
Abstract
Autophagy is a powerful tool that host cells use to defend against viral infection. Double-membrane vesicles, termed autophagosomes, deliver trapped viral cargo to the lysosome for degradation. Specifically, autophagy initiates an innate immune response by cooperating with pattern recognition receptor signalling to induce interferon production. It also selectively degrades immune components associated with viral particles. Following degradation, autophagy coordinates adaptive immunity by delivering virus-derived antigens for presentation to T lymphocytes. However, in an ongoing evolutionary arms race, viruses have acquired the potent ability to hijack and subvert autophagy for their benefit. In this Review, we focus on the key regulatory steps during viral infection in which autophagy is involved and discuss the specific molecular mechanisms that diverse viruses use to repurpose autophagy for their life cycle and pathogenesis.
Similar content being viewed by others
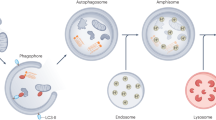


Introduction
Autophagy is an evolutionarily conserved degradative process that is required to maintain host health and facilitates the capture and clearance of invading pathogens by the immune system1,2. Eukaryotic cells deliver dangerous and unwanted cytoplasmic material to lysosomes for degradation via three major routes: microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy3. During microautophagy, invaginations of the lysosomal membrane directly take up cytoplasmic material4 or the material directly enters multivesicular bodies (MVBs) of late endosomes5. During CMA, a cytoplasmic chaperone mediates lysosomal-associated membrane protein 2A (LAMP2A)-dependent uptake of unfolded proteins6. Macroautophagy involves a specialized double-membrane vesicle (DMV) known as the autophagosome (Box 1). Macroautophagy is the best-characterized form of autophagy, and we will hereafter refer to it simply as autophagy.
Autophagy begins with the sequestration of cytoplasmic proteins and damaged organelles into a cup-shaped double-membrane termed the isolation membrane (or phagophore), which is derived from various cellular compartments7. The immature isolation membrane grows to envelop the engulfed contents, thus forming an autophagosome, which subsequently fuses with the lysosome to form autolysosomes. At this stage, the contents undergo degradation to enable their recycling upon nutrient depletion8. Acidification is a key regulatory step in autophagosome maturation and requires the activity of vacuolar ATPases.
Autophagy is involved in various physiological processes, including starvation, cell differentiation and development, and degradation of aberrant structures, which ultimately maintains cellular homeostasis9. Moreover, autophagy is part of host stress responses such as the unfolded protein response (UPR)10. Given that viral infection and replication cause cell stress, autophagy is a frequent by-product of infection. However, autophagy during viral infection is not merely a passive process. As clearance of cytoplasmic components is a major function of autophagy, the innate immune system activates autophagy to degrade and dispose of invading viruses11. Furthermore, at later stages of infection, autophagy facilitates antigen processing and thereby the induction of adaptive immune responses12,13. There is a form of selective autophagy, xenophagy, that specifically recognizes intracellular microorganisms and targets them to autophagosomes for degradation14. Despite the seemingly hazardous conditions in autophagosomes for viruses, some viruses convert the autophagosome to their home for replication. The autophagosome provides a membrane-bound, protected environment to generate their progeny, and viruses can use autophagy-generated metabolites and energy for replication. Another type of autophagy, termed lipophagy, is a degradation pathway for lipid droplets in cells, and viruses can also hijack this process15. Lipid droplets are an ideal platform for virion assembly, and viruses can directly activate lipophagy to maintain the high level of ATP required for viral replication. In summary, current evidence suggests that viruses have evolved diverse strategies to either combat or utilize autophagy to promote their own life cycle. In this Review, we provide an overview of the function of autophagy as an antiviral defence mechanism and then focus on how viruses subvert and exploit multiple steps of the autophagic pathway to evade immune responses and facilitate viral replication.
Autophagy and innate antiviral immunity
Pattern recognition receptors, autophagy and antiviral interferon responses
The innate immune system is the first line of defence against invading microorganisms, including viruses. Germline-encoded receptors called pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs), including virus-specific features, and activate the type I interferon (IFN) pathway to establish an antiviral milieu16. PRRs are found in different subcellular locations, which enables the innate immune system to detect infection throughout the viral life cycle.
The intracellular endosomal Toll-like receptors (TLRs) are primary detectors of viral PAMPs, including double-stranded (ds)RNA (TLR3), single-stranded (ss)RNA (TLR7 and TLR8) and DNA with unmethylated CpG sites (TLR9) (Fig. 1). Most TLRs recruit the adaptor myeloid differentiation primary response protein MYD88, whereas TLR3 and TLR4 recruit another adaptor, TIR domain-containing adaptor molecule 1 (TRIF; also known as TICAM1). Both adaptors activate nuclear factor-κB (NF-κB) for the synthesis of inflammatory cytokines and IFN regulatory factors (IRFs) for triggering IFN production16. TLR stimulation induces binding of MYD88 or TRIF to Beclin 1, which disrupts the Beclin 1–B cell lymphoma-2 (BCL-2) interaction and ultimately activates autophagy (Fig. 1). Conversely, autophagy can inhibit TLR signalling by promoting the selective degradation of TRIF17 (Fig. 1). Furthermore, plasmacytoid dendritic cells (pDCs) that are deficient for autophagy protein 5 (ATG5) showed reduced TLR7-dependent production of IFNs during infection with vesicular stomatitis virus (VSV) or Sendai virus (SeV)18. ATG5 also contributed to TLR9-induced IFNα production in pDCs infected with herpes simplex virus type 1 (HSV-1)19. During viral infection, TLR activation tends to induce autophagy to improve IFN production, whereas the negative regulation of autophagy helps terminate TLR signalling.

In host cells, various pattern-recognition receptors (PRRs) recognize viral pathogen-associated molecular patterns (PAMPs), which leads to the activation of transcription factors and the induction of the interferon (IFN) response. Endosomal Toll-like receptors (TLRs) recognize viral nucleic acids and recruit the adaptor proteins TIR domain-containing adaptor molecule 1 (TRIF) and myeloid differentiation primary response protein MYD88, relaying signals to the nuclear factor-κB (NF-κB) and mitogen-associated protein kinase (MAPK) pathways. Binding to MYD88 or TRIF causes Beclin 1 to dissociate from the B cell lymphoma-2 (BCL-2) inhibitory complex, resulting in the induction of autophagy. TRIF is targeted by selective autophagy by the tripartite motif-containing protein 32 (TRIM32)–TAX1-binding protein 1 (TAX1BP1) complex for degradation. JUN N-terminal kinase (JNK) phosphorylates BCL-2 to initiate Beclin 1-mediated autophagy. During RNA virus infection, retinoic acid-inducible gene I (RIG-I) senses double-stranded (ds)RNA and signals through mitochondrial antiviral-signalling protein (MAVS) to activate interferon regulatory factor 3 (IRF3), leading to the production of IFN. To suppress RIG-I signalling, the autophagy protein 5 (ATG5)–ATG12 complex disrupts the interaction between RIG-I and MAVS. Interferon-induced, dsRNA-activated protein kinase (PKR) phosphorylates eukaryotic translation initiation factor 2 subunit 1 (eIF2α) to induce autophagy. During DNA virus infection, the DNA sensor cyclic GMP–AMP (cGAMP) synthase (cGAS) produces the secondary messenger cGAMP, which activates stimulator of interferon genes protein (STING), IRF3 and IFN expression. Furthermore, cGAS competes with Rubicon for Beclin 1 binding, thus triggering autophagy. Beclin 1 also suppresses cGAMP production and activates ULK1 to phosphorylate STING for its degradation. ATG9 inhibits the aggregation of STING on Golgi apparatus-derived compartments to suppress the DNA sensing. STING undergoes desumoylation and chaperone-mediated autophagy (CMA)-mediated degradation. Finally, extracellular IFN is recognized by the interferon receptor (IFNAR) and activates JAK–STAT (Janus kinase–signal transducer and activator of transcription) signalling. JAK1 and non-receptor tyrosine-protein kinase TYK2 phosphorylate insulin receptor substrate 1 (IRS1) and IRS2, leading to the activation of the PI3K–AKT–mTOR (phosphoinositide 3-kinase–AKT–mechanistic target of rapamycin) pathway and forkhead box 3 transcription factor (FOXO3) as well as the expression of autophagy-related genes. ISGs, interferon-stimulated genes; MDA5, interferon-induced helicase C domain-containing protein 1; SENP2, sentrin-specific protease 2; TBK1, tank-binding kinase 1; TUFM, elongation factor Tu, mitochondrial; VPS34, vacuolar protein sorting 34.
