
Abstract
Acute inflammation is essential for initiating and coordinating the body’s response to injuries and infections. However, in acute inflammatory diseases, inflammation is not resolved but propagates further, which can ultimately lead to tissue damage such as in sepsis, acute respiratory distress syndrome and deep vein thrombosis. Currently, clinical protocols are limited to systemic steroidal treatments, fluids and antibiotics that focus on eradicating inflammation rather than modulating it. Strategies based on stem cell therapeutics and selective blocking of inflammatory molecules, despite showing great promise, still lack the scalability and specificity required to treat acute inflammation. By contrast, polymeric particle systems benefit from uniform manufacturing at large scales while preserving biocompatibility and versatility, thus providing an ideal platform for immune modulation. Here, we outline design aspects of polymeric particles including material, size, shape, deformability and surface modifications, providing a strategy for optimizing the targeting of acute inflammation.
Similar content being viewed by others

Introduction
Inflammation plays an essential part in the immune response against harmful stimuli and injury through recognition and containment of invading pathogens and toxins. Overly responsive or uncontrolled inflammation can lead to tissue damage and organ dysfunction1,2,3, and is associated with numerous human disorders, such as acute lung and liver injury, sepsis, asthma, inflammatory bowel disease, rheumatoid arthritis and neurodegenerative diseases2,3. Acute inflammation, regulated by the innate immune system, is responsible for the initial recognition of an inflammatory stimulus and focuses on the rapid containment of the offending pathogen or injury. Such a response aims at accelerating inflammatory resolution and typically lasts on a scale of hours to days (Fig. 1a). If the acute inflammation response is excessive or fails to contain the inflammatory stimulus, the response is shifted to a chronic (pathological) phase characterized by prolonged inflammatory episodes and can last on a scale of weeks to years3,4,5. Although chronic inflammation has mainly been associated with cells from the adaptive immune system, the innate arm of the immune system also has a role, because, in chronic inflammatory diseases, repeated acute inflammatory episodes propagate tissue damage2,3,5,6. Recognizing the persistent role of acute inflammation in chronic diseases has, in part, contributed to the growing interest in developing therapeutics aimed at suppressing this acute response. In this Review, we summarize crucial aspects of the acute inflammation response and discuss particle-based therapies developed towards modulating or resolving this process.

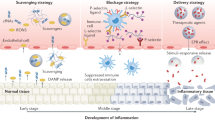
a | Neutrophil recruitment and functions in inflammation. Neutrophils slowly roll along the endothelium by expressing P-selectin glycoprotein ligand 1 (PSGL1) and L-selectin, which bind to their corresponding ligands on the endothelium. Once at the site of inflammation, lymphocyte function-associated antigen 1 (LFA1) expressed on the neutrophil locks with intercellular adhesion molecule 1 (ICAM1) on the endothelium to initiate transmigration to the infected or inflamed tissue space. At the site of infection, neutrophils phagocytose pathogens or release reactive oxygen species (ROS), granules or neutrophil extracellular traps (NETs) to prevent pathogenic spread. Neutrophils then commit to apoptosis, initiating migration of other immune cell types. Monocytes and macrophages clean up dead cellular materials (pathogens and neutrophils) by efferocytosis and ultimately migrate to the liver and lymph nodes to remove and process pathogenic materials. Additionally, post efferocytosis monocytes shift to a pro-resolution phenotype to promote tissue restoration. b | Non-particle-based therapeutics for modulating inflammation. Stem cell therapies: transplanted mesenchymal stem cells (MSCs) can secrete immunosuppressive cytokines to promote regulatory T (Treg) cell production and inhibit T helper 1 (TH1) and T helper 2 (TH2) cell differentiation or suppress immune cell recruitment and activation. Cytokines and antibody-based therapies: blocking antibodies or decoy receptors bind to inflammatory cytokines to inhibit their activity and dampen systemic inflammation. Vasculature blocking: biological agents prevent transmigration of immune cells by blocking specific leukocyte adhesion molecules on vascular endothelial cells, halting the inflammatory response. Targeted immune cell blocking: therapeutics can directly inhibit immune cells from pathological activation by blocking specific receptors on their surface. mAb, monoclonal antibody.
Acute inflammation
Acute inflammatory cascade
Acute inflammation is initiated by either pathogenic infections or exogenously by mechanical trauma, ischaemia-reperfusion or chemicals2,3. Pattern recognition receptors (PRRs) are proteins circulating in the blood or expressed on innate immune cells7. Pathogens entering the body through punctured skin, orally or inhalation, are recognized by PRRs through pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), which trigger an inflammatory response1,8. Within the infected tissue space, resident macrophages, dendritic cells (DCs) and neutrophils are the first cells to interact with invading pathogens. Activated macrophages and DCs function as antigen-presenting cells, phagocytose foreign bodies, migrate to lymph nodes and present the processed antigen to lymphocytes3,9. Concurrently, activated endothelial cells release inflammatory cytokines, including tumour necrosis factor (TNF), several interleukins (IL-1, IL-6, IL-8, IL-12 and IL-17) and interferon-γ (IFNγ), which accumulate in the bloodstream, calling white blood cells (WBCs; also known as leukocytes) into action10.
Neutrophils are the first circulating WBCs recruited to the infected tissue space and they have an essential role in pathogen clearance and inflammation resolution. Representing about 60–70% of all circulating WBCs in humans, neutrophils locate the inflammation by following the release of cytokines and chemokines, then slowly rolling along the endothelium mediated by weak adhesive interactions with endothelial surface-expressed proteins, such as upregulated selectin molecules2,9,11,12. Once at the site of inflammation, enhanced integrin expression on the endothelium firmly adheres the neutrophils through chemokine-activated lymphocyte function-associated antigen 1 (LFA1) on the neutrophil surface to initiate transmigration12. At this stage, activated neutrophils shed P-selectin glycoprotein ligand 1 (PSGL1) and L-selectin (also known as CD62L) from their membrane surface13,14. Once inside, neutrophils deploy several mechanisms to contain pathogenic infections, including phagocytosis, the release of reactive oxygen species (ROS), degranulation and the production of neutrophil extracellular traps (NETs)2. Once the infection has been contained, released granules, NETs and apoptotic neutrophils recruit monocytes and adaptive immune cells to initiate inflammation resolution2,15 (Fig. 1a).
Although it is typically associated with an invading pathogen, inflammation can also arise from sterile, non-pathogenic events. Sterile inflammations such as mechanical injury, blood clots or chemical irritants can cause damage at the cellular or tissue scale, initiating the release of DAMPs1. The released DAMPs are sensed by resident immune and endothelial cells, leading to inflammatory cytokine production (TNF and IL-1)1,2. Similar to pathogenic infections, these immunostimulatory molecules initiate neutrophil recruitment to the injured site2. Despite the absence of pathogens in sterile injuries, neutrophils still employ similar tactics to contain the inflammation, followed by monocytes clearing out any remaining necrotic cells and apoptotic neutrophils in a process called efferocytosis1,16.
Excess release of granules, ROS and NETs by an overabundance of neutrophils could still cause damage in surrounding host cells1,2, leading to acute pathological conditions such as acute lung and liver injuries or chronic inflammation.
Inflammatory disease propagation
A failure to contain the acute inflammatory response might stem from prolonged infection, foreign body presence or an underlying genetic condition4,10. Prolonged neutrophil mobilization alongside host tissue damage leads to an increased release of inflammatory cytokines, also known as a cytokine storm17. A cytokine storm can cause a positive feedback loop, bringing other immune cells to the site of inflammation, thereby further increasing inflammation and organ damage18. Some of the detrimental effects of a cytokine storm include changes in immune cell proliferation, such as excess granulocyte and reduced lymphocyte production, preventing inflammation resolution19. Cytokine storms, in particular, have been related to tissue damage in patients with COVID-19 (ref.18), as indicated by the many therapeutic approaches against COVID-19 that are focused on minimizing the occurrence of these storms20. Finally, greater neutrophil infiltration further increases vascular permeability, allowing pathogens to enter the bloodstream, which ultimately causes systemic inflammation21.
The innate immune response is not limited to cellular components, but is also facilitated by protein mediators10,22. In the blood and interstitial fluid, the complement system becomes activated in the presence of pathogens through the classical, lectin or alternative pathway22. The complement system, consisting of distinct plasma proteins, plays a part in targeting or marking foreign materials by coating their surface (also known as opsonization), which in turn can elicit severe immune responses termed complement activation-related pseudo allergy (CARPA)23,24.
The complement system opsonizes foreign bodies and enzymatically cleaves C3 to the anaphylatoxins C3a and C5a, causing downstream inflammation22. Complement activation combined with pathogen or injury-generated systemic inflammatory cytokines can induce tissue factor expression on endothelial cells, initiating the coagulation pathway10. Once triggered, the coagulation pathway is difficult to contain by standard feedback mechanisms, such as antithrombin, activated protein C or tissue factor pathway inhibitor, and can thus result in distant intravascular coagulation and eventually multi-organ failure10.
Acute inflammatory disease treatments
Most clinical treatments for acute inflammatory diseases centre on infection control through antibiotics, pain management and supportive care, involving steroidal and nonsteroidal anti-inflammatory therapies21,25,26,27,28. However, these approaches have limitations, including increased risk of secondary infections with prolonged use owing to their lack of specificity, coupled with their systemic (oral or intravenous) mode of delivery27,29,30,31,32,33,34. Alternatively, intravenous cellular and monoclonal antibody therapies have emerged as promising agents to target and treat the damaging effects of inflammation35,36,37,38,39 (Fig. 1b).
Stem cell therapy
Stem cell therapies against inflammation typically involve transplantation of autologous mesenchymal stem cells (MSCs), which are multipotent cells derived from bone marrow, adipose tissue, dental pulp or umbilical cord tissue. Following implantation, MSCs secrete immune-suppressive cytokines (IL-10 and transforming growth factor-β (TGFβ)) to promote regulatory T (Treg) cell production and inhibit differentiation of T helper 1 (TH1) and T helper 2 (TH2) cells, aiding in containing the pathological inflammation40,41,42,43. Finally, MSCs can suppress neutrophil recruitment and activation through secretion of superoxide dismutase 3 (refs.41,43,44). Several clinical trials using MSCs have shown promising results in improving the symptoms of inflammatory disorders, including rheumatoid arthritis, multiple sclerosis and acute respiratory distress syndrome (ARDS)45. For example, intravenously injected human umbilical cord MSCs considerably reduced the concentration of inflammatory cytokines in a COVID-19 patient with ARDS, leading to the patient’s recovery46, with similar results shown in COVID-19 patients with pneumonia (ChiCTR2000029990)47,48.
