315 (2007).
Eugenin, E. A., Gaskill, P. J. & Berman, J. W. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell. Immunol. 254, 142–148 (2009).
Hashimoto, M. et al. Potential role of the formation of tunneling nanotubes in HIV-1 spread in macrophages. J. Immunol. 196, 1832 (2016).
Generous, A. R. et al. Trans-endocytosis elicited by nectins transfers cytoplasmic cargo, including infectious material, between cells. J. Cell Sci. 132, jcs235507 (2019).
Kumar, A. et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Sci. Rep. 7, 40360 (2017).
Panasiuk, M., Rychłowski, M., Derewońko, N. & Bieńkowska-Szewczyk, K. Tunneling Nanotubes as a novel route of cell-to-cell spread of herpesviruses. J. Virol. 92, e00090-18 (2018).
Cudmore, S., Cossart, P., Griffiths, G. & Way, M. Actin-based motility of vaccinia virus. Nature 378, 636–638 (1995).
Cudmore, S., Reckmann, I., Griffiths, G. & Way, M. Vaccinia virus: a model system for actin–membrane interactions. J. Cell Sci. 109, 1739 (1996).
Chen, Y.-H. et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 160, 619–630 (2015). This paper shows that enterovirus genomes are packaged in clusters into membrane-bound vesicles and released non-lytically from infected cells.
Cuevas, J. M., Duran-Moreno, M. & Sanjuan, R. Multi-virion infectious units arise from free viral particles in an enveloped virus. Nat. Microbiol. 2, 17078 (2017). This paper shows that multi-virion clusters form spontaneously via protein–lipid interactions, promoting co-transmission of different genetic variants to a target cell.
Feng, Z. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367–371 (2013).
McKnight, K. L. et al. Protein composition of the hepatitis A virus quasi-envelope. Proc. Natl Acad. Sci. USA 114, 6587 (2017).
Bird, S. W., Maynard, N. D., Covert, M. W. & Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl Acad. Sci. USA 111, 13081 (2014).
Robinson, S. M. et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 10, e1004045 (2014).
Santiana, M. et al. Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe 24, 208–220.e8 (2018). This paper shows that vesicle-cloaked enteric viruses are shed non-lytically from infected cells in clusters that enhance the multiplicity of infection and disease severity in the subsequent host.
Hutchinson, E. C. et al. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 5, 4816 (2014). This paper shows that the influenza virion architecture includes a substantial number of components of host exosomes.
Bou, J.-V., Geller, R. & Sanjuán, R. Membrane-associated enteroviruses undergo intercellular transmission as pools of sibling viral genomes. Cell Rep. 29, 714–723.e4 (2019).
Marshall, N., Priyamvada, L., Ende, Z., Steel, J. & Lowen, A. C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 9, e1003421 (2013).
Souriant, S. et al. Tuberculosis exacerbates HIV-1 infection through IL-10/STAT3-dependent tunneling nanotube formation in macrophages. Cell Rep. 26, 3586–3599.e7 (2019). This paper shows that M. tuberculosis and HIV-1 co-infection promotes TNT formation in infected macrophages and enhances cell-to-cell spread of HIV-1.
Hoffmann, M. et al. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 585, 588–590 (2020). This paper shows that heterogeneity in host entry co-factor usage by SARS-CoV-2 leads to differential efficacy of the entry inhibitor chloroquine.
Knierim, D., Menzel, W. & Winter, S. Immunocapture of virions with virus-specific antibodies prior to high-throughput sequencing effectively enriches for virus-specific sequences. PLoS ONE 14, e0216713 (2019).
Martinez-Hernandez, F. et al. Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 8, 15892 (2017).
Acknowledgements
The authors thank members of the Lakdawala laboratory for critical reading of this review. S.S.L. is funded by National Institutes of Health (NIH) grant (1R01AI139063-01A1), the American Lung Association Biomedical Research grant and a New Initiative Award from the Charles E. Kaufman Foundation, a supporting organization of The Pittsburgh Foundation. J.E.J. is a recipient of a Catalyst Award from the University of Pittsburgh Center for Evolutionary Biology and Medicine and a T32 (T32 AI049820) from the University of Pittsburgh.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article and contributed to discussion of the content. J.E.J., V.L. and S.S.L wrote the article. All authors reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Microbiology thanks N. Altan-Bonnet and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Pleomorphic viruses
-
A population of viruses exhibiting irregularity of size, shape or protein copy number.
- Receptor avidity
-
The strength or affinity of the interaction between a viral receptor-binding protein and its target ligand on a host cell.
- Multiplicity of infection
-
The ratio of the number of infectious units of a virus to the number of cells.
- Epistatic mutations
-
Mutations whose effects are dependent upon the presence or absence of one or more mutations in other genes.
- Plaque-forming unit
-
(PFU). The concentration of viruses capable of lysing host cells and forming a visible plaque, per unit volume.
- Syncytia
-
A multinucleate cell arising from fusion of several cells.
- Minimal infectious unit
-
The minimal concentration of virus particles required to initiate infection.
- Exosomes
-
Extracellular vesicles produced in the host cell carrying proteins and RNA that are released into the extracellular space and can fuse with neighbouring cells.
Rights and permissions
About this article

Cite this article
Jones, J.E., Le Sage, V. & Lakdawala, S.S. Viral and host heterogeneity and their effects on the viral life cycle. Nat Rev Microbiol 19, 272–282 (2021). https://doi.org/10.1038/s41579-020-00449-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-020-00449-9
This article is cited by
-
Wording the trajectory of the three-year COVID-19 epidemic in a general population – Belgium
BMC Public Health (2024)
-
Revealing viral diversity in the Napahai plateau wetland based on metagenomics
Antonie van Leeuwenhoek (2024)
-
Personalizing Oncolytic Immunovirotherapy Approaches
Molecular Diagnosis & Therapy (2024)
-
Can iron chelators ameliorate viral infections?
BioMetals (2024)
-
Mapping global zoonotic niche and interregional transmission risk of monkeypox: a retrospective observational study
Globalization and Health (2023)