The RIG-I like receptors (RLRs) retinoic acid-inducible gene I (RIG-I; also known as DDX58) and melanoma differentiation-associated protein 5 (MDA5; also known as IFIH1) sense viral RNAs in the cytosol and signal through interaction with mitochondrial antiviral-signalling (MAVS) via their caspase-recruiting domain (CARD), activating interferon regulatory factor 3 (IRF3), IRF7 and NF-κB20. Although autophagy enhances IFN production in virally infected cells, it also can limit RLR-triggered IFN production. ATG5 deficiency increases the production of IFN in response to RNA viruses such as VSV and inhibits viral replication21. The ATG5–ATG12 complex and its binding protein (elongation factor Tu, mitochondrial (TUFM)) suppress dsRNA-induced production of IFN by disrupting RIG-I signalling22 (Fig. 1). In addition, interferon-induced, dsRNA-activated protein kinase (EIF2AK2; also known as PKR) binds to Beclin 1 within the vacuolar protein sorting 34 (VPS34) complex, which initiates the formation of autophagosomes. PKR-mediated phosphorylation of the eukaryotic translation initiation factor 2 subunit 1 (EIF2S1; also known as eIF2α) also induces autophagy23 (Fig. 1). The RLRs provide an example of how autophagy can negatively regulate immune signalling.
The cytosolic DNA sensor cyclic GMP–AMP (cGAMP) synthase (cGAS) recognizes dsDNA during infection with bacteria or DNA viruses24 and produces cGAMP. Subsequently, cGAMP binds to and activates the endoplasmic reticulum (ER)-associated stimulator of interferon genes protein (STING), leading to IRF3 activation and IFN production (Fig. 1). During HSV-1 infection, the DNA-sensing pathway and autophagy pathway converge: Beclin 1 interacts with and impairs the nucleotidyl transferase activity of cGAS, decreasing the amount of cGAMP available to stimulate STING25. Conversely, cGAS competes with the autophagy inhibitor Rubicon for Beclin 1 binding, thus triggering autophagy and stimulating the degradation of cytosolic viral DNA to avoid persistent immune stimulation25. In addition, cGAMP leads to the dissociation of ULK1 from 5′-AMP-activated protein kinase catalytic subunit α2 (AMPK), and activated ULK1 then phosphorylates STING, leading to its degradation26 (Fig. 1). Loss of ATG9 causes aggregation of STING on Golgi apparatus-derived compartments and increases STING-dependent production of IFNs27. During the late phase of HSV-1 infection, STING is also targeted by CMA-mediated degradation to terminate IFN signalling28. Additionally, cGAS-dependent signalling induces autophagy, which promotes pathogen clearance and functions as a negative feedback loop to turn off signalling.
Downstream of PRR-detection of viral infection, newly synthesized IFN is secreted and binds to the IFN receptor (IFNAR), leading to the JAK1 and non-receptor tyrosine-protein kinase TYK2-mediated phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2, which activate transcription of IFN-stimulated genes (ISGs) to restrict viral infection29 (Fig. 1). Additionally, JAK1 and TYK2 can phosphorylate insulin receptor substrate 1 (IRS1) and IRS230,31, which activate the PI3K–AKT–mTOR (mechanistic target of rapamycin) pathway. Activated AKT has several targets, including forkhead box transcription factor 3 (FOXO3), which activates the transcription of a variety of autophagy-related genes32 (Fig. 1). Upon viral infection, crosstalk between PRRs and autophagy leads to the activation and/or inactivation of various intracellular signalling pathways, which generates an optimal antiviral milieu.
Autophagy-mediated restriction of viral replication
Autophagy can be harnessed to degrade viral components, viral particles or even host factors required for viral replication; thus, autophagy functions as an important innate antiviral response (Box 2). However, this autophagic degradation of virions, which is also known as virophagy, can be subverted by several viruses.
During Sindbis virus (SINV) infection, Beclin 1 protects against SINV-mediated encephalitis33. Moreover, ATG5 deficiency results in delayed SINV clearance and accumulation of the autophagy receptor p62 (also called sequestosome 1 (SQSTM1)). Interestingly, p62 binds to a SINV capsid protein and targets the viral capsid to the autophagosome34 (Fig. 2). There, SMURF1, an E3-ubiquitin ligase, is required for the colocalization of p62 with the SINV capsid protein and for virophagy35. Recently, Fanconi anaemia group C protein (FANCC) was found to interact with the SINV capsid protein to facilitate virophagy36,37 (Fig. 2). SMURF1 and FANCC also target HSV-1 for virophagy, suggesting that those two proteins commonly function as virophagic factors35,36.

Autophagy targets intruding viruses by directly eliminating them in autophagosomes. Adaptor protein p62 binds to ubiquitin-coated viral particles that are subsequently delivered to the autophagic machinery. For example, p62 binds to Sindbis virus (SINV) capsid protein and mediates selective autophagy by interacting with LC3 through its LC3-interacting region (LIR). Two host factors, E3 ubiquitin-protein ligase SMURF1 and Fanconi anaemia group C protein (FANCC), are involved in the virophagy of SINV and herpes simplex virus type 1 (HSV-1). During hepatitis C virus (HCV) infection, the host endoplasmic reticulum (ER) transmembrane protein SCOTIN associates with HCV non-structural protein 5A (NS5A) and leads to its degradation, restricting HCV replication. Poliovirus infection and membrane rupture are detected by galectin 8, targeting the virus to autophagosomes. Poliovirus uses HRAS-like suppressor 3 (PLA2G16) to escape from autophagic degradation. HIV-1 virion infectivity factor (Vif) is targeted by histone deacetylase 6 (HDAC6) for degradation. The autophagy protein 5 (ATG5)–ATG12–ATG16L1 complex is recruited in the replication complex and restricts norovirus (NoV) replication through interferon-γ (IFNγ)-inducible GTPases. In antigen-presenting cells (APCs), autophagy delivers intracellular and extracellular antigens to the endolysosome, where they are loaded onto major histocompatibility complex (MHC) class II molecules for presentation to CD4+ T cells. EBV nuclear antigen 1 (EBNA1) is the dominant CD4+ T cell antigen and is primarily processed in the autophagosome. HIV-1 Gag is also targeted by the autophagosome for processing. Autophagy regulates the internalization of MHC class I molecules from the cell membrane via AP2-associated kinase 1 (AAK1). UL138 from human cytomegalovirus (HCMV) and gB from HSV-1 are presented on MHC class I molecules in an autophagy-dependent pathway, when antigen peptide transporter (TAP)-dependent presentation is blocked. Autophagy also promotes an alternative pathway of class I presentation called cross-presentation. It modulates trafficking and processing of phagocytosed antigens from the endosome to MHC I, and autophagy induces antigen packaging in donor cells for release to APCs. 2A, protease 2A; CVB3, coxsackievirus B3; EBV, Epstein–Barr virus; IAV, influenza A virus; pp65, 65 kDa phosphoprotein; TCR, T cell receptor; Ub, ubiquitin.
Picornaviruses, including poliovirus, are detected by galectin 8, which restricts viral infection by initiating the autophagic degradation of the viral RNA genome38. Specifically, when poliovirus punctures the endosomal membrane to release its genome into the cytoplasm, β-galactosides are exposed and trigger galectin 8. This leads to the detection of permeated endosomes and marks them for autophagic degradation. In turn, poliovirus uses the host protein HRAS-like suppressor 3 (PLA2G16) to evade this detection and enable genome delivery (Fig. 2). Another picornavirus, coxsackievirus B3 (CVB3), uses the viral protease 2A to cleave p62 and inhibit virophagy39 (Fig. 2).
The ability of hepatitis C virus (HCV) infection to induce autophagy has been widely demonstrated in cell culture and in the hepatocytes of chronically infected patients40. HCV can escape autophagic destruction, and it is thought to use autophagy for its own benefit (see below). However, a recent study demonstrated that an ER transmembrane protein, SHISA5 (also known as SCOTIN), interacts with the viral non-structural protein 5A (NS5A), which leads to its autophagic degradation and suppresses viral replication41 (Fig. 2).
Human immunodeficiency virus 1 (HIV-1) is also subject to autophagic degradation. To overcome innate immunity, HIV-1 uses the virion infectivity factor Vif, which promotes the degradation of the HIV-1 restriction factor APOBEC3G42. In turn, histone deacetylase 6 (HDAC6) forms a complex with APOBEC3G and mediates autophagy-dependent Vif degradation, thereby inhibiting HIV-1 replication43 (Fig. 2). In CD4+ T cells, autophagy selectively degrades the transactivator Tat, a protein that increases viral transcription through its ubiquitin-independent interaction with p6244 (Fig. 2). In Langerhans cells, the restriction factor tripartite motif-containing protein 5α (TRIM5α) mediates assembly of the autophagy-activating complexes and targets langerin-bound, engulfed HIV-1 to autophagic degradation45.