Despite showing promise, stem cell therapies have intrinsic limitations; isolating and expanding MSCs for each donor is time-consuming, making them unfit for large-scale manufacturing as would be needed for widespread, acute inflammatory illnesses such as COVID-19. MSCs as inflammatory therapeutics also require many viable cells (10 million per dose, with multiple doses required), which can prove challenging to access for autologous transplantation49. To address these limitations, allogeneic MSCs can be sourced in high numbers from donors50. Alternatively, exosomes secreted from MSCs can be leveraged as alternatives to live-cell therapeutics as they carry several immunotherapeutic components of the parent MSCs, including cytokines, signalling lipids and mRNA51.
Cytokine- and antibody-based therapy
Proteins can be designed to target cytokines, such as TNF, IL-1 and IL-6, or to dampen systemic inflammation in patients with immune disorders, by blocking antibodies or by inhibiting cytokines through receptor binding52. Cytokine inhibitors have shown excellent efficacy in treating inflammation in clinical trials53; for example, the anti-TNF antibody Humira in the treatment of rheumatoid arthritis (NCT00049751)54.
Antibodies can be deployed for targeted vasculature blocking to prevent the transmigration of immune cells and thus stop inflammatory activation55,56. Bimosiamose, for example, is a small-molecule inhibitor of adhesion proteins L-selectin, E-selectin and P-selectin, which showed a promising reduction of inflammation in chronic obstructive pulmonary disease, psoriasis and asthma patients during phase II clinical trials55,56. However, cytokine-targeting therapeutics lack specificity52, with reduced haematopoiesis as one of the most common side effects, ultimately leading to increased infections owing to reduced WBC populations52.
Targeted immune cell blocking
Immune cells can further be prevented from interacting with inflammation by specific immune cell blocking. Obstructing complement receptors (C5aR)57, cytokine receptors (such as IL-1R and IL-6R)52, B cell activation receptors (CD20)58,59 and adhesion receptors (PSGL1, α4β7 integrin, αEβ7 integrin and very late antigen 4 (VLA4))55,56,60 on the surface of immune cells can selectively inhibit inflammatory activation. For example, the small molecule CCX168 is a C5aR inhibitor that blocks binding to the activating complement protein C5a on neutrophils and has demonstrated efficacy in reducing inflammation in a phase III clinical trial (NCT02994927) for vasculitis (inflammation of blood vessels)57,61. Similar to cytokine targeting, targeted immune cell blocking can have systemic consequences owing to redundancies within the immunity cascade62. Furthermore, complexities associated to ligand-receptor-mediated responses can cause unintended downstream signalling effects62.
Cellular and antibody-based approaches, however, remain limited owing to the complexity of the inflammatory cascade, and they do not target the root cause of acute inflammation63. Alternatively, polymeric antibody-based approaches can mitigate overzealous inflammatory responses in acute lung injury (ALI), ARDS, sepsis, asthma, rheumatoid arthritis, type I diabetes, coagulopathic diseases and neurodegenerative diseases.
Promising particle-based therapeutics
Drug delivery systems can be applied to avoid adverse effects related to systemic treatments of inflammation. For example, lipid nanoparticles and lipid-based drug delivery systems, such as liposomes, show high biocompatibility and cargo stability, as demonstrated by the recent success of lipid-nanoparticle-based mRNA vaccines64. These carriers have mainly been deployed for priming immune cells in developing cancer vaccines and, recently, for vaccine against SARS-CoV-2 viral infections64,65,66. However, lipid systems are limited by poor structural stability, a limited range of potential cargos, low drug loading and poor circulation time67,68,69. Alternatively, synthetic polymer-based drug carriers provide spatiotemporal control over release and reproducibility compared to other drug delivery formulations70,71. The choice of LNPs versus polymeric carriers depends on the use and type of drug cargo, the cellular target and the in vivo delivery route72.
Liposomal particles were first used as drug delivery vehicles in cancer therapy to improve drug delivery efficiency to solid tumours; however, immunotoxicity remains a serious side effect for intravenous formulations73. Surface modifications of particle-based drug carriers, such as the grafting of polyethylene glycol (PEG) chains on particles, often referred to as PEGylation, were intended to help particles evade the immune system, while also increasing efficiency and reducing side effects of cancer therapeutics74. However, immunotoxicity issues persist in intravenous (as opposed to intramuscular) injections, leading researchers to re-evaluate the therapeutic potential of immune cell‒particle interactions75,76,77.
The tendency of particulate therapies to interact with immune cells can be independently exploited as a therapeutic strategy. For example, in an in vivo model of melanoma, mice treated with poly(lactic-co-glycolic acid) (PLGA) particles loaded with a chemotherapeutic drug (paclitaxel) in combination with the CXCL1 chemokine, had significant reduction in tumour size compared to all control groups78. Interestingly, neutrophil uptake of the particles followed by chemotaxis to the CXCL1-treated tumour was responsible for the decrease in tumour burden, pointing to the potential applicability of particle-based therapeutics in immune-cell-related diseases78. Therefore, particle-based therapeutics have been explored for tissue-localized and systemic treatment of inflammation (Fig. 2 and Table 1).

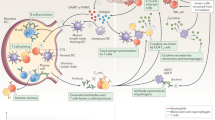
a | Tissue-resident immune cells, such as macrophages, dendritic cells, memory T cells and B cells, are directly targeted through local tissue immunomodulation. Particle administration can passively reprogram inflammatory cells for immunosuppressive purposes. b | Polymeric particles are designed with ligands or antibodies that target leukocyte adhesion molecules overexpressed in inflamed vasculature. The ligand-mediated anchoring of particles to the endothelium permits accumulation of drug carriers to pathological areas, halting inflammation. Targeted particles can also competitively block binding sites used in immune cell migration, preventing immune cell accumulation at sites of inflammation. c | Polymeric particles can divert or reroute circulating blood-resident cells, such as neutrophils and monocytes, from inflammation sites through particle–cell association and cell phenotypic changes after particle uptake. WBC, white blood cell.
Tissue-specific targeting
Local tissue immunomodulation can be achieved by stimulating or activating tissue-resident immune cells, for example, by vaccination79,80,81,82,83,84. However, particle-based vaccine therapeutics can also be designed to target and passively reprogram inflammatory DCs within a draining lymph node, acting as regulatory vaccines for immune suppression in autoimmune diseases81,85,86 (Fig. 2a).
DC-based immunomodulatory therapeutics mostly rely on autologous transplantation of exogenously tolerized DCs, which can be costly and plagued by variability of the transplanted DC populations85. Injecting specialized polymeric particle-based therapeutics circumvents the limitations of autologous transplantation by directly conditioning and reprogramming DC populations within the host’s lymphoid organs87. One approach by which polymeric particles modulate tissue relies on tissue draining; for example, a steroid hormone can be co-delivered with immunosuppressive cytokines loaded into a PLGA nanoparticle for the treatment of rheumatoid arthritis85. This immunosuppressive approach reprogrammed DCs within the draining lymph node, effectively regressing rheumatoid arthritis symptoms in mice85.
Similar particle-based treatments containing immunosuppressive cytokines and autoantigens within PLGA particles were shown to retrain DCs in mouse models of type 1 diabetes and multiple sclerosis, reducing disease presentation and progression86,88,89,90. More specifically, co-delivery of autoantigens and cytokines through a PLGA-based particle therapeutic provided the antigen-specific immune tolerance necessary to reduce disease presentation and progression in a mouse model of multiple sclerosis88. The three main routes for therapeutic particle delivery are intravenous, mucosal and direct tissue injection (subcutaneous or lymphoid) (Fig. 3). Subcutaneously injected particles traffic to lymph nodes through tissue-resident antigen-presenting cells and ultimately induce tolerance by DCs, following natural peripheral tolerance pathways88. Conversely, intravenously injected particles affect tolerance through liver-resident antigen-presenting cells (Kupffer cells) and macrophage scavengers in the spleen88. Similar to subcutaneous injection, particle-based therapeutics directly injected into the lymph node stimulate an increase in the expansion of antigen-specific effector T cells89 (Box 1).

The effectiveness of particle-based therapeutics strongly depends on the route of administration. a | Intravenous routes allow systemic delivery, but pose challenges including quick clearance and complement activation. Grafting of polyethylene glycol (PEGylation) reduces protein corona formation allowing for improved targeting and reduced clearance. b | Mucosal drug delivery allows direct targeting of inflamed tissue. Here, particles are designed to target or travel through mucous membranes of diseased tissue. c | Direct tissue drug delivery can be applied to target lymphoid tissue by intramuscular, subcutaneous or intranodal injections. VTC, vascular-targeted carrier.
Polymeric particles can not only be designed to reprogram immune cells, but can also be exclusively immunosuppressive and directly target sites of inflammation, for example, by being loaded with immunosuppressive drugs81,91,92,93. This approach is beneficial given that most immunosuppressants used to treat inflammatory diseases can oversuppress the immune system, causing several side effects. For example, rapamycin acts as an immunosuppressant, anti-inflammatory and anti-proliferative agent that can lead to diarrhoea, headaches, myelosuppression and hyperlipidaemia93. Encapsulating rapamycin and similar drugs in polymeric particles could prevent undesirable side effects, allowing efficient drug delivery at sites of inflammation in rheumatoid arthritis, multiple sclerosis and graft-versus-host disease93,94.
Particle-based therapeutics with surface decoration of targeting ligands can improve targeting of tissue-specific inflammation sites following intravenous administration. For example, localized infection-related inflammation in the lungs of mice can be treated with intravenously injected PLGA particles that bind specifically to inflamed cells (tumour necrosis factor receptor 1 (TNFR1) on macrophages and intercellular adhesion molecule 1 (ICAM1) on endothelium) at the diseased site95,96,97. To target antigen-presenting cells such as lung-resident macrophages, mesoporous silica nanoparticles can be coated with ligands, including TNFR1, to induce endocytosis96. As the particles degrade, they slowly release immunosuppressant agents to reduce inflammatory signals96. This approach can be used for a range of diseases, including autoimmune and acute inflammatory diseases95,96,98,99.
Vasculature targeting
Vascular endothelial cells are major participants and regulators of inflammatory reactions. During acute inflammation, the endothelium rapidly changes its phenotype to support various stages of the inflammatory response. Primarily, activated endothelial cells facilitate the capture and extravasation of leukocytes to infected or damaged tissue100. During the inflammatory response, leukocyte adhesion molecules are overexpressed at the injured or inflamed endothelium100,101. Thus, particles can be designed to target the remodelled endothelium and underlying signalling pathways, for example, by surface modification with antibodies or cell surface proteins against leukocyte adhesion molecules, including selectins, ICAM1 and vascular cell adhesion molecule (VCAM1), which are known to be involved in leukocyte recognition, adhesion and extravasation100,101.