Autophagy also has antiviral activity independently of degradation. Norovirus (NoV) is a cause of human non-bacterial epidemic gastroenteritis46. In a mouse model of murine norovirus (MNV) infection, IFNγ-mediated antiviral defence requires the ATG5–ATG12–ATG16L1 complex that functions in autophagosome formation47. In ATG16L1 hypomorphic mice, MNV infection provoked a phenotype that resembled Crohn’s disease48. Interestingly, the induction, fusion and degradative activities of autophagy were not required; instead, IFNγ-inducible GTPases, which were targeted to MNV replication complexes by LC3, inhibited viral replication49 (Fig. 2).
Collectively, multiple host pathways detect viral infection to initiate autophagy-mediated innate defences. However, viruses have evolved to evade autophagy and to harness autophagy for their own life cycles.
Autophagy-mediated viral antigen presentation
The adaptive immune response is initiated by the presentation of protein fragments on major histocompatibility complex (MHC) molecules in antigen-presenting cells (APCs), which are then recognized by T cells. MHC class I molecules present intracellular antigens after they have been processed by the proteasome and transported into the ER by the antigen peptide transporter (TAP). By contrast, MHC class II molecules load extracellular antigens after lysosomal degradation in late endosomal MHC class II-containing compartments (MIICs). There is an additional mechanism for loading exogenous antigens onto MHC class I molecules in a process known as cross-presentation.
Autophagosomes constitutively form in MHC class II-positive dendritic cells, B cells and epithelial cells and fuse with multivesicular MIICs to deliver cytoplasmic proteins for MHC class II presentation and antiviral immunity50. Epstein–Barr virus (EBV) establishes a persistent infection by maintaining a latent state in memory B cells. EBV nuclear antigen 1 (EBNA1) is incorporated into autophagosomes and degraded before being presented on MHC class II molecules13 (Fig. 2). Blocking of autophagy with a PI3K inhibitor or through knockdown of ATG12 reduced MHC class II presentation to CD4+ T cells. HIV-1 is also targeted to autophagosomes, processed by the autophagic machinery and presented on MHC class II molecules. HIV-1 Gag-derived proteins colocalize and interact with LC3, and autophagy promotes Gag processing51 (Fig. 2). In turn, the HIV-1 envelope protein activates mTOR to shut down autophagy and impede the immune function of dendritic cells52.
Autophagy limits MHC class I presentation by mediating internalization and degradation of MHC class I molecules. The AP2-associated kinase 1 (AAK1) associates with LC3 to facilitate this internalization53 (Fig. 2). Dendritic cells that are deficient for ATG5, ATG7 or VPS34 had increased surface expression of MHC class I and induced CD8+ T cell activation53,54. Autophagy can also contribute to antigen presentation when TAP is inhibited. Both HSV-1 and human cytomegalovirus (HCMV) encode TAP-blocking proteins55. Although macrophages infected with HSV-1 initially used the conventional MHC class I presentation pathway, a second pathway involving a vacuolar compartment was triggered later during infection. Specifically, a previously unknown type of autophagosome originated from the nuclear envelope and mediated presentation of HSV-1 antigens on MHC class I molecules56 (Fig. 2). Similarly, during latent infection, the HCMV protein UL138 is presented to CD8+ T cells in an autophagy-dependent pathway57,58 and colocalizes with LAMP2-positive late endosomal structures57 (Fig. 2). This suggests that autophagic viral cargos and antigens can access the MHC class I presentation machinery.
Autophagy also contributes to the delivery and processing of extracellular antigens for cross-presentation by MHC class I molecules in a cell-specific manner59,60,61. The autophagic machinery modulates endosomes to enhance cross-presentation and assists in packaging antigens for release by donor cells, including virus-infected, tumour or dying cells, to neighbouring dendritic cells62. Autophagy was also involved in cross-presentation of viral antigens during vaccination, which was promoted by a stress-dependent initiation of autophagy in dendritic cells63. Autophagy-dependent exocytosis transfers extracellular antigens in LC3-coated vesicles from donor cells to dendritic cells (Fig. 2). For example, mouse embryonic fibroblasts that are infected with influenza A virus (IAV) provide antigens to dendritic cells to stimulate IAV-specific CD8+ T cells64. Recently, the CD8+ T cell-specific antigens of HCMV 65 kDa phosphoprotein (pp65) were shown to be cross-presented by human B cells through a pathway involving autophagosomes rather than the TAP-dependent conventional secretory pathway60 (Fig. 2).
During viral infection, the host uses autophagy for optimal antigen processing for MHC class II presentation and, when TAP is inhibited, MHC class I presentation. In addition, autophagy-mediated exocytosis of antigens in vesicular compartments supports cross-presentation on MHC class I molecules.
Viral evasion of autophagic degradation
As previously discussed, autophagy can restrict viral infections. Persisting viruses, however, have evolved various strategies to escape or inhibit multiple steps of the autophagic pathway. HSV-1-encoded neurovirulence factor ICP34.5 interacts with Beclin 1 to inhibit autophagy23 (Fig. 3). ICP34.5 also counteracts the antiviral role of PKR by recruiting host phosphatase PP1α to dephosphorylate eIF2α65 (Fig. 3). TANK-binding kinase 1 (TBK1) was recently discovered as a target of ICP34.5 (ref.66). TBK1 regulates autophagosome maturation by phosphorylating the autophagy receptors p62 and optineurin. Thus, inhibition of TBK1 by ICP34.5 represents an additional strategy of HSV-1 to suppress autophagy (Fig. 3). Late during the HSV-1 life cycle, the RNA-binding tegument protein Us11 binds to PKR to prevent phosphorylation of eIF2α65 (Fig. 3).

Viruses interfere with autophagosome formation and fusion with the lysosome. Herpes simplex virus type 1 (HSV-1) neurovirulence factor ICP34.5 directly targets Beclin 1 to block autophagosome formation. ICP34.5 also recruits the phosphatase PP1α to dephosphorylate eukaryotic translation initiation factor 2 subunit 1 (eIF2α) and blocks tank-binding kinase 1 (TBK1)-mediated autophagosome maturation. HSV-1 tegument protein Us11 interacts with interferon-induced, double-stranded RNA-activated protein kinase (PKR) to prevent the phosphorylation of eIF2α and regulates autophagosome formation. Human cytomegalovirus (HCMV) encodes a functional homologue of ICP34.5 called TRS1, which can bind to Beclin 1 and inhibit autophagy. γ-Herpesviruses carry viral homologues of B cell lymphoma-2 (vBCL-2), which attenuate autophagosome formation by interacting with Beclin 1. vBCL-2 evades JUN N-terminal kinase (JNK) phosphorylation, allowing it to bind to Beclin 1 with high affinity. Kaposi’s sarcoma-associated herpesvirus (KSHV) expresses another host protein homologue, vFLIP, that prevents autophagy protein 3 (ATG3) from processing LC3 during phagosome elongation. KSHV K7 interacts with Rubicon and inhibits fusion of the autophagosome with the lysosome. Viruses also control autophagosome formation and remodel vesicles for viral replication. Poliovirus induces autophagy to form a double-membrane vesicle (DMV) for replication. The poliovirus proteins 2BC and 3A increase LC3 lipidation and DMV formation. The foot-and-mouth disease virus (FMDV) capsid protein VP1 uses the adaptor p62 for autophagosome formation. Mouse hepatitis virus (MHV) hijacks the LC3-coated EDEMsosome by accumulating endoplasmic reticulum (ER) degradation-enhancing α-mannosidase-like protein 1 (EDEM1) and OS-9 in DMVs. Flaviviruses (dengue virus (DENV), West Nile virus (WNV) and Zika virus (ZIKV)) use ER-derived membranes for viral replication. ZIKV non-structural protein 4A (NS4A) and NS4B activate autophagy by inhibiting AKT and mechanistic target of rapamycin complex 1 (mTORC1). Furthermore, the viral protease NS3 cleaves reticulophagy regulator 1 (FAM134B) to block reticulophagy so that ZIKV can use the ER as a replication site. Influenza A virus (IAV) M2 binds to and relocalizes LC3 to the plasma membrane. A highly pathogenic IAV H5N1 strain modulates the AKT–tuberin (TSC2)–mTOR pathway to induce autophagy. Human parainfluenza virus type 3 (HPIV3) also directly inhibits the interaction between syntaxin 17 (STX17) and synaptosomal-associated protein 29 (SNAP29) by expressing phosphoprotein P, resulting in the accumulation of autophagosomes. HPIV3 matrix protein M interacts with elongation factor Tu, mitochondrial (TUFM), inducing mitophagy to inhibit the interferon response. Hepatitis C virus (HCV) has multiple targets to regulate the autophagic pathway. The RNA-dependent RNA polymerase NS5B binds to ATG5. By differentially inducing the expression of Rubicon and ultraviolet radiation resistance-associated gene protein (UVRAG), HCV NS4B temporally regulates the autophagic flux to enhance viral replication. Host immunity-related GTPase family M protein (IRGM) is also targeted by HCV for Golgi apparatus fragmentation and viral replication. Viral proteins inducing or inhibiting autophagy are displayed in red and blue, respectively. ARF, ADB ribosylation factor 1; GBF1, Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1; IRS1, insulin receptor substrate 1; MHV68, murine γ-herpesvirus 68; PAS, pre-autophagosomal structure; PI3K, phosphoinositide 3-kinase; PtdIns3P, phosphatidylinositol-3-phosphate; Ub, ubiquitin; VPS34, vacuolar protein sorting 34; WIPI, WD-repeat domain phosphoinositide-interacting protein.
HCMV also counteracts autophagy and has a functional homologue of ICP34.5 called TRS1 (ref.67). Unlike ICP34.5, the PKR-binding domain of TRS1 is not involved in autophagy inhibition. Instead, TRS1 binds to Beclin 1 through its N-terminal region, and this binding is essential to inhibit autophagy. Recently, another HCMV protein, IRS1, was also found to block autophagy by interacting with Beclin 1 (ref.68) (Fig. 3).
To antagonize autophagy, γ-herpesviruses, including Kaposi’s sarcoma-associated herpesvirus (KSHV) and murine γ-herpesvirus 68 (MHV68), have viral homologues of BCL-2, ORF16 and M11 (ref.69). These viral BCL-2 (vBCL-2) proteins mimic their cellular counterparts (cBCL-2) and attenuate autophagy through direct interaction with Beclin 1 (ref.70). Structural and biochemical analyses demonstrate that vBCL-2 has a markedly higher affinity for Beclin 1 and inhibits autophagosome formation more efficiently than cBCL-2 (ref.71). Moreover, vBCL-2 lacks the regulatory loop of cBCL-2 that is phosphorylated by JUN N-terminal kinase (JNK)72; therefore, it can continuously associate with Beclin 1 (ref.73) (Fig. 3). These data collectively show that vBCL-2 has evolved to be a highly potent autophagy inhibitor. Although EBV also encodes two vBCL-2 proteins (BHRF1 and BALF1), it is unknown whether they inhibit autophagy74. The K7 protein of KSHV promotes Rubicon–Beclin 1 interaction and inhibits the enzymatic activity of VPS34, which blocks the fusion of autophagosomes with lysosomes75 (Fig. 3). In addition, KSHV vFLIP, a homologue of the cellular FLICE-like inhibitor protein (FLIP; also known as ORF71), suppresses autophagy76 by preventing ATG3 from binding and processing LC3 during autophagosome elongation (Fig. 3). Interestingly, two KSHV vFLIP-derived peptides are individually sufficient for binding and releasing ATG3 from cellular FLIP, which results in robust autophagy and autophagic cell death76.
HIV-1 also inhibits autophagy to prevent the sequestration of HIV-1 proteins within autophagosomes and their lysosomal degradation. Although HIV-1 infection induces autophagosome formation and HIV-1 Gag colocalizes with LC3, HIV-1 Nef blocks autophagosome maturation by interacting with Beclin 1 (ref.51) (Fig. 3). In infected macrophages, the interaction between HIV-1 and TLR8 and the Beclin 1-dependent dephosphorylation and nuclear translocation of transcription factor EB (TFEB) activates autophagy and lysosome biogenesis77. HIV-1 Nef interacts with Beclin 1 to sequester TFEB in the cytosol, thus inhibiting maturation of autophagosomes. These examples demonstrate the importance of deregulating the autophagy pathway to facilitate viral persistence.
Autophagy supporting viral replication
Viral manipulation of autophagosomes for replication
RNA viruses exploit autophagy for their replication. Double-membrane compartments formed during autophagy can provide a physical platform for the viral replication machinery, locally concentrate essential intermediates and protect viral RNAs from detection by innate immune sensors and degradation.
The first example of viral reshaping of intracellular membranes came from polioviruses, which lack a membrane envelope78. The autophagy-inducer rapamycin increased poliovirus replication, whereas silencing of genes that are essential for autophagosome formation decreased it79. Electron microscopy of poliovirus-infected cells revealed DMVs that resembled autophagosomes and provided scaffolds for viral RNA replication. Further, poliovirus infection led to the accumulation of LC3 in puncta, and the expression of the viral proteins 2BC and 3A led to LC3 lipidation and DMV formation, which provides a mechanistic link between autophagy and poliovirus replication (Fig. 3). Other picornaviruses, such as CVB3 and foot-and-mouth disease virus (FMDV), also use autophagy for replication80,81. When CVB3-infected cells were treated with inhibitors of autophagosome maturation, virion production was increased and p62 protein levels remained unchanged82, suggesting that viral replication increases the generation of autophagic membrane structures without progression to lysosomal degradation. FMDV also induces ATG5-dependent autophagosome formation and redistributes LC3 to punctate vesicles. However, this induction does not require the PI3K activity of VPS34 and occurs very early during infection as ultraviolet-inactivated FMDV can still trigger autophagosome formation81. FMDV-induced autophagosomes contain the viral capsid protein VP1 that colocalizes with p62 (ref.83), suggesting that autophagosome formation is induced during FMDV entry (Fig. 3). In summary, picornaviruses use multiple mechanisms to modulate autophagy for their replication.
Coronaviruses (CoVs), including the severe acute respiratory syndrome (SARS)-CoV and mouse hepatitis virus (MHV), also induce the formation of DMVs. Several controversial studies concluded that CoVs induce autophagy but do not require the complete autophagy pathway84,85,86. Interestingly, MHV sequesters the non-lipidated, LC3-coated EDEMsosome (a vesicle involved in ER-associated degradation (ERAD)), into its replication and transcription complexes87. MHV also causes accumulation of two ERAD regulatory proteins, ER degradation-enhancing α-mannosidase-like protein 1 (EDEM1) and OS-9, in the DMVs, thereby hijacking the ERAD machinery for viral replication (Fig. 3).
Flaviviruses take advantage of the fact that autophagy and ER processes are frequently connected88. Initially, the induction of autophagy during flavivirus infections was thought to be a consequence of the ER stress-derived UPR. However, dengue virus (DENV) and West Nile virus (WNV) non-structural proteins can induce autophagy independently of the UPR89,90. In human neural progenitor cells, Zika virus (ZIKV) infection led to ER rearrangement and the formation of vesicular clusters thought to be the sites of viral RNA replication and virion assembly91,92. ZIKV-infected primary fibroblasts contained multi-membrane structures that resembled autophagic vesicles93. Moreover, the increase of lipidated LC3 in ZIKV-infected placentae and lower titres of ZIKV in ATG16-deficient fetuses in mice support the proviral function of autophagy during ZIKV infection94. Co-expressed in fetal neural stem cells, ZIKV non-structural proteins NS4A and NS4B decreased AKT phosphorylation and subsequently inhibited mTOR activation, which induced autophagy95 (Fig. 3). Recently, flaviviruses were shown to use the viral protease NS3 to directly cleave the ER-localized autophagic receptor reticulophagy regulator 1 (FAM134B; also known as RETREG1), thereby inhibiting FAM134B-mediated ER degradation (reticulophagy)96,97 (Fig. 3).
HCV induces autophagosome formation but blocks lysosomal fusion, which benefits viral replication and virion production. The HCV RNA-dependent RNA polymerase NS5B interacts with ATG5 early during infection98 (Fig. 3). HCV infection induces the expression of two autophagy regulatory proteins with different kinetics, ultraviolet radiation resistance-associated gene protein (UVRAG) and Rubicon, which stimulate and inhibit the maturation of autophagosomes, respectively99. HCV NS4B induces Rubicon early during infection to inhibit the maturation of autophagosomes, resulting in accumulation of autophagosomes in support of HCV replication99. Overexpression of UVRAG was shown to suppress HCV replication. Recently, an IFN-inducible GTPase, immunity-related GTPase family M protein (IRGM), was shown to regulate autophagy and the formation of various intracellular membrane compartments100. Upon HCV infection, IRGM interacts with Golgi apparatus-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1) and promotes AMPK-mediated GBF1 phosphorylation, thereby activating GTPase ADB ribosylation factor 1 (ARF1) for Golgi apparatus fragmentation and facilitating viral replication101 (Fig. 3). Moreover, HCV triggers IRGM-mediated phosphorylation of ULK1 (ref.102) (Fig. 3). These studies suggest that HCV dynamically modulates autophagy to coordinate viral replication.