Vascular-targeted carriers are advantageous for their ability to localize and accumulate at specific disease sites throughout the vasculature, providing controlled release of therapeutics and preventing systemic side effects102,103 (Fig. 2b). For example, PLGA and poly(lactic acid)-poly(ethylene glycol) (PLA-PEG) spheres coated with biotinylated antibodies against the selectins VCAM1 and ICAM1 (refs.104,105) (Fig. 3a) can target inflammation markers and exhibit selective adhesion towards inflamed endothelium both in vitro and in vivo104,105. Likewise, vascular-targeted carriers are often designed with dual adhesion receptors, simulating the multistep adhesion process of WBCs and improving particle adhesion properties106,107. The ability of vascular-targeted carriers to bind to leukocyte adhesion molecules can also be leveraged to competitively block excessive and unregulated immune cell trafficking, as occurs in conditions such as ARDS108. Although such an approach seems counterintuitive, preventing immune cell migration to pathological areas has already shown promising results at preventing tissue damage and accumulation of inflammatory cytokines in preclinical studies in ALI mice109.
Limitations of vascular-targeted particles remain in terms of performance and functionality. In particular, diseased blood conditions and the physical characteristics of particles, such as size and ligand density, can affect their ability to target the inflamed endothelium, ultimately hindering full therapeutic potential110,111,112,113.
Circulating white blood cell targeting
Given that most immune cells or cell precursors involved in pathologic inflammation circulate through the bloodstream, intravenously injected immunotherapeutics are among the most prevalent therapeutic options27,29,39. Polymeric particles can be used to block circulating WBCs from excessive tissue migration during severe inflammation, diverting these inflammatory cells away from the injured tissue109,114,115,116. The primary mechanism by which polymeric particles achieve immunomodulation in the bloodstream is by interaction with WBCs. Phagocytosis of particles by WBCs affects cell physiology, including cytokine release, surface protein expression and gene expression108,114,115,117, which leads to the alteration of cell trafficking and signalling (Fig. 2c). Degradable particles alleviate the possibility of long-term particle accumulation, and their byproducts may further provide anti-inflammatory or therapeutic effects. PLG-based particles for example, degrade into lactate, which reduces the inflammatory signals of DCs and macrophages118, as shown by drug-free PLG- or PLA-based particles in mice models of spinal cord injury and sepsis114,115.
Particles fabricated from high-density lipoprotein-mimicking peptide-phospholipid scaffolds (HPPS) can also be loaded with the anti-inflammatory drug curcumin and designed to specifically target and redirect monocytes116. The latter are targeted by scavenger receptor class B type 1, a receptor that strongly interacts with high-density lipoproteins. In a mouse model of multiple sclerosis, treatment with curcumin-loaded HPPS led to a reduction in monocyte accumulation and morbidity at the injury site, further highlighting the potential of targeting innate immune cells, such as monocytes, to treat inflammatory diseases116. Further optimization of the physicochemical properties of particles, known to affect phagocytosis of WBCs, will be required to ensure recognition of different subsets of immune cells within the blood119,120.
Particle design optimization
For the treatment of inflammatory conditions, the material, size, shape, surface chemistry and deformability of polymeric particles can be modified to ensure efficient interaction with immune cells (Fig. 4).

a | Materials can be designed to be biodegradable, anti-inflammatory, biomimetic or stimuli-responsive. b | Microparticles are phagocytosed and target the vascular wall, whereas nanoparticles permeate the vasculature. c | Polymeric materials can be formulated into a variety of shapes to target specific cell populations. d | Functional groups on polymeric materials can be conjugated with targeting ligands or stealth polymeric chains. In addition, the polymer can be positively or negatively charged. e | Particle rigidity can be modified to selectively target specific cell populations and endothelium in the blood. Soft particles are ideal for marginating along the endothelium in blood flow compared to rigid particles. RBC, red blood cell; PEG, polyethylene glycol; MMP, matrix metalloproteinase.
Material
Material selection affects the release of byproducts and particle accumulation, thereby influencing immune cell modulation and inflammatory signals (Fig. 4a). For example, phagocytosis of non-degradable polystyrene by neutrophils can induce inflammatory neutrophil phenotypes108. By contrast, degradable PLG particles confer anti-inflammatory traits to DCs and monocytes121. Degradable polymeric materials are ideal for particle-based therapeutics of inflammatory diseases, because they are easily modified, optimized, produced in large quantities, and most importantly, do not accumulate in the body.
PLGA has been widely explored for polymeric nanoparticle design in the context of inflammation owing to its ease of synthesis, manipulation and biocompatible degradation products (lactic and glycolic acids) which can be metabolized in vivo122. PLGA has been incorporated into several US Food and Drug Administration (FDA)-approved particle-based therapeutics and is thus considered a safe material123. In small quantities, the primary degradation byproduct of PLGA, lactic acid, has anti-inflammatory properties on both macrophages and DCs114,121. Depending on the molecular weight and thus degradation rate of the specific PLGA polymer, these anti-inflammatory properties may vary121. For example, low-molecular-weight PLGA (10 kDa) that degrades quickly will have inherently anti-inflammatory properties, whereas a high-molecular-weight PLGA polymeric particle will take longer to degrade and may have initial inflammatory properties prior to degradation121,124,125. However, PLGA particles can be used to deliver anti-inflammatory therapeutics such as nonsteroidal anti-inflammatory drugs (NSAIDs) to further reduce inflammation and to overcome any immediate side effects of PLGA degradation126.
In treating neurological inflammation, particles comprised of polymerized phosphatidylserine, a marker for apoptotic cells, reduced inflammation of activated microglial cells and macrophages in vitro127. Although in vitro studies of these biomimetic particles have proved promising, in vivo experiments have only been completed in a myocardial infarction mouse model using phosphatidylserine loaded liposomes, resulting in improved angiogenesis and scar formation128.
Polymeric particles can also be fabricated from polymerized anti-inflammatory compounds. Degradable polymers can be functionalized with a range of anti-inflammatory agents, including aspirin, naproxen and ibuprofen129,130. The resulting compound can then be synthesized into a particle by single oil–water emulsion117,131. Intravenous injection of the resulting polymerized salicylic acid (PolyAspirin) particles have shown to alleviate lung inflammation in an endotoxin and a bacterial mouse model of ALI and ARDS, respectively117. PolyAspirin particles more efficiently diverted neutrophils from the inflamed lungs and further reduced inflammatory cytokines compared to non-treated polystyrene and PLGA particles117. One suggested mechanism is that interactions between neutrophils and PolyAspirin particles prevent the initial accumulation of neutrophils in the lungs, and the degradation of PolyAspirin may ameliorate inflammation117.
Degradable polymeric particles can also be designed to degrade at the site of inflammation, typically within the inflamed tissue space, by incorporating stimuli-responsive properties. Site-dependent degradation at low pH or through cleavage by matrix metalloproteinases (MMPs) can be implemented in particles, allowing the stimulus-triggered release of incorporated anti-inflammatory drugs132. Furthermore, vanillyl alcohol, an antioxidant and anti-inflammatory agent, can be incorporated into copolyoxalate through hydrogen-peroxide-sensitive peroxalate ester bonds, resulting in a polymer that can easily be formulated into a particle-based therapeutic125. The particle then degrades through hydrogen peroxide scavenging when exposed to nitric oxide, a molecule heavily expressed by the endothelium during inflammation125. Similarly, naproxen, an anti-inflammatory drug, can be modified with the ROS scavenging linker, phenylboronic acid (PBA), and then conjugated onto dextran133. The modified PBA–dextran can then be formulated into nanoparticles together with a pH-sensitive acetylated dextran, resulting in significantly reduced cytokine release from stimulated macrophages in vitro133. Crosslinked poly-amino acid-conjugated polyethylene glycol (PAAP) is another example of a degradable polymer that breaks down at low pH and can be loaded with anti-inflammatory proteins, such as DNAse1, to degrade NETs134. ROS-responsive poly(propylene sulfide) (PPS) microparticles loaded with curcumin have shown in vivo efficacy after being intramuscularly injected at the site of ischaemia in a diabetic mouse model135. When exposed to ROS, the PPS particles degraded, leading to ROS scavenging and curcumin release. Here, the curcumin functioned synergistically with PPS particles; however, non-loaded PPS particles also had therapeutic properties, suggesting that ROS scavenging may be enough to reduce inflammation135. Overall, versatile stimuli-responsive materials have great potential for treating inflammatory diseases as well as for immunotherapeutic applications136.
Size
The size customizability of particulates makes them particularly interesting for immunotherapeutics, as they can be designed to mimic pathogens and airborne particles while driving specific immune responses124,137 (Fig. 4b). Upon particle administration, microparticles (≥1 μm) are rapidly cleared through phagocytosis by macrophages, including rat alveolar macrophages, murine peritoneal macrophages and human spleen macrophages138,139,140. By contrast, nanoparticle uptake can occur through multiple mechanisms, including phagocytosis, pinocytosis, caveolae-, clathrin- and scavenger receptor-mediated endocytosis139,140. Nanoparticles have a low risk of capillary occlusion, and they travel passively across permeable vasculature, which is a common feature of inflammation141. The passive transport of nanoparticles makes them ideal candidates for accumulation within the inflamed tissue. For example, PEGylated polyalkylcyanoacrylate nanoparticles injected into an experimental autoimmune encephalomyelitis rat model, can accumulate in the central nervous system142. The passive passage of nanoparticles through the blood–brain barrier is attributed to an increase in cerebrovascular permeability, characteristic of such animal model of brain inflammation. Another example is the spleen, where nanoparticle accumulation is greater for 500 nm and 100 nm than for 20 nm polystyrene particles, as shown in an endotoxin-induced systemic inflammation mouse model143. Optimum particle size is also crucial for particle retention to inflammation sites that do not intend to reach the vasculature or lymphatic system. For example, intra-articular injection of poly(d,l)-lactic (PLA) particles in a mouse model of arthritis showed that 300 nm and 3 μm particles could rapidly diffuse out of the inflamed joint, hindering long-term accumulation and thus, therapeutic benefit144.
Particle accumulation and retention at inflammation sites through vascular immunotargeting is also dependent on the carrier size. For example, vascular-targeted nanoparticles often lack high targeting efficiency owing to poor vascular wall localization110,111,112,113,145. Although micrometre-sized particles can exhibit higher vascular targeting, they are more susceptible to immune cell uptake and dangerous capillary occlusion compared to nanoparticles110,145,146.
Particle association with inflammatory cells could also provide therapeutic benefits for unresolved inflammatory conditions through immune cell rerouting109. For intravenously injected therapies, particle–cell association can be improved by exploring microspheres as therapeutic carriers. Micrometre-sized carriers display enhanced migration out of the red blood cell core in blood flow, consequently co-localizing these particles with leukocytes that are also enriched near the blood vessel wall110,112. Polymeric microparticles successfully modulated inflammation through cell–particle interaction in multiple inflammatory mouse models109,147; daily intravenous injection of drug-free polymeric microparticles target inflammatory monocytes in the circulation and redirect their migration out of the injured site147. Microparticles can further prevent neutrophil adhesion to inflamed tissue in vitro, where selectin-targeted polystyrene (≥2 μm) particles reduced neutrophil adhesion to activated human umbilical vein endothelial cells (HUVECs) more efficiently compared to nanoparticles at equal concentration108. Therefore, therapeutic use of particles requires an optimum size range design, depending on the type of inflammatory condition, desired immune response and route of particle administration.