IAV also triggers the accumulation of autophagosomes for viral replication103. The virus matrix 2 (M2) ion channel protein is pivotal for IAV assembly and budding104. It also regulates autophagy through its LC3-interacting region (LIR)105. IAV relocates LC3 to the plasma membrane in an M2-dependent manner (Fig. 3), and disruption of the M2–LC3 interaction decreases virion budding and stability. NS1, a multifunctional protein of IAV, also stimulates autophagy indirectly by increasing the synthesis of HA and M2 (ref.106). A highly pathogenic avian H5N1 strain of IAV can also induce autophagy by inhibiting mTOR107. This virus reduces the phosphorylation of tuberin (TSC2), an upstream inhibitor of mTOR, and this modulation of the AKT–TSC2–mTOR pathway and the subsequent autophagic cell death might explain the high mortality of this virus (Fig. 3).
Finally, human parainfluenza virus type 3 (HPIV3) blocks autophagosome maturation and triggers the accumulation of autophagosomes108. Fusion of autophagosomes with lysosomes depends on binding of the adaptor protein synaptosomal-associated protein 29 (SNAP29) to syntaxin 17 (STX17), which is located on the outer membrane of complete autophagosomes, and to the SNAP receptor (SNARE) vesicle-associated membrane protein 8 (VAMP8) on the lysosome109. HPIV3 phosphoprotein (P) directly binds to the SNARE domains of SNAP29 and inhibits the interaction between STX17 and SNAP29 (Fig. 3), ultimately preventing autophagosome–lysosome fusion108. Furthermore, HPIV3 matrix protein (M) also interacts with TUFM and binds LC3 to induce TUFM-mediated mitophagy and inhibit the subsequent IFN response110 (Fig. 3).
Viruses are under high pressure to block autophagosome–lysosome fusion; however, the mechanisms for achieving this vary considerably among viruses. Thus, how a virus interacts with the host autophagic machinery is often a reflection of the unique requirements that an individual virus has for its replication.
Viral regulation of lipophagy for replication
All eukaryotic cells store lipids inside lipid droplets, which are organelles with a neutral lipid core of triglycerides surrounded by a monolayer of cholesteryl esters and coat proteins111,112. Lipid droplets were initially identified as a lipid reservoir that contributes to both energy metabolism and membrane biogenesis. Storage of cellular lipids is regulated by lysosomal degradation in a process termed lipophagy111. In starving cells, lipophagy degrades lipid droplets to supply mitochondria with fatty acids, which are oxidized to generate acetyl-CoA113. How autophagy selectively identifies and sequesters lipid droplets remains unknown; however, lipid droplets that are targeted for degradation are decorated with LC3, similar to autophagosomes111. Although lipophagy has been primarily studied in the context of lipid metabolism-related diseases, there are also multiple viruses that exploit lipophagy.
DENV infection increases the number of lipid droplets per cell, and, thus, pharmacological inhibition of droplet formation markedly reduces DENV replication. DENV-induced lipid droplets contain viral capsid proteins, suggesting that they provide a platform for nucleocapsid formation and viral replication114. Moreover, DENV infection induces lipophagy, which depletes stored triglycerides and increases β-oxidation and energy production for viral replication (Fig. 4). In autophagy-deficient cells, adding exogenous free fatty acids rescues DENV replication, but the drug etomoxir, which prevents the transport of fatty acids into the mitochondria, blocks this process115. These results suggest that lipophagy increases the release of free fatty acids and the amount of ATP, which is crucial for DENV replication. Furthermore, DENV induces AMPK kinase activity, which in turn inhibits mTOR complex 1 (mTORC1), and this regulation is required for virus-induced lipophagy116.

Dengue virus (DENV) infection activates 5′-AMP-activated protein kinase catalytic subunit α2 (AMPK), which in turn inhibits mechanistic target of rapamycin 1 (mTORC1) to induce lipophagy. Degradation of lipid droplets by DENV-mediated lipophagy contributes to β-oxidation and ATP production in mitochondria, which provides energy for viral replication. DENV viral protease NS3 sequesters fatty acid synthase (FASN) to the sites of DENV replication to increase fatty acid biosynthesis. Hepatitis C virus (HCV) also induces the formation of autophagosome-containing lipid cargos. HCV-induced autophagosomes comprise caveolin 1, caveolin 2 and annexin A2, which promote HCV assembly. Viruses also hijack the exocytosis pathway for the secretion of viral particles. Poliovirus uses phosphatidylserine (PS)-enriched autophagosome-like vesicles for non-lytic release. Coxsackievirus B3 (CVB3) exits cells through autophagosomes that contain the exosome marker flotillin 1. HCV is also released through exocytic pathways that are regulated by autophagy. Enveloped double-stranded DNA viruses replicating in the nucleus obtain their second envelope from endoplasmic reticulum (ER) and Golgi apparatus membranes. Varicella zoster virus (VZV) exits cells with autophagic membranes that contain lipidated LC3 and the endocytosis marker RAB11.
Similar to DENV, HCV also induces the production of lipid-filled autophagosomes117. HCV inhibits autophagosome maturation, and this leads to the accumulation of cholesterol. Lipid droplets are important for the assembly of HCV virions. HCV-induced autophagosomes contain lipid raft-associated proteins, such as caveolin 1, caveolin 2 and annexin A2 (ref.118) (Fig. 4), and depletion of cholesterol, a major component of lipid rafts, reduces viral replication119. Lipophagy is less well studied than other aspects of autophagy during viral infection, but it likely has a crucial role in viral replication.
Viral exocytosis and autophagy
Vesicle exocytosis involves the release of cellular contents from the trans-Golgi network (TGN) or fusion of recycling endosomes with the plasma membrane120. Exocytosis and autophagy both involve membrane trafficking and fusion events and share molecular machineries, such as GTPases for tethering and SNARE proteins for fusion. Moreover, exosomes that originate from MVBs contain lipidated LC3, suggesting that autophagosomes are redirected to MVBs for exocytosis. Similar to their use of autophagic membranes, viruses can hijack the secretion machinery for exocytosis.
The enveloped dsDNA herpesviruses undergo two envelope-acquisition events during lytic replication120. The second envelope originates from ER and Golgi apparatus membranes, and at the cell surface, this outer membrane fuses with the plasma membrane121. Two herpesviruses are known to use autophagic membranes for their second envelope acquisition. EBV causes the accumulation of LC3-coated membranes during lytic replication in B cells122. If LC3 lipidation is inhibited, EBV genomes accumulate in the cytosol, likely owing to impaired second envelope acquisition. Similarly, varicella zoster virus (VZV) exits cells with autophagic membranes123. VZV infection increases the autophagic flux, whereas inhibition of autophagy reduces the spread of infectious virus. Specifically, both lipidated LC3 and RAB11 are found on some infectious VZV particles (Fig. 4), and the viral glycoprotein gE colocalizes with LC3 in the cytoplasm123.
As discussed above, picornaviruses accumulate autophagic membranes to support their viral replication and release. As non-enveloped RNA viruses, picornaviruses are packaged inside phosphatidylserine-enriched autophagosome-like vesicles and are released non-lytically within these membranes, enabling multiple viral transmissions124 (Fig. 4). Indeed, ATG-silencing inhibited the non-lytic spread of poliovirus125, and autophagic membranes have similar functions for the release of other picornaviruses79,83. Mature CVB3 seems to exit cells in autophagic membranes80, as CVB3-containing vesicles are associated with both lipidated LC3 and the exosome marker flotillin 1 (ref.80) (Fig. 4).
Flaviviruses use autophagic membranes both for replication and for exocytosis through MVBs126,127 (Fig. 4). Autophagic membranes enable viral particles to reach the extracellular space either in exosomes or viral envelopes. However, it is unclear how this trafficking is coordinated.
Autophagy and viral pathogenesis
Ideally, cells use autophagy to control viral replication, destroy viral particles and activate immune signalling, all of which should prevent disease. However, the success of many viruses depends on the subversion and sequestration of host autophagic responses. For example, in neurons, autophagy is involved in HSV-1 replication and reactivation of latent infection128. During reactivation in neurons, HSV-1 can relocate to the central nervous system, and, there, the expression of the autophagy inhibitor ICP34.5 is involved in causing fatal encephalitis129,130. Strikingly, 50% of mice infected with wild-type HSV-1 developed neuropathogenesis, whereas an ICP34.5-mutant caused no symptoms131. ICP34.5-mediated inhibition of Beclin 1-dependent autophagy is a crucial regulatory step for HSV-1 pathogenesis in the central nervous system.
Numerous cancer risk factors, such as ageing, obesity and chronic inflammation, disturb autophagy132. Accordingly, as improved autophagy correlates with tumour-suppression, its downregulation by viral infection may facilitate cancer development. Viruses that are associated with cancer development, including EBV, hepatitis B virus (HBV), HCV and KSHV, have evolved multiple strategies to avoid autophagy-mediated elimination, which can facilitate tumour progression. During reactivation, EBV replication and transcription activator (Rta) transiently activates autophagy through extracellular-signal-regulated kinase (ERK) signalling133, but early lytic viral products block autophagy and prevent degradation134. In Burkitt’s lymphoma cells, the inhibition of autophagy increases EBV transcription and replication, which links the modulation of autophagy to EBV-associated malignancies135. The HBV small surface protein increases the risk of liver cancer by triggering the UPR and autophagy. Moreover, HBV X protein (HBx) binds to VPS34 and stimulates autophagy initiation. HBx also interacts with V-ATPase and impairs lysosome acidification, resulting in hepatocyte death136. Similar to HBV, HCV infection frequently leads to chronic liver diseases, liver cirrhosis and hepatocellular carcinoma. HCV-infected hepatocytes have high UPR levels, which activates autophagy, sustains viral replication and induces disease progression137,138. As mentioned before, the KSHV vFLIP and vBCL-2 target ATG3 and Beclin 1, respectively, and these interactions block autophagosome formation and vesicle nucleation, thereby supporting KSHV replication and disease progression.
As previously discussed, many RNA viruses enhance autophagy for viral replication and disease progression. For example, autophagy is involved in ZIKV transmission from mother to fetus94,139. Genetic or pharmacological inhibition of autophagy reduced ZIKV infection in the fetus, suggesting that blocking autophagy can be a promising treatment to limit the vertical transmission of ZIKV94.
Autophagy is also involved in DENV pathogenesis. Sub-neutralizing antibodies from prior DENV infection may improve secondary DENV infection via facilitating viral binding and entry into Fcγ receptor-bearing cells, in a process termed antibody-dependent enhancement140. Infection that involves antibody-dependent enhancement induces the expression of ATG5 and ATG12 earlier in the infection process compared with direct DENV infection141. Induction of autophagy in antibody-enhanced DENV infection facilitates viral replication142. Antibody-dependent enhancement primes the autophagic machinery before viral entry, leading to greater DENV pathogenesis.
In pancreatic cells, CVB3 modulates autophagy to induce MVB formation143. A genetic deficiency of autophagy in pancreatic acinar cells led to the reduction of CVB3 infection and protected mice from disease, suggesting the temporary inhibition of autophagy as a possible therapeutic approach144.
Collectively, hosts attempt to balance the induction of autophagy to avoid viral infection and pathogenesis. As obligate parasites, viruses can either strongly block or activate the autophagy pathway, which can upset this balance and lead to pathogenesis.
Conclusions
As a catabolic pathway of mammalian cells, autophagy controls viral infections at multiple levels by causing the destruction of viruses, regulating inflammatory responses and promoting antigen presentation. Moreover, viruses manipulate autophagy for their immune evasion, replication and release from infected cells. The broad mechanisms that viruses use to interfere with autophagy are striking. Viruses that establish either acute or persistent infections often target similar host pathways, but they do so in unique ways and with different functional outcomes. Further, a virus may block certain aspects of autophagy while requiring others. For example, herpesviruses have genes that potently block autophagosome maturation, yet autophagosomes are the source of their outer membrane. Many RNA viruses exist in an intriguing balance in which they both cannot live with autophagy and cannot live without it. Each virus uses unique strategies to fine-tune autophagy signalling so that it can simultaneously escape destruction while harnessing the structural and nutrient benefits that autophagy provides. However, the exact functions of ATG proteins and the detailed mechanisms that govern autophagy during viral infection are still being elucidated. Further research is needed to clarify the detailed functions of autophagy for immunity against viral infections and for viral pathogenesis.

References
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Mizushima, N., Yoshimori, T. & Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell. Dev. Biol. 27, 107–132 (2011).
Mijaljica, D., Prescott, M. & Devenish, R. J. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7, 673–682 (2011).
Sahu, R. et al. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell. 20, 131–139 (2011).
Orenstein, S. J. & Cuervo, A. M. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin. Cell. Dev. Biol. 21, 719–726 (2010).
Shibutani, S. T. & Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 24, 58–68 (2014).
Lamb, C. A., Yoshimori, T. & Tooze, S. A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell. Biol. 14, 759–774 (2013).
Mizushima, N. & Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell. Biol. 12, 823–830 (2010).
Senft, D. & Ronai, Z. A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends. Biochem. Sci. 40, 141–148 (2015).
Deretic, V., Saitoh, T. & Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737 (2013).
Romao, S., Gannage, M. & Munz, C. Checking the garbage bin for problems in the house, or how autophagy assists in antigen presentation to the immune system. Semin. Cancer. Biol. 23, 391–396 (2013).
Paludan, C. et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596 (2005). In this study, autophagy-mediated lysosomal processing contributes to MHC class II-restricted surveillance of EBV to generate EBNA1-specific CD4+ T cells.
Levine, B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162 (2005).
Heaton, N. S. & Randall, G. Dengue virus and autophagy. Viruses 3, 1332–1341 (2011).
Lee, M. S. & Min, Y. J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480 (2007).
Yang, Q. et al. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 13, e1006600 (2017).
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N. & Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401 (2007). This study reveals that autophagy occurs in pDCs and is required for recognition of viral infection and production of type I IFNs, highlighting the importance of autophagy in innate immunity against viral pathogenesis.
Yordy, B., Tal, M. C., Hayashi, K., Arojo, O. & Iwasaki, A. Autophagy and selective deployment of Atg proteins in antiviral defense. Int. Immunol. 25, 1–10 (2013).
Chiang, J. J., Davis, M. E. & Gack, M. U. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. 25, 491–505 (2014).
Jounai, N. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 104, 14050–14055 (2007).
Lei, Y. et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 36, 933–946 (2012).
Talloczy, Z. et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 99, 190–195 (2002).
Sun, L. J., Wu, J. X., Du, F. H., Chen, X. & Chen, Z. J. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Liang, Q. M. et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15, 228–238 (2014). In this study, the interaction between cGAS and Beclin 1 suppresses cGAMP synthesis and enhances the autophagic degradation of cytosolic DNA to prevent persistent immune activation.
Konno, H., Konno, K. & Barber, G. N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698 (2013).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 106, 20842–20846 (2009).
Hu, M. M. et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 45, 555–569 (2016).
Kessler, D. S., Veals, S. A., Fu, X. Y. & Levy, D. E. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev. 4, 1753–1765 (1990).
Platanias, L. C., Uddin, S., Yetter, A., Sun, X. J. & White, M. F. The type I interferon receptor mediates tyrosine phosphorylation of insulin receptor substrate 2. J. Biol. Chem. 271, 278–282 (1996).
Burfoot, M. S. et al. Janus kinase-dependent activation of insulin receptor substrate 1 in response to interleukin-4, oncostatin M, and the interferons. J. Biol. Chem. 272, 24183–24190 (1997).
Mammucari, C., Schiaffino, S. & Sandri, M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 4, 524–526 (2008).
Liang, X. H. et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596 (1998). This study is the first to identify one of the most important mammalian autophagy proteins, Beclin 1, as a BCL-2-binding protein upon viral infection.
Orvedahl, A. et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127 (2010).
Orvedahl, A. et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117 (2011). This study identifies molecular determinants of selective autophagy in mammalian cells using a small interfering RNA (siRNA) screen and identifies SMURF1 as a mediator of viral autophagy and mitophagy.
Sumpter, R. Jr. et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 165, 867–881 (2016).
Sumpter, R. Jr. & Levine, B. Selective autophagy and viruses. Autophagy 7, 260–265 (2011).
Staring, J. et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 541, 412–416 (2017). This study uncovers how host and virus compete during viral entry: activation of a pore-activated (galectin 8-mediated) autophagic degradation pathway and recruitment of the host factor PLA2G16 by the virus to enable viral genome release.
Shi, J. et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFkB signaling. Autophagy 9, 1591–1603 (2013).
Ait-Goughoulte, M. et al. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 82, 2241–2249 (2008).
Kim, N. et al. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 7, e10631 (2016).
Marin, M., Rose, K. M., Kozak, S. L. & Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9, 1398–1403 (2003).
Valera, M. S. et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 12, 53 (2015).
Sagnier, S. et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 89, 615–625 (2015).