Shape
Phagocytic cells internalize pathogens and airborne particles of various size and shape (Fig. 4c). Despite considerable progress in understanding the mechanisms of cellular recognition of conventional spherical carriers, our knowledge about the effect of the carrier’s shape on phagocytosis and the subsequent immune response remains limited. Early reports describe that the particle axis of elongated polystyrene particles modulates the mechanism of macrophage uptake148. Macrophages proceed with phagocytosis only if the first point of contact is at the minor axis (that is, the smaller side) of elongated particles. Such axis-dependent uptake was attributed to actin remodelling, which is necessary for engulfing particles, indicating that the minor axis of elongated particles favoured actin cup formation rather than cell spreading on the particles148. Thus, owing to the high energy requirement for actin remodelling, high-aspect-ratio carriers such as elongated particles show reduced phagocytosis by macrophages compared to spherical carriers, ultimately increasing carrier residence time at sites of particle delivery149,150,151.
When macrophages are exploited as therapeutic targets, other particle shapes can be explored; for example, low-aspect-ratio spherical and disk-shaped polystyrene particles are phagocytosed by macrophages at a faster rate than are elongated particles152.
Unlike macrophages, both primary human and mouse neutrophils preferentially internalize rod-shaped particles over spherically shaped ones. Here, the selective particle-neutrophil uptake is independent of material type, and increasing aspect ratios of the particles increase phagocytosis120. The observed higher internalization of rod-shaped particles by neutrophils was associated with possible neutrophil-specific phagocytic mechanisms120, probably linked to the role of neutrophils as the primary human defence against bacterial infection, many of whom have elongated shapes. Such a shape-dependent internalization is an excellent opportunity to engineer particle-based therapies for neutrophilic inflammatory disorders.
The advantages of non-spherical polymeric carriers for anti-inflammatory therapies go beyond their morphology-dependent and cell-type-specific uptake. In particular, rod-shaped and disk-shaped particles demonstrate greater particle margination within the blood compared to spherical carriers153,154,155. These geometries partially counteract hydrodynamic forces in the bloodstream, enabling a large contact area between carriers and cells, thus being attractive for vascular-targeted drug delivery. For example, rod-shaped and spherical polystyrene particles coated with anti-VCAM1 were designed to evaluate the effect of particle shape on binding affinity. Targeted elongated particles showed greater targeting efficiency than targeted spheres in inflamed brain endothelial cells in vitro156. ICAM1-targeted rod-shaped polystyrene nanoparticles also showed preferential accumulation in the endothelium of the brain and lungs of healthy mice compared to targeted spherical particles, providing opportunities to enhance selective organ targeting using shape effects157.
Benefits of non-spherical particles for inflammatory therapies also include overcoming cardiopulmonary reactions caused by both complement activation and responsive macrophages. Unlike with spherical particles, rod-shaped and disk-shaped polystyrene particles did not induce cardiopulmonary distress post-intravenous injection in pigs158. Hence, shape modification of polymeric particles is an appealing strategy to leverage particle-based therapies for desired immune responses, and to circumvent adverse effects of such treatments.
Surface modifications
Surface chemistry and coatings of particulate systems substantially affect the carrier’s interaction with immune cells, including particle clearance and therapeutic effect. Intravenously injected particles are inevitably tagged by plasma proteins that form a protein corona. Particle parameters such as surface charge and hydrophobicity have an essential role in protein corona formation and composition, which dictates subsequent cellular interactions (Fig. 4d). In general, hydrophobic particles showcase higher protein absorption than hydrophilic ones. Likewise, surface charge affects the level of absorption of plasma proteins. For example, increasing the negative surface charge of polystyrene nanoparticles boosts protein absorption, but not protein corona species159. The composition of protein corona can vary among particles with different levels of surface hydrophobicity and cationic or anionic surface charges, ultimately governing uptake by phagocytes160,161. Typically, proteins adsorbed onto particles behave as opsonins, enhancing particle internalization161.
Rapid particle clearance is a major challenge for designing particle-based immunotherapies that aim to reach the vascular wall or inflamed or damaged tissue. The cellular uptake of carriers can be mitigated by modifying their surface with a hydrophilic polymer, such as PEG161,162,163. PEGylated particle formulations can evade uptake by immune cells, extending their blood circulation time161,162,163 (Fig. 3a). Besides intravenous delivery, PEGylated particles also showcase longer residence time through other routes of administration, such as the pulmonary route. Pulmonary delivery of non-spherical polymeric hydrogels functionalized with PEG coatings reduced mouse alveolar macrophage uptake in vitro and in vivo. Accordingly, PEGylated particles showed increased retention in the lungs and minimal inflammatory response for at least a month164.
Although PEG is often recognized as immunologically safe and allergies caused by this compound are rare, some PEGylated drug formulations can trigger complement activation and, in a small portion of patients, lead to severe anaphylaxis165,166,167,168,169. Additionally, the widespread use of PEGylation in pharmaceutical research has led to the discovery of PEG-specific antibodies that compromise its potential efficacy67,170. For example, increased clearance of PEGylated particles from blood circulation in mouse and rats have been reported after repeated doses, particularly for liposome carriers67,170,171,172. Thus, PEG alternatives are being explored, including biodegradable polymers such as poly(glutamic acid) (PGA) and ionic liquid coatings173,174.
The immune evasive effects of particle PEGylation have mainly been explored for macrophage and monocyte uptake and less for neutrophils, despite their important role in immune response modulation and pathogen removal163,175. Surprisingly, PEGylation of carriers had the opposite effect on particle uptake in human blood; PEGylated polystyrene or PLGA particles showed increased uptake by human neutrophils compared to their non-PEGylated counterparts175. It was determined that factors present in the human plasma contribute to the lost immune evasive properties of PEGylated particles175.
Surface modification further includes decorating particles with vascular-targeted ligands to localize and accumulate carriers at sites of inflammation or injury. Defining the optimal ligand surface density is essential to prevent suboptimal targeting of the endothelium or nonspecific targeting effects. In general, high ligand density on the particle surface increases the probability of encountering the specific binding partners and reduces particle-detaching forces owing to increased multivalent interactions107,176,177. However, excessive ligand density may inhibit optimal carrier binding to target cells owing to antibody steric hindrances or overcrowding.
Optimal ligand surface density is also crucial in controlling selective binding of vascular-targeted carriers to pathological vasculature while minimizing binding to healthy tissue sites107. For example, carriers targeted with excessively high anti-ICAM1 surface density face a high off-target risk owing to the ubiquitous basal expression of ICAM1 on the vascular wall in healthy tissues102. In a mouse model of ALI, low density of ICAM1 on poly(4-vinylphenol) (PVPh) nanoparticles increased selective binding of these nanoparticles to inflamed pulmonary tissue relative to healthy vasculature177. By contrast, high ICAM1 density resulted in nanoparticle binding to both healthy and injured endothelium177. In vitro, in vivo and in silico binding assays showed that a low ligand density minimizes binding to areas with low receptor expression but maximizes binding to surfaces with highly expressed receptors107. In the case of inflammation, receptor expression increases at the vascular wall, improving the likelihood that particles decorated with a low density of ligands finding the receptors to enable adhesive and multivalent interaction107.
In summary, the examples above illustrate the heterogeneous nature of particle surface chemistry on broad aspects of blood circulation and particle uptake. Specifically, the differences observed in immune cell populations affect the design of particle-based immunotherapies. Likewise, it showcases the importance of designing safe formulations to minimize exacerbation of inflammatory and allergic responses (Table 2).
Deformability
Particle deformability provides a tuneable factor in particle-based therapeutic design. By adjusting the polymer content in the polymer precursor solution or the functionality of the polymer building block, the degree of crosslinking can be modulated178. Therefore, the particle’s elasticity and flexibility can be tuned to improve leukocyte avoidance, vascular localization, vascular navigation and biodegradation178,179,180. Thus, recent work has sought to understand the role of particle deformability in designing particle-based drug carriers.
Studies investigating the effect of particle elasticity with regards to drug carrier design work across a range of moduli, typically from around 10 kPa up to approximately 1,000 kPa (ref.178). Within this range, a softer particle benefits from a longer circulation time, thereby avoiding clearance by leukocytes. Conversely, comparatively stiffer particles exhibit a shorter circulation time and increased phagocytosis181,182,183. For example, stiff (3,000 kPa) PEG-based nanoparticles are engulfed at a faster rate by J774 macrophages compared to softer (10 kPa) ones in vitro. Soft particles also have a higher persistence in the blood for up to four hours, after which this difference is substantially reduced181. Similarly, micrometre-sized, rigid polyacrylamide beads having a threefold-higher modulus have a greater propensity of being phagocytosed by bone-marrow-derived macrophages in vitro compared to softer beads182. Bone-marrow-derived monocytes exhibit similar behaviour, with up to threefold-reduced uptake of soft (1.3 kPa) disk-shaped particles compared to their rigid counterparts (15 kPa)183. These studies suggest that deformability is an important parameter to consider, especially in avoiding leukocyte clearance for vascular-targeted approaches to immunomodulation.
In addition to leukocyte–particle interactions, the role of elasticity in modulating particle accumulation at specific sites has been explored179,180. Soft PEG-based particles (20–100 kPa) outperform stiffer particles (300–500 kPa) in an in vitro blood flow system under various shear rates; at low rates (500 s−1 or less), softer particles adhere to the endothelium at the same or greater rate compared to their rigid counterparts179,180. This trend is reversed at high-shear (1,000 s−1 or greater) conditions179. Additionally, soft hydrogel microparticles can shuttle nanoparticles to the vascular wall184; for example, intravenous delivery of nanoparticles to the endothelium of mice can be enhanced by loading them into deformable microparticles184. Deformable particles in particular are better suited for immunomodulatory approaches that do not rely on cellular uptake for activity, especially in the case of loading and delivering smaller particles to a site of inflammation such as in vascular-targeted approaches.
Deformable particles can also be used to mimic cells, such as platelets, for therapeutic applications. For example, despite showing great potential for treating coagulopathic diseases (in which clotting does not occur fast enough)185,186, platelet transfusions can still result in immunogenic side effects187. Poly(N-isopropylacrylamide-co-acrylic-acid) microgel particles (1 µm) conjugated to a fibrin antibody can mimic the size, morphology and fibrin binding of platelets187. These platelet-like particles increase clot formation and stability in traumatic brain injuries, preventing post-traumatic neuroinflammation. Additionally, they have a longer shelf life compared to natural platelets, with potential applicability for treating other haemorrhagic bleeding disorders that lead to downstream inflammation187.
Deformability mainly influences particle circulation and uptake; however, it could also be tuned to achieve stimuli-responsive properties. For example, deformable materials designed to degrade at specific sites with low pH or high MMP concentrations enable selective protein or drug release188.