Ribeiro, C. M. et al. Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 540, 448–452 (2016). In this study, a known HIV-1 restriction factor, TRIM5α, is connected to the autophagic degradation pathway through the C-type lectin langerin.
Baldridge, M. T., Turula, H. & Wobus, C. E. Norovirus regulation by host and microbe. Trends Mol. Med. 22, 1047–1059 (2016).
Hwang, S. et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 11, 397–409 (2012).
Cadwell, K. et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145 (2010).
Biering, S. B. et al. Viral replication complexes are targeted by LC3-guided interferon-inducible GTPases. Cell Host Microbe 22, 74–85.e7 (2017). This study represents an example of how autophagy (the LC3-conjugation system) and the innate immune response (IFN-inducible GTPases) cooperate to control viral infection.
Dengjel, J. et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 102, 7922–7927 (2005).
Kyei, G. B. et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell. Biol. 186, 255–268 (2009).
Blanchet, F. P. et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32, 654–669 (2010).
Loi, M. et al. Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8(+) T cell responses. Cell Rep. 15, 1076–1087 (2016).
Parekh, V. V. et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8alpha(+) dendritic cells. Proc. Natl Acad. Sci. USA 114, E6371–E6380 (2017).
Verweij, M. C. et al. Viral inhibition of the transporter associated with antigen processing (TAP): a striking example of functional convergent evolution. PLoS Pathog. 11, e1004743 (2015).
English, L. et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487 (2009). This study indicates that autophagy benefits the host by providing an additional pathway for the degradation of endogenous HSV-1 proteins for antigen presentation.
Tey, S. K. & Khanna, R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 120, 994–1004 (2012).
Tey, S. K., Goodrum, F. & Khanna, R. CD8+ T-cell recognition of human cytomegalovirus latency-associated determinant pUL138. J. Gen. Virol. 91, 2040–2048 (2010).
Shibutani, S. T., Saitoh, T., Nowag, H., Munz, C. & Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16, 1014–1024 (2015).
Dasari, V. et al. Autophagy and proteasome interconnect to coordinate cross-presentation through MHC class I pathway in B cells. Immunol. Cell. Biol. 94, 964–974 (2016).
Cruz, F. M., Colbert, J. D., Merino, E., Kriegsman, B. A. & Rock, K. L. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 35, 149–176 (2017).
Mintern, J. D. et al. Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy 11, 906–917 (2015).
Ravindran, R. et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343, 313–317 (2014). In this study, an unknown mechanism of vaccine-mediated protection reveals that stress-response dependent initiation of autophagy enhances antigen presentation to T cells.
Smed-Sorensen, A. et al. Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog. 8, e1002572 (2012).
Mulvey, M., Poppers, J., Sternberg, D. & Mohr, I. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 77, 10917–10928 (2003).
Kanai, R. et al. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 86, 4420–4431 (2012).
Chaumorcel, M. et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 86, 2571–2584 (2012).
Mouna, L. et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 12, 327–342 (2016).
Cuconati, A. & White, E. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 16, 2465–2478 (2002).
Pattingre, S. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 (2005). This study reveals that cellular and viral (KSHV) BCL-2 interact with Beclin 1 and inhibit the Beclin 1-associated class III PI3K activity, resulting in the inhibition of autophagy.
Ku, B. et al. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS. Pathog. 4, e25 (2008).
Wei, Y., Sinha, S. & Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951 (2008).
Yamamoto, K., Ichijo, H. & Korsmeyer, S. J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 19, 8469–8478 (1999).
Altmann, M. & Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 3, e404 (2005).
Liang, Q. et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 87, 12499–12503 (2013).
Lee, J. S. et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell. Biol. 11, 1355–1362 (2009).
Campbell, G. R., Rawat, P., Bruckman, R. S. & Spector, S. A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS. Pathog. 11, e1005018 (2015).
Dales, S., Eggers, H. J., Tamm, I. & Palade, G. E. Electron microscopic study of the formation of poliovirus. Virology. 26, 379–389 (1965). This study is the first to represent virus-mediated membrane-enclosed bodies in the cytoplasm upon poliovirus infection; later studies reveal these as DMVs.
Jackson, W. T. et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, e156 (2005).
Robinson, S. M. et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS. Pathog. 10, e1004045 (2014).
Berryman, S. et al. Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J. Virol. 86, 12940–12953 (2012).
Wong, J. et al. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153 (2008).
O’Donnell, V. et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology. 410, 142–150 (2011).
Prentice, E., Jerome, W. G., Yoshimori, T., Mizushima, N. & Denison, M. R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279, 10136–10141 (2004).
de Haan, C. A. & Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 4, 276–279 (2008).
Zhao, Z. J. et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3, 581–585 (2007).
Reggiori, F. et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell. Host. Microbe. 7, 500–508 (2010).
Paul, D. & Bartenschlager, R. Flaviviridae replication organelles: oh, what a tangled web we weave. Annu. Rev. Virol. 2, 289–310 (2015).
Miller, S., Kastner, S., Krijnse-Locker, J., Buhler, S. & Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 282, 8873–8882 (2007).
Blazquez, A. B., Martin-Acebes, M. A. & Saiz, J. C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in west nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 5, e797 (2014).
Cortese, M. et al. Ultrastructural characterization of zika virus replication factories. Cell Rep. 18, 2113–2123 (2017).
Offerdahl, D. K., Dorward, D. W., Hansen, B. T. & Bloom, M. E. Cytoarchitecture of zika virus infection in human neuroblastoma and aedes albopictus cell lines. Virology. 501, 54–62 (2017).
Hamel, R. et al. Biology of zika virus infection in human skin cells. J. Virol. 89, 8880–8896 (2015).
Cao, B., Parnell, L. A., Diamond, M. S. & Mysorekar, I. U. Inhibition of autophagy limits vertical transmission of zika virus in pregnant mice. J. Exp. Med. 214, 2303–2313 (2017).
Liang, Q. et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell. Stem. Cell. 19, 663–671 (2016).
Lennemann, N. J. & Coyne, C. B. Dengue and zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 13, 322–332 (2017).
Khaminets, A. et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015). This study shows that reticulon protein FAM134B targets the ER to autophagosomes and regulates ER structure and turnover as a determinant for reticulophagy.
Guevin, C. et al. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology. 405, 1–7 (2010).
Wang, L., Tian, Y. & Ou, J. H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS. Pathog. 11, e1004764 (2015).
Gregoire, I. P. et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 7, e1002422 (2011).
Hansen, M. D. et al. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 114, 3462–3471 (2017).
Chauhan, S., Mandell, M. A. & Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell. 58, 507–521 (2015). This study, together with references 100 and 101, identifies IRGM as a host factor targeted by RNA viruses for their optimal infection. Data analysis reveals that RNA viruses commonly target IRGM in the autophagic pathway. HCV utilizes IRGM for the induction of autophagy and membrane remodelling.
Zhou, Z. et al. Autophagy is involved in influenza A virus replication. Autophagy 5, 321–328 (2009).
Chen, B. J., Leser, G. P., Jackson, D. & Lamb, R. A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 82, 10059–10070 (2008).
Beale, R. et al. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell. Host. Microbe. 15, 239–247 (2014).
Zhirnov, O. P. & Klenk, H. D. Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J. Virol. 87, 13107–13114 (2013).
Ma, J. H., Sun, Q., Mi, R. F. & Zhang, H. B. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J. Genet. Genom. 38, 533–537 (2011).
Ding, B. et al. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15, 564–577 (2014).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Ding, B. et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell. Host. Microbe. 21, 538–547.e4 (2017).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009). In this study, lipid droplets and autophagic components associate during nutrient deprivation, and inhibition of autophagy increases triglyceride in lipid droplets, suggesting a critical function for autophagy in lipid metabolism.
Thiele, C. & Spandl, J. Cell biology of lipid droplets. Curr. Opin. Cell. Biol. 20, 378–385 (2008).
Liu, K. & Czaja, M. J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11 (2013).
Samsa, M. M. et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 (2009). This study reports that DENV infection increases the number of lipid droplets and accumulates mature capsid protein on the surface of lipid droplets for viral replication.
Heaton, N. S. & Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8, 422–432 (2010).
Jordan, T. X. & Randall, G. Dengue virus activates the AMP kinase-mTOR axis to stimulate a proviral lipophagy. J. Virol. 91, e02020–16 (2017).
McLauchlan, J. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta. 1791, 552–559 (2009).
Kim, J. Y., Wang, L., Lee, J. & Ou, J. J. Hepatitis C virus induces the localization of lipid rafts to autophagosomes for its RNA replication. J. Virol. 91, e00541–00517 (2017).