Outlook
Polymeric particle-based therapeutics are extremely versatile and are therefore an excellent tool for designing treatment strategies against inflammatory diseases. By designing the material, size and shape of particles, sites and cell subtypes can be specifically targeted to distinct inflammatory diseases. Particle-based therapeutics have substantially improved the clinical efficacy of a variety of therapies, including therapies for endometriosis, cancer, growth failure, gum disease and mood disorders123. Despite a range of clinically available polymeric-particle-based therapeutics and the plethora of literature on the topic, only 12 PLGA particle-based formulations have been approved by the FDA over the past 30 years123,189. This stark contrast between research and clinical approval stems mainly from translational inconsistencies between animal models and humans.
Although our understanding of inflammatory pathways is constantly evolving, the exact relation between particle design and subsequent inflammatory responses remains to be investigated. For example, the immune cells work in concert with complement pathways to respond to all invading foreign materials, including particles designed as immunotherapeutics24. Despite this known involvement, in vitro assays are limited owing to the difficulties of recapitulating inflammatory signalling pathways in a test tube24. Additionally, complement reactions vary across species, making it challenging to develop reliable in vivo assays for clinical translation24. Pigs have high CARPA reactions to particle-based therapeutics and have become an expensive but reliable model for a variety of applications24,190. Slower infusion rates, coupled with optimized surface properties, can help to prevent CARPA reactions190. However, screening assays need to be developed to investigated how to prevent CARPA reactions.
The design of new particle-based therapeutics further poses challenges in terms of regulatory approval. Unlike systemic, carrier-free therapeutics, particle-based medicines are typically composed of polymeric vehicles, therapeutic agents and surface modifications191,192,193. A slight change in any of these components can considerably alter particle function, biodistribution and toxicity, which makes regulatory evaluation challenging. For this reason, the National Cancer Institute instigated the establishment of the Nanotechnology Characterization Laboratory (NCL) to develop standardized assays to characterize particle-based therapeutics and related toxicities191. The NCL was established to streamline the clinical trial and FDA approval process; however, these procedures are designed for cancer therapeutics and not for generalized therapies191,192,193. Importantly, NCL guidelines, such as prolonged evasion of the mononuclear phagocyte system, inherently exclude particle-based therapeutics that are designed to target circulating phagocytes193. Despite these translational hurdles, the consistent progress made by the scientific community and a streamlined approval process could revolutionize particle-based therapeutics.
Particle technologies are practical solutions for several severe inflammatory diseases. The customizable nature and the physical and chemical attributes of particles fit the demand for innovative clinical applications, including the treatment of system-wide inflammation and vaccine development64,117,194,195,196. The synthesis of polymers can now be fine-tuned; however, the clinical translation of polymeric-based particles remains limited owing to a lack of scaling-up technologies for the fabrication of non-spherical particles in large quantities. The physicochemical parameters governing laboratory-batch particle fabrication are often very complex, and so large-scale production workflows need to be developed. Fortunately, promising large-scale processes are currently being explored for complex-shaped particle fabrication, including lithography-based and microfluidics technologies197,198. Although more work is needed to overcome these obstacles, the rapid expansion of particle-based medicine can offer state-of-the-art solutions to global problems199.
References
Chen, G. Y. & Nuñez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013).
Navegantes, K. C. et al. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Transl Med. 15, 36 (2017).
Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 8, S3 (2006).
Lawrence, T. & Gilroy, D. W. Chronic inflammation: a failure of resolution? Int. J. Exp. Pathol. 88, 85–94 (2007).
Su, Y., Gao, J., Kaur, P. & Wang, Z. Neutrophils and macrophages as targets for development of nanotherapeutics in inflammatory diseases. Pharmaceutics 12, 1222 (2020).
Amarante-Mendes, G. P. et al. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 9, 2379 (2018).
Mogensen, T. H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273 (2009).
Koltsova, E. K. et al. Dynamic T cell–APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Invest. 122, 3114–3126 (2012).
Castellheim, A., Brekke, O. L., Espevik, T., Harboe, M. & Mollnes, T. E. Innate immune responses to danger signals in systemic inflammatory response syndrome and sepsis. Scand. J. Immunol. 69, 479–491 (2009).
George-Gay, B. & Parker, K. Understanding the complete blood count with differential. J. Perianesth. Nurs. 18, 96–117 (2003).
Basit, A. et al. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L200–L207 (2006).
Ivetic, A., Hoskins Green, H. L. & Hart, S. J. L-selectin: a major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 10, 1068 (2019).
Davenpeck, K. L., Brummet, M. E., Hudson, S. A., Mayer, R. J. & Bochner, B. S. Activation of human leukocytes reduces surface P-selectin glycoprotein ligand-1 (PSGL-1, CD162) and adhesion to P-selectin in vitro. J. Immunol. 165, 2764 (2000).
Li, Y. et al. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal. 17, 147 (2019).
Doran, A. C., Yurdagul, A. & Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 20, 254–267 (2020).
Mangalmurti, N. & Hunter, C. A. Cytokine storms: understanding COVID-19. Immunity 53, 19–25 (2020).
Song, P., Li, W., Xie, J., Hou, Y. & You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 509, 280–287 (2020).
Fathi, N. & Rezaei, N. Lymphopenia in COVID-19: therapeutic opportunities. Cell Biol. Int. 44, 1792–1797 (2020).
Nabil, A. et al. Current coronavirus (SARS-CoV-2) epidemiological, diagnostic and therapeutic approaches: an updated review until June 2020. EXCLI J. 19, 992–1016 (2020).
Brown, K. A. et al. Neutrophils in development of multiple organ failure in sepsis. Lancet 368, 157–169 (2006).
Gralinski, L. E. et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9, e01753-18 (2018).
Szebeni, J. Complement activation-related pseudoallergy: a stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 61, 163–173 (2014).
Maisha, N., Coombs, T. & Lavik, E. Development of a sensitive assay to screen nanoparticles in vitro for complement activation. ACS Biomater. Sci. Eng. 6, 4903–4915 (2020).
Barnes, P. J. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br. J. Pharmacol. 148, 245–254 (2006).
Li, P., Zheng, Y. & Chen, X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to anti-TNF biologics. Front. Pharmacol. 8, 460 (2017).
Tabas, I. & Glass, C. K. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339, 166–172 (2013).
Vane, J. R. & Botting, R. M. Anti-inflammatory drugs and their mechanism of action. Inflamm. Res. 47 (Suppl. 2), S78–S87 (1998).
Bjarnason, I., Hayllar, J., MacPherson, A. J. & Russell, A. S. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 104, 1832–1847 (1993).
Bongartz, T. et al. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295, 2275–2285 (2006).
Borman, Z. A., Côté-Daigneault, J. & Colombel, J. F. The risk for opportunistic infections in inflammatory bowel disease with biologics: an update. Expert Rev. Gastroenterol. Hepatol. 12, 1101–1108 (2018).
Canalis, E. Mechanisms of glucocorticoid-induced osteoporosis. Curr. Opin. Rheumatol. 15, 454–457 (2003).
Gabriel, S. E., Jaakkimainen, L. & Bombardier, C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs: a meta-analysis. Ann. Intern. Med. 115, 787–796 (1991).
Howard, P. A. & Delafontaine, P. Nonsteroidal anti-inflammatory drugs and cardiovascular risk. J. Am. Coll. Cardiol. 43, 519–525 (2004).
Bao, Z., Ye, Q., Gong, W., Xiang, Y. & Wan, H. Humanized monoclonal antibody against the chemokine CXCL-8 (IL-8) effectively prevents acute lung injury. Int. Immunopharmacol. 10, 259–263 (2010).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Leckie, M. J. et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356, 2144–2148 (2000).
Shenkar, R., Coulson, W. F. & Abraham, E. Anti-transforming growth factor-beta monoclonal antibodies prevent lung injury in hemorrhaged mice. Am. J. Respir. Cell Mol. Biol. 11, 351–357 (1994).
Taylor, P. C. & Feldmann, M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 578–582 (2009).
Sargent, A. & Miller, R. H. MSC therapeutics in chronic inflammation. Curr. Stem Cell Rep. 2, 168–173 (2016).
Jiang, W. & Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 53, e12712 (2020).
Chaudhry, A. et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578 (2011).
Augello, A. et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 35, 1482–1490 (2005).
Davies, L. C., Heldring, N., Kadri, N. & Le Blanc, K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cell 35, 766–776 (2017).
Wang, L.-T. et al. Human mesenchymal stem cells (MSCs) for treatment towards immune- and inflammation-mediated diseases: review of current clinical trials. J. Biomed. Sci. 23, 76 (2016).
Zhang, Q. et al. Case report: human umbilical cord mesenchymal stem cells as a therapeutic intervention for a critically ill COVID-19 patient. Front. Med. 8, 691329 (2021).
Leng, Z. et al. Transplantation of ACE2– mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis. 11, 216–228 (2020).
Golchin, A., Seyedjafari, E. & Ardeshirylajimi, A. Mesenchymal stem cell therapy for COVID-19: present or future. Stem Cell Rev. Rep. 16, 427–433 (2020).
Regmi, S., Pathak, S., Kim, J. O., Yong, C. S. & Jeong, J.-H. Mesenchymal stem cell therapy for the treatment of inflammatory diseases: challenges, opportunities, and future perspectives. Eur. J. Cell Biol. 98, 151041 (2019).
Zhang, J. et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res. Ther. 6, 234 (2015).
Phinney, D. G. & Pittenger, M. F. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cell 35, 851–858 (2017).
Rider, P., Carmi, Y. & Cohen, I. Biologics for targeting inflammatory cytokines, clinical uses, and limitations. Int. J. Cell Biol. 2016, 9259646 (2016).
Vilcek, J. & Feldmann, M. Historical review: cytokines as therapeutics and targets of therapeutics. Trends Pharmacol. Sci. 25, 201–209 (2004).
Weinblatt, M. E. et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 48, 35–45 (2003).
Mackay, C. R. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat. Immunol. 9, 988–998 (2008).
Rossi, B. & Constantin, G. Anti-selectin therapy for the treatment of inflammatory diseases. Inflamm. Allergy Drug Targets 7, 85–93 (2008).
Horiuchi, T. & Tsukamoto, H. Complement-targeted therapy: development of C5- and C5a-targeted inhibition. Inflamm. Regen. 36, 11 (2016).
Preshaw, P. M. Host modulation therapy with anti-inflammatory agents. Periodontol. 2000 76, 131–149 (2018).
Taylor, R. P. & Lindorfer, M. A. Drug insight: the mechanism of action of rituximab in autoimmune disease–the immune complex decoy hypothesis. Nat. Clin. Pract. Rheumatol. 3, 86–95 (2007).
Park, S. C. & Jeen, Y. T. Anti-integrin therapy for inflammatory bowel disease. World J. Gastroenterol. 24, 1868–1880 (2018).
Jayne, D. R. W. et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J. Am. Soc. Nephrol. 28, 2756–2767 (2017).
De Bosscher, K., Haegeman, G. & Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 10, 497–504 (2010).
Mandavilli, A. Coronavirus can set off a ‘cytokine storm.’ These drugs may calm it. New York Times https://www.nytimes.com/2020/06/11/health/coronavirus-cytokine-storm.html (2020).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Grippin, A. J., Sayour, E. J. & Mitchell, D. A. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology 6, e1290036 (2017).
Liu, J., Miao, L., Sui, J., Hao, Y. & Huang, G. Nanoparticle cancer vaccines: design considerations and recent advances. Asian J. Pharm. Sci. 15, 576–590 (2020).
Ishida, T. et al. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 105, 305–317 (2005).
Naseri, N., Valizadeh, H. & Zakeri-Milani, P. Solid lipid nanoparticles and nanostructured lipid carriers: structure, preparation and application. Adv. Pharm. Bull. 5, 305–313 (2015).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
Song, R. et al. Current development of biodegradable polymeric materials for biomedical applications. Drug Des. Devel Ther. 12, 3117–3145 (2018).
Ikoba, U. et al. Nanocarriers in therapy of infectious and inflammatory diseases. Nanoscale 7, 4291–4305 (2015).
Eloy, J. O. et al. Liposomes as carriers of hydrophilic small molecule drugs: strategies to enhance encapsulation and delivery. Colloids Surf. B 123, 345–363 (2014).
Hannon, G., Lysaght, J., Liptrott, N. J. & Prina-Mello, A. Immunotoxicity considerations for next generation cancer nanomedicines. Adv. Sci. 6, 1900133 (2019).
Cagel, M., Grotz, E., Bernabeu, E., Moretton, M. A. & Chiappetta, D. A. Doxorubicin: nanotechnological overviews from bench to bedside. Drug Discov. Today 22, 270–281 (2017).
Nardo, D., Henson, D., Springer, J. E. & Venditto, V. J. in Nanomaterials for Clinical Applications (eds Pippa, N. & Demetzos, C.) 159–211 (Elsevier, 2020).
Kierstead, P. H. et al. The effect of polymer backbone chemistry on the induction of the accelerated blood clearance in polymer modified liposomes. J. Control. Release 213, 1–9 (2015).
Laverman, P. et al. Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection. J. Pharmacol. Exp. Ther. 298, 607 (2001).
Hao, J. et al. Neutrophils, as “Trojan horses”, participate in the delivery of therapeutical PLGA nanoparticles into a tumor based on the chemotactic effect. Drug Deliv. 27, 1–14 (2020).
Gu, P. et al. Rational design of PLGA nanoparticle vaccine delivery systems to improve immune responses. Mol. Pharm. 16, 5000–5012 (2019).
Bailey, B. A., Ochyl, L. J., Schwendeman, S. P. & Moon, J. J. Toward a single-dose vaccination strategy with self-encapsulating PLGA microspheres. Adv. Healthc. Mater. 6, 1601418 (2017).
Cifuentes-Rius, A., Desai, A., Yuen, D., Johnston, A. P. R. & Voelcker, N. H. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat. Nanotechnol. 16, 37–46 (2021).
Hamdy, S. et al. Enhanced antigen-specific primary CD4+ and CD8+ responses by codelivery of ovalbumin and Toll-like receptor ligand monophosphoryl lipid A in poly(d,l-lactic-co-glycolic acid) nanoparticles. J. Biomed. Mater. Res. Part A 81, 652–662 (2007).
Son, S. et al. Sugar-nanocapsules imprinted with microbial molecular patterns for mRNA vaccination. Nano Lett. 20, 1499–1509 (2020).
Zhu, J. et al. Mannose-modified PLGA nanoparticles for sustained and targeted delivery in hepatitis b virus immunoprophylaxis. AAPS PharmSciTech 21, 13 (2019).
Allen, R., Chizari, S., Ma, J. A., Raychaudhuri, S. & Lewis, J. S. Combinatorial, microparticle-based delivery of immune modulators reprograms the dendritic cell phenotype and promotes remission of collagen-induced arthritis in mice. ACS Appl. Bio Mater. 2, 2388–2404 (2019).
Lewis, J. S. et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model. ACS Biomater. Sci. Eng. 5, 2631–2646 (2019).
Schudel, A., Francis, D. M. & Thomas, S. N. Material design for lymph node drug delivery. Nat. Rev. Mater. 4, 415–428 (2019).
Casey, L. M. et al. Conjugation of transforming growth factor beta to antigen-loaded poly(lactide-co-glycolide) nanoparticles enhances efficiency of antigen-specific tolerance. Bioconjug. Chem. 29, 813–823 (2018).
Tostanoski, LisaH. et al. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep. 16, 2940–2952 (2016).
Lewis, J. S. et al. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin. Immunol. 160, 90–102 (2015).
Chen, X. & Gao, C. Influences of surface coating of PLGA nanoparticles on immune activation of macrophages. J. Mater. Chem. B 6, 2065–2077 (2018).
Huang, Y.-J., Hung, K.-C., Hung, H.-S. & Hsu, S.-h. Modulation of macrophage phenotype by biodegradable polyurethane nanoparticles: possible relation between macrophage polarization and immune response of nanoparticles. ACS Appl. Mater. Interf. 10, 19436–19448 (2018).
Kauffman, K. J. et al. Optimization of rapamycin-loaded acetalated dextran microparticles for immunosuppression. Int. J. Pharm. 422, 356–363 (2012).
Dhanabalan, K. M., Gupta, V. K. & Agarwal, R. Rapamycin–PLGA microparticles prevent senescence, sustain cartilage matrix production under stress and exhibit prolonged retention in mouse joints. Biomater. Sci. 8, 4308–4321 (2020).
Yang, Y. et al. Inflammation-targeting polymeric nanoparticles deliver sparfloxacin and tacrolimus for combating acute lung sepsis. J. Control. Release 321, 463–474 (2020).
García-Fernández, A. et al. Targeted-lung delivery of dexamethasone using gated mesoporous silica nanoparticles: a new therapeutic approach for acute lung injury treatment. J. Control. Release 337, 14–26 (2021).
Zhang, C. Y. et al. pH-responsive nanoparticles targeted to lungs for improved therapy of acute lung inflammation/injury. ACS Appl. Mater. Interf. 11, 16380–16390 (2019).
Zhao, J. et al. Multifunctional folate receptor-targeting and pH-responsive nanocarriers loaded with methotrexate for treatment of rheumatoid arthritis. Int. J. Nanomed. 12, 6735–6746 (2017).
Zhang, N., Chittasupho, C., Duangrat, C., Siahaan, T. J. & Berkland, C. PLGA nanoparticle−peptide conjugate effectively targets intercellular cell-adhesion molecule-1. Bioconjug. Chem. 19, 145–152 (2008).
Pober, J. S. & Sessa, W. C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 (2007).
Ley, K., Laudanna, C., Cybulsky, M. I. & Nourshargh, S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 (2007).
Howard, M. et al. Vascular targeting of nanocarriers: perplexing aspects of the seemingly straightforward paradigm. ACS Nano 8, 4100–4132 (2014).
Jin, K., Luo, Z., Zhang, B. & Pang, Z. Biomimetic nanoparticles for inflammation targeting. Acta Pharm. Sin. B 8, 23–33 (2018).
Eniola, A. O. A. O. & Hammer, D. A. D. A. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: effect of degradation on targeting activity. Biomaterials 26, 661–670 (2005).
Sakhalkar, H. S. et al. Leukocyte-inspired biodegradable particles that selectively and avidly adhere to inflamed endothelium in vitro and in vivo. Proc. Natl Acad. Sci. USA 100, 15895–15900 (2003).
Omolola Eniola, A. & Hammer, D. A. In vitro characterization of leukocyte mimetic for targeting therapeutics to the endothelium using two receptors. Biomaterials 26, 7136–7144 (2005).
Fromen, C. A. et al. Evaluation of receptor-ligand mechanisms of dual-targeted particles to an inflamed endothelium. Bioeng. Transl. Med. 1, 103–115 (2016).
Kelley, W. J., Onyskiw, P. J., Fromen, C. A. & Eniola-Adefeso, O. Model particulate drug carriers modulate leukocyte adhesion in human blood flows. ACS Biomater. Sci. Eng. 5, 6530–6540 (2019).
Fromen, C. A. et al. Neutrophil–particle interactions in blood circulation drive particle clearance and alter neutrophil responses in acute inflammation. ACS Nano 11, 10797–10807 (2017).
Charoenphol, P., Huang, R. B. & Eniola-Adefeso, O. Potential role of size and hemodynamics in the efficacy of vascular-targeted spherical drug carriers. Biomaterials 31, 1392–1402 (2010).
Gutierrez, M., Ojeda, L. S. & Eniola-Adefeso, O. Vascular-targeted particle binding efficacy in the presence of rigid red blood cells: implications for performance in diseased blood. Biomicrofluidics 12, 042217 (2018).
Müller, K., Fedosov, D. A. & Gompper, G. Margination of micro- and nano-particles in blood flow and its effect on drug delivery. Sci. Rep. 4, 4871 (2014).
Thompson, A. J. & Eniola-Adefeso, O. Dense nanoparticles exhibit enhanced vascular wall targeting over neutrally buoyant nanoparticles in human blood flow. Acta Biomater. 21, 99–108 (2015).
Casey, L. M. et al. Cargo-less nanoparticles program innate immune cell responses to Toll-like receptor activation. Biomaterials 218, 119333 (2019).
Park, J. et al. Intravascular innate immune cells reprogrammed via intravenous nanoparticles to promote functional recovery after spinal cord injury. Proc. Natl Acad. Sci. USA 116, 14947 (2019).
Lu, L. et al. Targeted immunomodulation of inflammatory monocytes across the blood-brain barrier by curcumin-loaded nanoparticles delays the progression of experimental autoimmune encephalomyelitis. Biomaterials 245, 119987 (2020).
Brannon, E. R. et al. Poly-salicylic acid polymer microparticle decoys therapeutically treat acute respiratory distress syndrome. Adv. Healthc. Mater. 11, 2101534 (2022).
Sangsuwan, R. et al. Lactate exposure promotes immunosuppressive phenotypes in innate immune cells. Cell. Mol. Bioeng. 13, 541–557 (2020).
Bisso, P. W., Gaglione, S., Guimarães, P. P. G., Mitchell, M. J. & Langer, R. Nanomaterial interactions with human neutrophils. ACS Biomater. Sci. Eng. 4, 4255–4265 (2018).
Safari, H. et al. Neutrophils preferentially phagocytose elongated particles — an opportunity for selective targeting in acute inflammatory diseases. Sci. Adv. 6, eaba1474 (2020).