Saxena, V., Lai, C. K., Chao, T. C., Jeng, K. S. & Lai, M. M. C. Annexin A2 is involved in the formation of hepatitis C virus replication complex on the lipid raft. J. Virol. 86, 4139–4150 (2012).
Munz, C. The autophagic machinery in viral exocytosis. Front. Microbiol. 8, 269 (2017). This review article presents the relationships between autophagy and exocytosis and how viruses exit infected cells in packages wrapped into autophagic membranes.
Johnson, D. C. & Baines, J. D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9, 382–394 (2011).
Nowag, H. et al. Macroautophagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine 1, 116–125 (2014).
Buckingham, E. M., Jarosinski, K. W., Jackson, W., Carpenter, J. E. & Grose, C. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J. Virol. 90, 8673–8685 (2016).
Chen, Y. H. et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 160, 619–630 (2015).
Bird, S. W., Maynard, N. D., Covert, M. W. & Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 111, 13081–13086 (2014).
Metz, P. et al. Dengue virus inhibition of autophagic flux and dependency of viral replication on proteasomal degradation of the autophagy receptor p62. J. Virol. 89, 8026–8041 (2015).
Shrivastava, S. et al. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J. Virol. 90, 1387–1396 (2015).
Orvedahl, A. et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell. Host. Microbe. 1, 23–35 (2007). In this study, a mutant HSV-1 lacking the Beclin 1-binding domain of ICP34.5 fails to inhibit autophagy and demonstrates impaired viral neurovirulence.
Cabrera, C. V. et al. Herpes simplex virus DNA sequences in the CNS of latently infected mice. Nature 288, 288–290 (1980).
Chou, J., Kern, E. R., Whitley, R. J. & Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250, 1262–1266 (1990).
Yordy, B., Iijima, N., Huttner, A., Leib, D. & Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12, 334–345 (2012).
Zhong, Z., Sanchez-Lopez, E. & Karin, M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166, 288–298 (2016).
Hung, C. H. et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 88, 12133–12145 (2014).
Granato, M. et al. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 88, 12715–12726 (2014).
De Leo, A. et al. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 6, e1876 (2015).
Liu, B. et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 10, 416–430 (2014).
Dash, S. et al. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses 8, 150 (2016).
Dash, S. et al. Autophagy in hepatocellular carcinomas: from pathophysiology to therapeutic response. Hepat. Med. 8, 9–20 (2016).
Delorme-Axford, E. et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl. Acad. Sci. USA 110, 12048–12053 (2013).
Screaton, G., Mongkolsapaya, J., Yacoub, S. & Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 15, 745–759 (2015).
Huang, X. W. et al. Antibody-dependent enhancement of dengue virus infection inhibits RLR-mediated Type-I IFN-independent signalling through upregulation of cellular autophagy. Sci. Rep. 6, 22303 (2016).
Fang, Y. T. et al. Autophagy facilitates antibody-enhanced dengue virus infection in human pre-basophil/mast cells. PLoS. ONE. 9, e110655 (2014).
Kemball, C. C. et al. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84, 12110–12124 (2010).
Alirezaei, M., Flynn, C. T., Wood, M. R. & Whitton, J. L. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell. Host. Microbe. 11, 298–305 (2012). This study shows that coxsackievirus replication and pathology is decreased in the absence of autophagy due to ATG5 deficiency and that there is a reduction of virus-mediated membrane vesicles.
Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 24, 9–23 (2014).
Hara, T. et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell. Biol. 181, 497–510 (2008).
Hosokawa, N. et al. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979 (2009).
Mercer, C. A., Kaliappan, A. & Dennis, P. B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5, 649–662 (2009).
Itakura, E., Kishi, C., Inoue, K. & Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 19, 5360–5372 (2008).
Matsunaga, K. et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell. Biol. 11, 385–396 (2009).
Fujita, N. et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 19, 2092–2100 (2008).
Russell, R. C. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell. Biol. 15, 741–750 (2013).
Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 59, 285–297 (2015).
Ge, L., Melville, D., Zhang, M. & Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2, e00947 (2013).
Hailey, D. W. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 (2010).
Hamasaki, M. et al. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 (2013).
Hayashi-Nishino, M. et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell. Biol. 11, 1433–1437 (2009).
Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C. & Rubinsztein, D. C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell. Biol. 12, 747–757 (2010).
Proikas-Cezanne, T., Takacs, Z., Donnes, P. & Kohlbacher, O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J. Cell. Sci. 128, 207–217 (2015).
Ichimura, Y. et al. A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 (2000).
Kirisako, T. et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell. Biol. 151, 263–276 (2000).
Tanida, I., Ueno, T. & Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell. Biol. 36, 2503–2518 (2004).
Mizushima, N. et al. A protein conjugation system essential for autophagy. Nature 395, 395–398 (1998). This paper is the first to describe the ubiquitylation-like conjugation system in yeast at the molecular level, discovering the E1-like enzyme ATG7.
Nakatogawa, H., Ichimura, Y. & Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178 (2007).
McEwan, D. G. et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell. 57, 39–54 (2015).
Kim, I., Rodriguez-Enriquez, S. & Lemasters, J. J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 (2007).
Kraft, C., Deplazes, A., Sohrmann, M. & Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell. Biol. 10, 602–610 (2008).
Hutchins, M. U., Veenhuis, M. & Klionsky, D. J. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J. Cell. Sci. 112, 4079–4087 (1999).
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N. & Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221 (2009).
Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 (2011).
Zheng, Y. T. et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916 (2009).
Kirkin, V. et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 33, 505–516 (2009).
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A. & Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012).
Acknowledgements
During the preparation of this Review article, the authors were supported by grants from the US National Institutes of Health (CA200422, CA180779, DE023926, AI073099, AI116585 and AI129496) and the Fletcher Jones Foundation. The authors sincerely apologize for not being able to cite all papers related to this topic owing to space limitations.
Author information
Authors and Affiliations
Contributions
Y.C. and J.U.J. substantially contributed to the discussion of content. All authors wrote the article and reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Multivesicular bodies
-
(MVBs). A specialized subset of endosomes that contain membrane-bound intraluminal vesicles.
- Unfolded protein response
-
(UPR). A cellular stress response activated in response to an accumulation of unfolded or aggregated proteins in the endoplasmic reticulum.
- Type I interferon
-
(IFN). A subset of interferons, including IFNα, IFNβ and IFNω, that have potent antiviral roles by binding to a common receptor (interferon receptor (IFNAR)).
- Plasmacytoid dendritic cells
-
(pDCs). A subset of dendritic cells that are distinct from conventional dendritic cells and that produce large amounts of interferon.
- IFN-stimulated genes
-
(ISGs). A subset of genes induced by the activation of the interferon receptor-mediated JAK–STAT pathway.
- E3-ubiquitin ligase
-
An enzyme catalysing transfer of ubiquitin from the activated site of an E2 enzyme to a primary amine on a lysine side chain or amino terminus of a protein.
- Restriction factor
-
An antiviral protein that is produced in the host and blocks viral replication.
- Langerhans cells
-
Immune cells found in the epidermis that function as antigen-presenting cells that bind antigens entering through the skin.
- Crohn’s disease
-
A chronic inflammatory disease of the gastrointestinal tract.
- Puncta
-
A structural marker that is involved in autophagosome formation by the dynamic process of LC3 association.
- LC3 lipidation
-
A conjugation of phosphatidylethanolamine (PE) to LC3 to form LC3–PE, which is recruited to autophagosomal membranes.
- ER-associated degradation
-
(ERAD). A process through which misfolded proteins are transported from the endoplasmic reticulum to the cytoplasm for proteasomal degradation.
- SNAP receptor
-
(SNARE). A key component of the cellular membrane fusion machinery that mediates vesicular fusion events in autophagosome–lysosome fusion.
- Lipid raft
-
A small heterogeneous cellular compartment enriched with sphingolipids and cholesterols.
Rights and permissions
About this article

Cite this article
Choi, Y., Bowman, J.W. & Jung, J.U. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol 16, 341–354 (2018). https://doi.org/10.1038/s41579-018-0003-6
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-018-0003-6
This article is cited by
-
Autophagy degrades immunogenic endogenous retroelements induced by 5-azacytidine in acute myeloid leukemia
Leukemia (2024)
-
Hypoxia enhances autophagy level of human sperms
Scientific Reports (2024)
-
DDRGK1-mediated ER-phagy attenuates acute kidney injury through ER-stress and apoptosis
Cell Death & Disease (2024)
-
The impact of nanomaterials on autophagy across health and disease conditions
Cellular and Molecular Life Sciences (2024)
-
Zika virus triggers autophagy to exploit host lipid metabolism and drive viral replication
Cell Communication and Signaling (2023)