Allen, R. P., Bolandparvaz, A., Ma, J. A., Manickam, V. A. & Lewis, J. S. Latent, immunosuppressive nature of poly(lactic-co-glycolic acid) microparticles. ACS Biomater. Sci. Eng. 4, 900–918 (2018).
Danhier, F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release 161, 505–522 (2012).
Park, K. et al. Injectable, long-acting PLGA formulations: analyzing PLGA and understanding microparticle formation. J. Control. Release 304, 125–134 (2019).
Nicolete, R., dos Santos, D. F. & Faccioli, L. H. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int. Immunopharmacol. 11, 1557–1563 (2011).
Jeong, D. et al. Porous antioxidant polymer microparticles as therapeutic systems for the airway inflammatory diseases. J. Control. Release 233, 72–80 (2016).
Baek, J.-S., Yeo, E. W., Lee, Y. H., Tan, N. S. & Loo, S. C. J. Controlled-release nanoencapsulating microcapsules to combat inflammatory diseases. Drug Des. Dev. Ther. 11, 1707–1717 (2017).
Nakagawa, Y. et al. Apoptotic cell-inspired polymeric particles for controlling microglial inflammation toward neurodegenerative disease treatment. ACS Biomater. Sci. Eng. 5, 5705–5713 (2019).
Harel-Adar, T. et al. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl Acad. Sci. USA 108, 1827 (2011).
Erdmann, L. & Uhrich, K. E. Synthesis and degradation characteristics of salicylic acid-derived poly(anhydride-esters). Biomaterials 21, 1941–1946 (2000).
Rosario-Meléndez, R., Yu, W. & Uhrich, K. E. Biodegradable polyesters containing ibuprofen and naproxen as pendant groups. Biomacromolecules 14, 3542–3548 (2013).
Rosario-Meléndez, R., Ouimet, M. A. & Uhrich, K. E. Formulation of salicylate-based poly(anhydride-ester) microspheres for short- and long-term salicylic acid delivery. Polym. Bull. 70, 343–351 (2013).
He, L. et al. Dual-stimuli responsive polymeric micelles for the effective treatment of rheumatoid arthritis. ACS Appl. Mater. Interf. 13, 21076–21086 (2021).
Lee, S., Stubelius, A., Hamelmann, N., Tran, V. & Almutairi, A. Inflammation-responsive drug-conjugated dextran nanoparticles enhance anti-inflammatory drug efficacy. ACS Appl. Mater. Interf. 10, 40378–40387 (2018).
Zhu, L. et al. Dynamically deformable protein delivery strategy disassembles neutrophil extracellular traps to prevent liver metastasis. Adv. Funct. Mater. 31, 2105089 (2021).
Poole, K. M. et al. ROS-responsive microspheres for on demand antioxidant therapy in a model of diabetic peripheral arterial disease. Biomaterials 41, 166–175 (2015).
Pacifici, N., Bolandparvaz, A. & Lewis, J. S. Stimuli-responsive biomaterials for vaccines and immunotherapeutic applications. Adv. Ther. 3, 2000129 (2020).
Malachowski, T. & Hassel, A. Engineering nanoparticles to overcome immunological barriers for enhanced drug delivery. Eng. Regen. 1, 35–50 (2020).
Champion, J. A., Walker, A. & Mitragotri, S. Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res. 25, 1815–1821 (2008).
Kuhn, D. A. et al. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 5, 1625–1636 (2014).
Pratten, M. K. & Lloyd, J. B. Pinocytosis and phagocytosis: the effect of size of a particulate substrate on its mode of capture by rat peritoneal macrophages cultured in vitro. Biochim. Biophys. Acta Gen. Subj. 881, 307–313 (1986).
Brusini, R., Varna, M. & Couvreur, P. Advanced nanomedicines for the treatment of inflammatory diseases. Adv. Drug Deliv. Rev. 157, 161–178 (2020).
Calvo, P. et al. Quantification and localization of PEGylated polycyanoacrylate nanoparticles in brain and spinal cord during experimental allergic encephalomyelitis in the rat. Eur. J. Neurosci. 15, 1317–1326 (2002).
Chen, K. H. et al. Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale 7, 15863–15872 (2015).
Pradal, J. et al. Effect of particle size on the biodistribution of nano- and microparticles following intra-articular injection in mice. Int. J. Pharm. 498, 119–129 (2016).
Namdee, K., Thompson, A. J., Charoenphol, P. & Eniola-Adefeso, O. Margination propensity of vascular-targeted spheres from blood flow in a microfluidic model of human microvessels. Langmuir 29, 2530–2535 (2013).
Huang, R. B., Mocherla, S., Heslinga, M. J., Charoenphol, P. & Eniola-Adefeso, O. Dynamic and cellular interactions of nanoparticles in vascular-targeted drug delivery (review). Mol. Membr. Biol. 27, 190–205 (2010).
Getts, D. R. et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci. Transl Med. 6, 219ra217 (2014).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl Acad. Sci. USA 103, 4930 (2006).
Champion, J. A. & Mitragotri, S. Shape induced inhibition of phagocytosis of polymer particles. Pharm. Res. 26, 244–249 (2009).
Yoo, J. W., Chambers, E. & Mitragotri, S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr. Pharm. Des. 16, 2298–2307 (2010).
Dasgupta, S., Auth, T. & Gompper, G. Shape and orientation matter for the cellular uptake of nonspherical particles. Nano Lett. 14, 687–693 (2014).
Sharma, G. et al. Polymer particle shape independently influences binding and internalization by macrophages. J. Control. Release 147, 408–412 (2010).
Cooley, M. et al. Influence of particle size and shape on their margination and wall-adhesion: implications in drug delivery vehicle design across nano-to-micro scale. Nanoscale 10, 15350–15364 (2018).
Namdee, K. et al. In vivo evaluation of vascular-targeted spheroidal microparticles for imaging and drug delivery application in atherosclerosis. Atherosclerosis 237, 279–286 (2014).
Thompson, A. J., Mastria, E. M. & Eniola-Adefeso, O. The margination propensity of ellipsoidal micro/nanoparticles to the endothelium in human blood flow. Biomaterials 34, 5863–5871 (2013).
Da Silva-Candal, A. et al. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 309, 94–105 (2019).
Kolhar, P. et al. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proc. Natl Acad. Sci. USA 110, 10753 (2013).
Wibroe, P. P. et al. Bypassing adverse injection reactions to nanoparticles through shape modification and attachment to erythrocytes. Nat. Nanotechnol. 12, 589–594 (2017).
Gessner, A., Lieske, A., Paulke, B. & Müller, R. Influence of surface charge density on protein adsorption on polymeric nanoparticles: analysis by two-dimensional electrophoresis. Eur. J. Pharm. Biopharm. 54, 165–170 (2002).
Pustulka, S. M., Ling, K., Pish, S. L. & Champion, J. A. Protein nanoparticle charge and hydrophobicity govern protein corona and macrophage uptake. ACS Appl. Mater. Interf. 12, 48284–48295 (2020).
Walkey, C. D., Olsen, J. B., Guo, H., Emili, A. & Chan, W. C. W. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 134, 2139–2147 (2012).
Perry, J. L. et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 12, 5304–5310 (2012).
Yang, Q. et al. Evading immune cell uptake and clearance requires PEG grafting at densities substantially exceeding the minimum for brush conformation. Mol. Pharm. 11, 1250–1258 (2014).
Shen, T. W. et al. Distribution and cellular uptake of PEGylated polymeric particles in the lung towards cell-specific targeted delivery. Pharm. Res. 32, 3248–3260 (2015).
Kozma, G. T. et al. Pseudo-anaphylaxis to polyethylene glycol (PEG)-coated liposomes: roles of anti-PEG IgM and complement activation in a porcine model of human infusion reactions. ACS Nano 13, 9315–9324 (2019).
Moghimi, S. M. et al. Complement activation cascade triggered by PEG–PL engineered nanomedicines and carbon nanotubes: the challenges ahead. J. Control. Release 146, 175–181 (2010).
Ganson, N. J. et al. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J. Allergy Clin. Immunol. 137, 1610–1613.e7 (2016).
Sellaturay, P., Nasser, S. & Ewan, P. Polyethylene glycol-induced systemic allergic reactions (anaphylaxis). J. Allergy Clin. Immunol. Pract. 9, 670–675 (2021).
Sellaturay, P., Nasser, S., Islam, S., Gurugama, P. & Ewan, P. W. Polyethylene glycol (PEG) is a cause of anaphylaxis to the Pfizer/BioNTech mRNA COVID-19 vaccine. Clin. Exp. Allergy 51, 861–863 (2021).
Suk, J. S., Xu, Q., Kim, N., Hanes, J. & Ensign, L. M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 99, 28–51 (2016).
Dams, E. T. et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 292, 1071–1079 (2000).
Ishida, T. et al. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 112, 15–25 (2006).
Knop, K., Hoogenboom, R., Fischer, D. & Schubert, U. S. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 49, 6288–6308 (2010).
Hamadani Christine, M., Goetz Morgan, J., Mitragotri, S. & Tanner Eden, E. L. Protein-avoidant ionic liquid (PAIL)–coated nanoparticles to increase bloodstream circulation and drive biodistribution. Sci. Adv. 6, eabd7563 (2020).
Kelley, W. J., Fromen, C. A., Lopez-Cazares, G. & Eniola-Adefeso, O. PEGylation of model drug carriers enhances phagocytosis by primary human neutrophils. Acta Biomater. 79, 283–293 (2018).
Haun, J. B. & Hammer, D. A. Quantifying nanoparticle adhesion mediated by specific molecular interactions. Langmuir 24, 8821–8832 (2008).
Zern, B. J. et al. Reduction of nanoparticle avidity enhances the selectivity of vascular targeting and PET detection of pulmonary inflammation. ACS Nano 7, 2461–2469 (2013).
Anselmo, A. C. & Mitragotri, S. Impact of particle elasticity on particle-based drug delivery systems. Adv. Drug Deliv. Rev. 108, 51–67 (2017).
Fish, M. B. et al. Exploring deformable particles in vascular-targeted drug delivery: softer is only sometimes better. Biomaterials 124, 169–179 (2017).
Guo, P. et al. Nanoparticle elasticity directs tumor uptake. Nat. Commun. 9, 130 (2018).
Anselmo, A. C. et al. Elasticity of nanoparticles influences their blood circulation, phagocytosis, endocytosis, and targeting. ACS Nano 9, 3169–3177 (2015).
Beningo, K. A. & Wang, Y. L. Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J. Cell Sci. 115, 849–856 (2002).
Key, J. et al. Soft discoidal polymeric nanoconstructs resist macrophage uptake and enhance vascular targeting in tumors. ACS Nano 9, 11628–11641 (2015).
Fish, M. B. et al. Deformable microparticles for shuttling nanoparticles to the vascular wall. Sci. Adv. 7, eabe0143 (2021).
Bachelani, A. M. et al. Assessment of platelet transfusion for reversal of aspirin after traumatic brain injury. Surgery 150, 836–843 (2011).
Huijben, J. A. et al. Variation in blood transfusion and coagulation management in traumatic brain injury at the intensive care unit: a survey in 66 neurotrauma centers participating in the Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury Study. J. Neurotrauma 35, 323–332 (2018).
Todd, J. et al. Platelet-like particles reduce coagulopathy-related and neuroinflammatory pathologies post-experimental traumatic brain injury. J. Biomed. Mater. Res. B 109, 2268–2278 (2021).
Shah, A. et al. Stimuli-responsive peptide-based biomaterials as drug delivery systems. Chem. Eng. J. 353, 559–583 (2018).
Lee, B. K., Yun, Y. & Park, K. PLA micro- and nano-particles. Adv. Drug Deliv. Rev. 107, 176–191 (2016).
Onwukwe, C. et al. Engineering intravenously administered nanoparticles to reduce infusion reaction and stop bleeding in a large animal model of trauma. Bioconjug. Chem. 29, 2436–2447 (2018).
Alper, J. US NCI launches nanotechnology plan. Nat. Biotechnol. 22, 1335–1336 (2004).
Crist, R. M. et al. Common pitfalls in nanotechnology: lessons learned from NCI’s nanotechnology characterization laboratory. Integr. Biol. 5, 66–73 (2013).
Dawidczyk, C. M. et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Control. Release 187, 133–144 (2014).
Yuk Simseok, A. et al. Nanocapsules modify membrane interaction of polymyxin B to enable safe systemic therapy of Gram-negative sepsis. Sci. Adv. 7, eabj1577 (2021).
Gao, W. & Zhang, L. Nanomaterials arising amid antibiotic resistance. Nat. Rev. Microbiol. 19, 5–6 (2021).
Lee, N.-Y., Ko, W.-C. & Hsueh, P.-R. Nanoparticles in the treatment of infections caused by multidrug-resistant organisms. Front. Pharmacol. 10, 1153 (2019).
Dendukuri, D., Tsoi, K., Hatton, T. A. & Doyle, P. S. Controlled synthesis of nonspherical microparticles using microfluidics. Langmuir 21, 2113–2116 (2005).
Xu, J. et al. Future of the particle replication in nonwetting templates (PRINT) technology. Angew. Chem. Int. Ed. 52, 6580–6589 (2013).
Tammam, S. N. et al. Repurpose but also (nano)-reformulate! The potential role of nanomedicine in the battle against SARS-CoV2. J. Control. Release 337, 258–284 (2021).
Mathaes, R., Winter, G., Siahaan, T. J., Besheer, A. & Engert, J. Influence of particle size, an elongated particle geometry, and adjuvants on dendritic cell activation. Eur. J. Pharm. Biopharm. 94, 542–549 (2015).
Jewell, C. M., Bustamante López, S. C. & Irvine, D. J. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl Acad. Sci. USA 108, 15745 (2011).
Sanchez-Cano, C. & Carril, M. Recent developments in the design of non-biofouling coatings for nanoparticles and surfaces. Int. J. Mol. Sci. 21, 1007 (2020).
Bobo, D., Robinson, K. J., Islam, J., Thurecht, K. J. & Corrie, S. R. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res. 33, 2373–2387 (2016).
Bernkop-Schnürch, A. & Dünnhaupt, S. Chitosan-based drug delivery systems. Eur. J. Pharm. Biopharm. 81, 463–469 (2012).
Fisher, A., Watling, M., Smith, A. & Knight, A. Pharmacokinetic comparisons of three nasal fentanyl formulations; pectin, chitosan and chitosan-poloxamer 188. Int. J. Clin. Pharmacol. Ther. 48, 138–145 (2010).
Jhaveri, J., Raichura, Z., Khan, T., Momin, M. & Omri, A. Chitosan nanoparticles-insight into properties, functionalization and applications in drug delivery and theranostics. Molecules 26, 272 (2021).
Gao, W. & Zhang, L. Coating nanoparticles with cell membranes for targeted drug delivery. J. Drug Target. 23, 619–626 (2015).
de Almeida, T. S., Júlio, A., Mota, J. P., Rijo, P. & Reis, C. P. An emerging integration between ionic liquids and nanotechnology: general uses and future prospects in drug delivery. Ther. Deliv. 8, 461–473 (2017).
Adawiyah, N., Moniruzzaman, M., Hawatulaila, S. & Goto, M. Ionic liquids as a potential tool for drug delivery systems. MedChemComm 7, 1881–1897 (2016).
Johansen, P., Mohanan, D., Martínez-Gómez, J. M., Kündig, T. M. & Gander, B. Lympho-geographical concepts in vaccine delivery. J. Control. Release 148, 56–62 (2010).
Blanco, E., Shen, H. & Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015).
Fam, S. Y. et al. Stealth coating of nanoparticles in drug-delivery systems. Nanomaterials 10, 787 (2020).
Liu, Y., Hardie, J., Zhang, X. & Rotello, V. M. Effects of engineered nanoparticles on the innate immune system. Semin. Immunol. 34, 25–32 (2017).
Rampado, R., Crotti, S., Caliceti, P., Pucciarelli, S. & Agostini, M. Recent advances in understanding the protein corona of nanoparticles and in the formulation of “stealthy” nanomaterials. Front. Bioeng. Biotechnol. 8, 166–166 (2020).
Ilinskaya, A. N. et al. Nanoparticle physicochemical properties determine the activation of intracellular complement. Nanomedicine 17, 266–275 (2019).
Yang, Q. & Lai, S. K. Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 7, 655–677 (2015).
Fahy, J. V. & Dickey, B. F. Airway mucus function and dysfunction. N. Engl. J. Med. 363, 2233–2247 (2010).
Johansson, M. E. Mucus layers in inflammatory bowel disease. Inflamm. Bowel Dis. 20, 2124–2131 (2014).
Lai, S. K., Wang, Y.-Y. & Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 61, 158–171 (2009).
Netsomboon, K. & Bernkop-Schnürch, A. Mucoadhesive vs. mucopenetrating particulate drug delivery. Eur. J. Pharm. Biopharm. 98, 76–89 (2016).
Hua, S., Marks, E., Schneider, J. J. & Keely, S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue. Nanomedicine 11, 1117–1132 (2015).
Maisel, K. et al. Nanoparticles coated with high molecular weight PEG penetrate mucus and provide uniform vaginal and colorectal distribution in vivo. Nanomedicine 11, 1337–1343 (2016).
Huckaby, J. T. & Lai, S. K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 124, 125–139 (2018).
Pereira de Sousa, I. et al. Nanoparticles decorated with proteolytic enzymes, a promising strategy to overcome the mucus barrier. Eur. J. Pharm. Biopharm. 97, 257–264 (2015).
Bonengel, S., Prüfert, F., Perera, G., Schauer, J. & Bernkop-Schnürch, A. Polyethylene imine-6-phosphogluconic acid nanoparticles — a novel zeta potential changing system. Int. J. Pharm. 483, 19–25 (2015).
Youshia, J. & Lamprecht, A. Size-dependent nanoparticulate drug delivery in inflammatory bowel diseases. Expert Opin. Drug Deliv. 13, 281–294 (2016).
Lamprecht, A., Yamamoto, H., Takeuchi, H. & Kawashima, Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J. Pharmacol. Exp. Ther. 315, 196–202 (2005).
Karn, P. R., Vanić, Z., Pepić, I. & Skalko-Basnet, N. Mucoadhesive liposomal delivery systems: the choice of coating material. Drug Dev. Ind. Pharm. 37, 482–488 (2011).
Makhlof, A., Tozuka, Y. & Takeuchi, H. pH-sensitive nanospheres for colon-specific drug delivery in experimentally induced colitis rat model. Eur. J. Pharm. Biopharm. 72, 1–8 (2009).
Mane, V. & Muro, S. Biodistribution and endocytosis of ICAM-1-targeting antibodies versus nanocarriers in the gastrointestinal tract in mice. Int. J. Nanomed. 7, 4223–4237 (2012).
Huff, R. D., Carlsten, C. & Hirota, J. A. An update on immunologic mechanisms in the respiratory mucosa in response to air pollutants. J. Allergy Clin. Immunol. 143, 1989–2001 (2019).
Turner, J. R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
McLennan, D. N., Porter, C. J. H. & Charman, S. A. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol. 2, 89–96 (2005).
Nestle, F. O., Di Meglio, P., Qin, J.-Z. & Nickoloff, B. J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 9, 679–691 (2009).
Randolph, G. J., Angeli, V. & Swartz, M. A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5, 617–628 (2005).
Acknowledgements
This work was supported by the National Science Foundation Graduate Research Fellowship Program (to E.R.B., M.V.G., J.K.L. and N.J.P.) by grant number NIH R01 HL145709 (to O.E.-A.), by grant numbers R01 AI139399 and R35GM125012 (to J.S.L.) and by grant T32GM099608 (to N.J.P.).
Author information
Authors and Affiliations
Contributions
E.R.B. and M.V.G. wrote the abstract, introduction and main sections of the manuscript, Box 1 and Fig. 3. E.R.B. created Table 2, Fig. 1a and Fig. 4, and edited Table 1. M.V.G. created Fig. 2 and edited Table 1. N.J.P. wrote Table 1, created Fig. 1b and edited the manuscript. J.K.L. wrote the deformability section. J.S.L. and O.E.-A. laid the framework for, wrote, edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
O.E.-A. serves as a consultant for Asalyxa Bio, working to develop ‘Polymer Particle for Neutrophil Injury’. O.E.-A. is listed as inventor on a recently filed patent application (US Provisional Application No. 62/870,879). The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Materials thanks Mitsuhiro Ebara and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Nanotechnology Characterization Laboratory: https://www.cancer.gov/nano/research/ncl
National Cancer Institute: https://www.cancer.gov/
Rights and permissions
About this article

Cite this article
Brannon, E.R., Guevara, M.V., Pacifici, N.J. et al. Polymeric particle-based therapies for acute inflammatory diseases. Nat Rev Mater 7, 796–813 (2022). https://doi.org/10.1038/s41578-022-00458-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41578-022-00458-5
This article is cited by
-
The therapeutic potential of immunoengineering for systemic autoimmunity
Nature Reviews Rheumatology (2024)
-
Polyethylene Glycol–Based Polymer-Drug Conjugates: Novel Design and Synthesis Strategies for Enhanced Therapeutic Efficacy and Targeted Drug Delivery
Applied Biochemistry and Biotechnology (2024)
-
Monolithic-to-focal evolving biointerfaces in tissue regeneration and bioelectronics
Nature Chemical Engineering (2024)
-
Glucocorticoid nanoformulations relieve chronic pelvic pain syndrome and may alleviate depression in mice
Journal of Nanobiotechnology (2023)
-
Cargo-free particles divert neutrophil-platelet aggregates to reduce thromboinflammation
Nature Communications (2023)
